
Accelerating the Mdx Heart Histo-Pathology through Physical Exercise

 , , ,
, , , 
Abstract
:1. Introduction
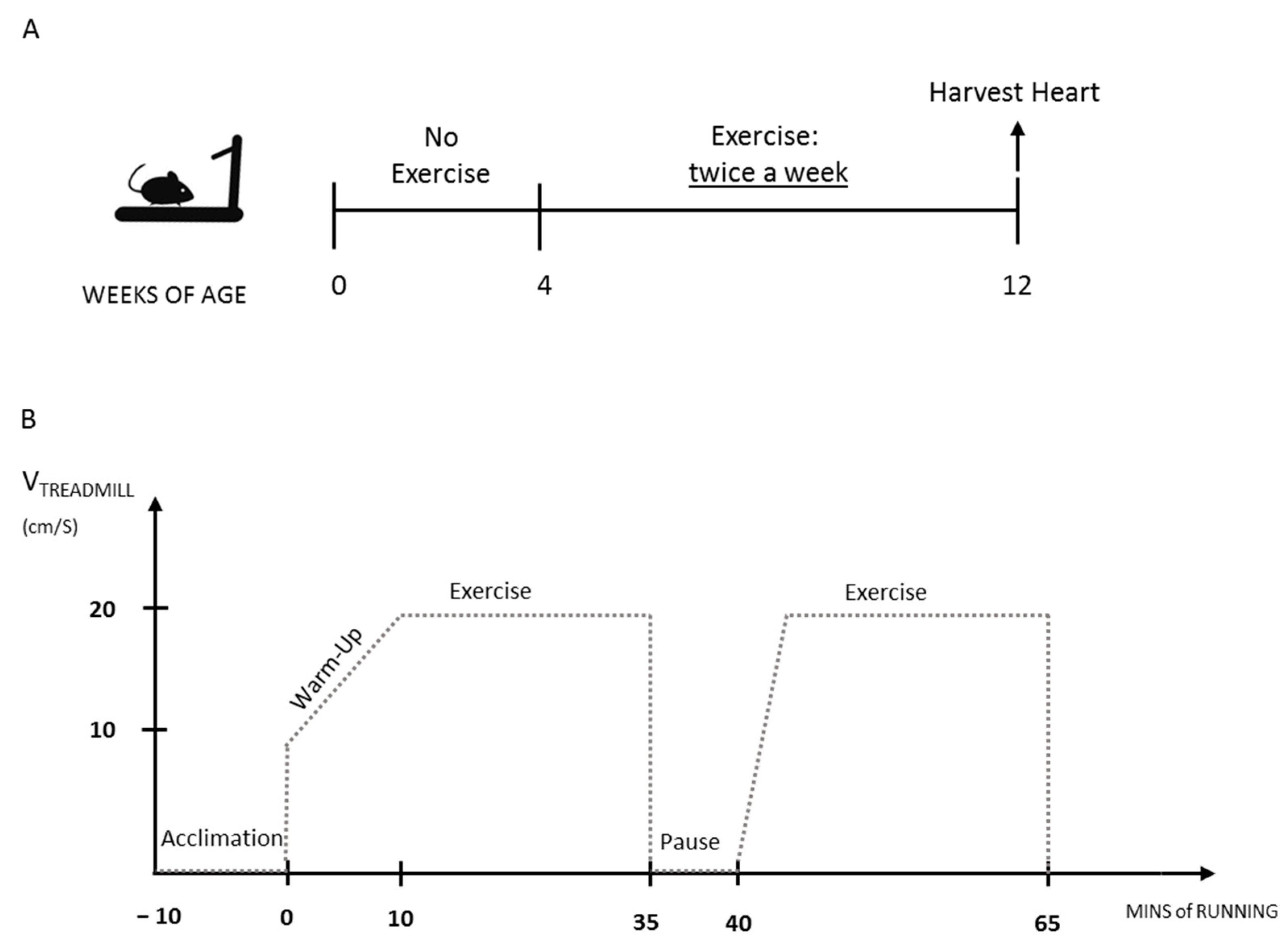
2. Materials and Methods
3. Results
3.1. Mdx Mice Display Late Onset Heart Fibrosis
3.2. The Exercise Protocol Accelerates Fibrosis Onset in Mdx Heart
3.3. The Exercised Mdx Mice Display Increased Cardiomyocyte Necrosis and Cell Infiltration
3.4. Inflammatory Cell Infiltration Is Increased in the Exercised Mdx Heart
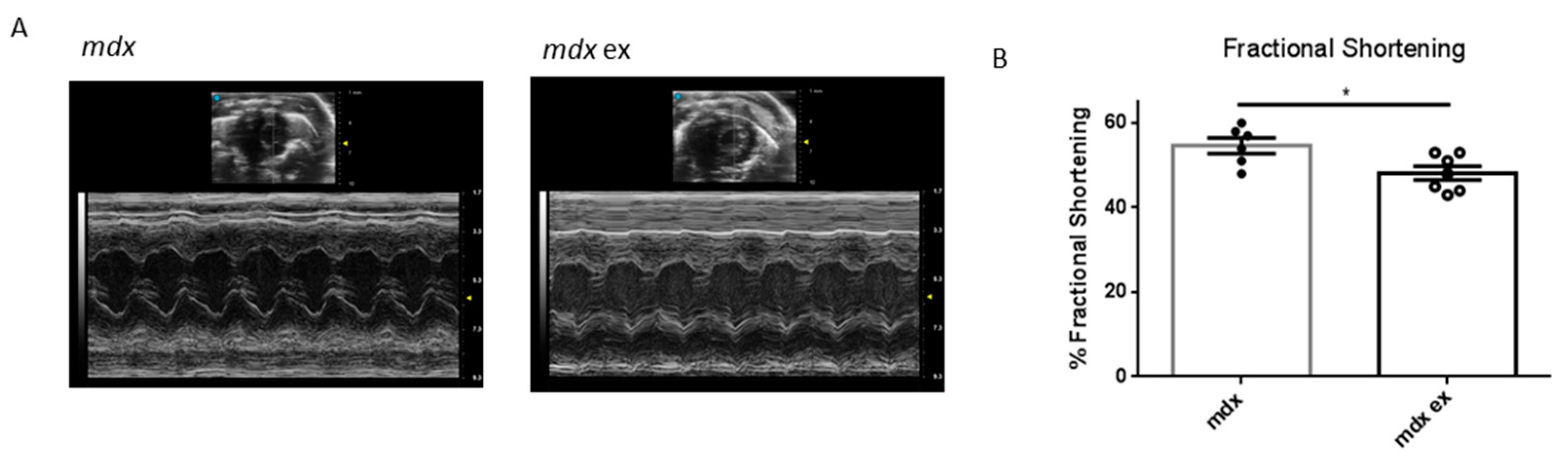
3.5. The Exercise Protocol Alters Mdx Heart Function
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meyers, T.A.; Townsend, D.W. Cardiac pathophysiology and the future of cardiac therapies in duchenne muscular dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [Green Version]
- Mosqueira, M.; Zeiger, U.; Förderer, M.; Brinkmeier, H.; Fink, R.H. Cardiac and respiratory dysfunction in duchenne muscular dystrophy and the role of second messengers. Med. Res. Rev. 2013, 33, 1174–1213. [Google Scholar] [CrossRef]
- D’Amario, D.; Gowran, A.; Canonico, F.; Castiglioni, E.; Rovina, D.; Santoro, R.; Spinelli, P.; Adorisio, R.; Amodeo, A.; Perrucci, G.; et al. Dystrophin Cardiomyopathies: Clinical Management, Molecular Pathogenesis and Evolution towards Precision Medicine. J. Clin. Med. 2018, 7, 291. [Google Scholar] [CrossRef] [Green Version]
- Buddhe, S.; Cripe, L.; Friedland-Little, J.; Kertesz, N.; Eghtesady, P.; Finder, J.; Hor, K.; Judge, D.P.; Kinnett, K.; McNally, E.M.; et al. Cardiac management of the patient with Duchenne muscular dystrophy. Pediatrics 2018, 142, S72–S81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J.I. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. DMM Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.; Faelan, C.; Patterson-Kane, J.C.; Rudmann, D.G.; Moore, S.A.; Frank, D.; Charleston, J.; Tinsley, J.; Young, G.D.; Milici, A.J. Duchenne and Becker Muscular Dystrophies: A Review of Animal Models, Clinical End Points, and Biomarker Quantification. Toxicol. Pathol. 2017, 45, 961–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007, 21, 2195–2204. [Google Scholar] [CrossRef] [Green Version]
- Landfeldt, E.; Thompson, R.; Sejersen, T.; McMillan, H.J.; Kirschner, J.; Lochmüller, H. Life expectancy at birth in Duchenne muscular dystrophy: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Isaac, C.; Wright, A.; Usas, A.; Li, H.; Tang, Y.; Mu, X.; Greco, N.; Dong, Q.; Vo, N.; Kang, J.; et al. Dystrophin and utrophin “double knockout” dystrophic mice exhibit a spectrum of degenerative musculoskeletal abnormalities. J. Orthop. Res. 2013, 31, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, Z.M.; Dorn, L.E.; Lowe, J.; Gertzen, M.D.; Ciccone, P.; Rastogi, N.; Odom, G.L.; Accornero, F.; Chamberlain, J.S.; Rafael-Fortney, J.A. Micro-dystrophin gene therapy prevents heart failure in an improved Duchenne muscular dystrophy cardiomyopathy mouse model. JCI Insight 2021, 6, e146511. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short telomeres and stem cell exhaustion model duchenne muscular dystrophy in mdx/mTR mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selsby, J.T.; Acosta, P.; Sleeper, M.M.; Barton, E.R.; Sweeney, H.L. Long-term wheel running compromises diaphragm function but improves cardiac and plantarflexor function in the mdx mouse. J. Appl. Physiol. 2013, 115, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Baltgalvis, K.A.; Call, J.A.; Cochrane, G.D.; Laker, R.C.; Yan, Z.; Lowe, D.A. Exercise training improves plantar flexor muscle function in mdx Mice. Med. Sci. Sports Exerc. 2012, 44, 1671–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogelman, B.; Putker, K.; Hulsker, M.; Tanganyika-de Winter, C.; van der Weerd, L.; Aartsma-Rus, A.; van Putten, M. Voluntary exercise improves muscle function and does not exacerbate muscle and heart pathology in aged Duchenne muscular dystrophy mice. J. Mol. Cell. Cardiol. 2018, 125, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Hourdé, C.; Joanne, P.; Medja, F.; Mougenot, N.; Jacquet, A.; Mouisel, E.; Pannerec, A.; Hatem, S.; Butler-Browne, G.; Agbulut, O.; et al. Voluntary physical activity protects from susceptibility to skeletal muscle contraction-induced injury but worsens heart function in mdx mice. Am. J. Pathol. 2013, 182, 1509–1518. [Google Scholar] [CrossRef]
- Costas, J.M.; Nye, D.J.; Henley, J.B.; Plochocki, J.H. Voluntary exercise induces structural remodeling in the hearts of dystrophin-deficient mice. Muscle Nerve 2010, 42, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Barbin, I.C.C.; Pereira, J.A.; Bersan Rovere, M.; de Oliveira Moreira, D.; Marques, M.J.; Santo Neto, H. Diaphragm degeneration and cardiac structure in mdx mouse: Potential clinical implications for Duchenne muscular dystrophy. J. Anat. 2016, 228, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdi, R.; Rolland, J.F.; Fraysse, B.; Litvinova, K.; Cozzoli, A.; Giannuzzi, V.; Liantonio, A.; Camerino, G.M.; Sblendorio, V.; Capogrosso, R.F.; et al. Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: Outcome of a large array of in vivo and ex vivo tests. J. Appl. Physiol. 2009, 106, 1311–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanarica, F.; Mantuano, P.; Conte, E.; Cozzoli, A.; Capogrosso, R.F.; Giustino, A.; Cutrignelli, A.; Cappellari, O.; Rolland, J.F.; De Bellis, M.; et al. Proof-of-concept validation of the mechanism of action of Src tyrosine kinase inhibitors in dystrophic mdx mouse muscle: In vivo and in vitro studies. Pharmacol. Res. 2019, 145, 1042600. [Google Scholar] [CrossRef]
- Capogrosso, R.F.; Mantuano, P.; Cozzoli, A.; Sanarica, F.; Massari, A.M.; Conte, E.; Fonzino, A.; Giustino, A.; Rolland, J.F.; Quaranta, A.; et al. Contractile efficiency of dystrophic mdx mouse muscle: In vivo and ex vivo assessment of adaptation to exercise of functional end points. J. Appl. Physiol. 2017, 122, 828–843. [Google Scholar] [CrossRef]
- Bouchentouf, M.; Benabdallah, B.F.; Mills, P.; Tremblay, J.P. Exercise improves the success of myoblast transplantation in mdx mice. Neuromuscul. Disord. 2006, 16, 518–529. [Google Scholar] [CrossRef]
- da Bizario, J.C.S.; Cerri, D.G.; Rodrigues, L.C.; Oliveira, G.L.V.; Nomizo, A.; de Araujo, D.D.; Fukuhara, P.S.; Ribeiro, J.C.; de Castro, F.A.; Costa, M.C.R. Imatinib mesylate ameliorates the dystrophic phenotype in exercised mdx mice. J. Neuroimmunol. 2009, 212, 93–101. [Google Scholar] [CrossRef]
- Hyzewicz, J.; Ruegg, U.T.; Takeda, S. Comparison of Experimental Protocols of Physical Exercise for mdx Mice and Duchenne Muscular Dystrophy Patients. J. Neuromuscul. Dis. 2015, 2, 325–342. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Yoshida, K.; Takeda, S.; Dohi, N.; Ikeda, S.I. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 2002, 520, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Zelikovich, A.S.; Quattrocelli, M.; Salamone, I.M.; Kuntz, N.L.; McNally, E.M. Moderate exercise improves function and increases adiponectin in the mdx mouse model of muscular dystrophy. Sci. Rep. 2019, 9, 5770. [Google Scholar] [CrossRef]
- Taniguti, A.P.T.; Pertille, A.; Matsumura, C.Y.; Neto, H.S.; Marques, M.J. Prevention of muscle fibrosis and myonecrosis in mdxmice by suramin, a TGF-β1 blocker. Muscle Nerve 2011, 43, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Schill, K.E.; Altenberger, A.R.; Lowe, J.; Periasamy, M.; Villamena, F.A.; Rafael-Fortney, J.A.; Devor, S.T. Muscle damage, metabolism, and oxidative stress in mdx mice: Impact of aerobic running. Muscle Nerve 2016, 54, 110–117. [Google Scholar] [CrossRef]
- Frinchi, M.; Morici, G.; Mudó, G.; Bonsignore, M.R.; Di Liberto, V. Beneficial role of exercise in the modulation of mdx muscle plastic remodeling and oxidative stress. Antioxidants 2021, 10, 558. [Google Scholar] [CrossRef]
- Betts, C.A.; Saleh, A.F.; Carr, C.A.; Hammond, S.M.; Coenen-Stass, A.M.L.; Godfrey, C.; McClorey, G.; Varela, M.A.; Roberts, T.C.; Clarke, K.; et al. Prevention of exercised induced cardiomyopathy following Pip-PMO treatment in dystrophic mdx mice. Sci. Rep. 2015, 5, 8986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiore, P.F.; Benedetti, A.; Sandonà, M.; Madaro, L.; De Bardi, M.; Saccone, V.; Puri, P.L.; Gargioli, C.; Lozanoska-Ochser, B.; Bouché, M. Lack of PKCθ promotes regenerative ability of muscle stem cells in chronic muscle injury. Int. J. Mol. Sci. 2020, 21, 932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoletti, R.; Maffei, A.; Madaro, L.; Notte, A.; Stanganello, E.; Cifelli, G.; Carullo, P.; Molinaro, M.; Lembo, G.; Bouché, M. Protein kinase Cθ is required for cardiomyocyte survival and cardiac remodeling. Cell Death Dis. 2010, 1, e45. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Di Maggio, R.; Benedetti, A.; Morroni, J.; Bouche, M.; Lozanoska-Ochser, B. Splenic Ly6Chi monocytes are critical players in dystrophic muscle injury and repair. JCI Insight 2020, 5, e130807. [Google Scholar] [CrossRef] [Green Version]
- Lozanoska-Ochser, B.; Benedetti, A.; Rizzo, G.; Marrocco, V.; Di Maggio, R.; Fiore, P.; Bouche, M. Targeting early PKCθ-dependent T-cell infiltration of dystrophic muscle reduces disease severity in a mouse model of muscular dystrophy. J. Pathol. 2018, 244, 323–333. [Google Scholar] [CrossRef]
- Sciarretta, S.; Yee, D.; Nagarajan, N.; Bianchi, F.; Saito, T.; Valenti, V.; Tong, M.; Del Re, D.P.; Vecchione, C.; Schirone, L.; et al. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 1999–2010. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Maejima, Y.; DelRe, D.P.; Nagarajan, N.; Yee, D.; Liu, T.; Magnuson, M.A.; Volpe, M.; Frati, G.; et al. MTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 2015, 11, 125–136. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A. Use of Treadmill and Wheel Exercise for Impact on Mdx Mice Phenotype SOP; Wellstone Muscular Dystrophy Center: Washington, DC, USA, 2008. [Google Scholar]
- Au, C.G.; Butler, T.L.; Sherwood, M.C.; Egan, J.R.; North, K.N.; Winlaw, D.S. Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. Int. J. Exp. Pathol. 2011, 92, 57–65. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mele, A.; Mantuano, P.; De Bellis, M.; Rana, F.; Sanarica, F.; Conte, E.; Morgese, M.G.; Bove, M.; Rolland, J.F.; Capogrosso, R.F.; et al. A long-term treatment with taurine prevents cardiac dysfunction in mdx mice. Transl. Res. 2019, 204, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Wang, L.; Li, Z.; Yang, R.; Liang, Y.; Sun, Y.; Yu, Q.; Ghartey-Kwansah, G.; Sun, Y.; Wu, Y.; et al. A systematic comparison of exercise training protocols on animal models of cardiovascular capacity. Life Sci. 2019, 217, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Gargiulo, G.; Pironti, G.; Franzone, A.; Scudiero, L.; de Laurentis, M.; Magliulo, F.; Ilardi, F.; Carotenuto, G.; Schiattarella, G.G.; et al. Cardiovascular effects of treadmill exercise in physiological and pathological preclinical settings. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, 1983–1989. [Google Scholar] [CrossRef] [Green Version]
- Kemi, O.J.; Loennechen, J.P.; Wisløff, U.; Ellingsen, Y. Intensity-controlled treadmill running in mice: Cardiac and skeletal muscle hypertrophy. J. Appl. Physiol. 2002, 93, 1301–1309. [Google Scholar] [CrossRef] [Green Version]
- Bellafiore, M.; Battaglia, G.; Bianco, A.; Palma, A. Expression pattern of angiogenic factors in healthy heart in response to physical exercise intensity. Front. Physiol. 2019, 10, 238. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xu, M.; Bryant, J.L.; Ma, J.; Xu, X. Exercise-induced myokine FNDC5/irisin functions in cardiovascular protection and intracerebral retrieval of synaptic plasticity. Cell Biosci. 2019, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxid. Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| End-Diastolic Diameter [mm] | End-Systolic Diameter [mm] | Anterior Wall Thickness [mm] | Posterior Wall Thickness [mm] | Fractional Shortening [%] | Heart Rate (bpm) | Heart/Body Weight [mg/g] | |
|---|---|---|---|---|---|---|---|
| mdx | 2.77 ± 0.07 | 1.27 ± 0.08 | 1.13 ± 0.04 | 1.08 ± 0.02 | * 54.42 ± 1.89 | 452 ± 25.2 | 7.0 ± 0.24 |
| mdx ex | 2.76 ± 0.06 | 1.44 ± 0.03 | 1.06 ± 0.04 | 1.11 ± 0.03 | * 48.14 ± 1.61 | 443 ± 43.8 | 6.8 ± 0.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morroni, J.; Schirone, L.; Vecchio, D.; Nicoletti, C.; D’Ambrosio, L.; Valenti, V.; Sciarretta, S.; Lozanoska-Ochser, B.; Bouchè, M. Accelerating the Mdx Heart Histo-Pathology through Physical Exercise. Life 2021, 11, 706. https://doi.org/10.3390/life11070706
Morroni J, Schirone L, Vecchio D, Nicoletti C, D’Ambrosio L, Valenti V, Sciarretta S, Lozanoska-Ochser B, Bouchè M. Accelerating the Mdx Heart Histo-Pathology through Physical Exercise. Life. 2021; 11(7):706. https://doi.org/10.3390/life11070706
Chicago/Turabian StyleMorroni, Jacopo, Leonardo Schirone, Daniele Vecchio, Carmine Nicoletti, Luca D’Ambrosio, Valentina Valenti, Sebastiano Sciarretta, Biliana Lozanoska-Ochser, and Marina Bouchè. 2021. "Accelerating the Mdx Heart Histo-Pathology through Physical Exercise" Life 11, no. 7: 706. https://doi.org/10.3390/life11070706
APA StyleMorroni, J., Schirone, L., Vecchio, D., Nicoletti, C., D’Ambrosio, L., Valenti, V., Sciarretta, S., Lozanoska-Ochser, B., & Bouchè, M. (2021). Accelerating the Mdx Heart Histo-Pathology through Physical Exercise. Life, 11(7), 706. https://doi.org/10.3390/life11070706