
The Interplay of Mitophagy and Inflammation in Duchenne Muscular Dystrophy


{kind=link}
Abstract
:1. Introduction
1.1. Duchenne Muscular Dystrophy
1.2. Chronic Inflammation in DMD
1.3. Impaired Mitophagy in DMD
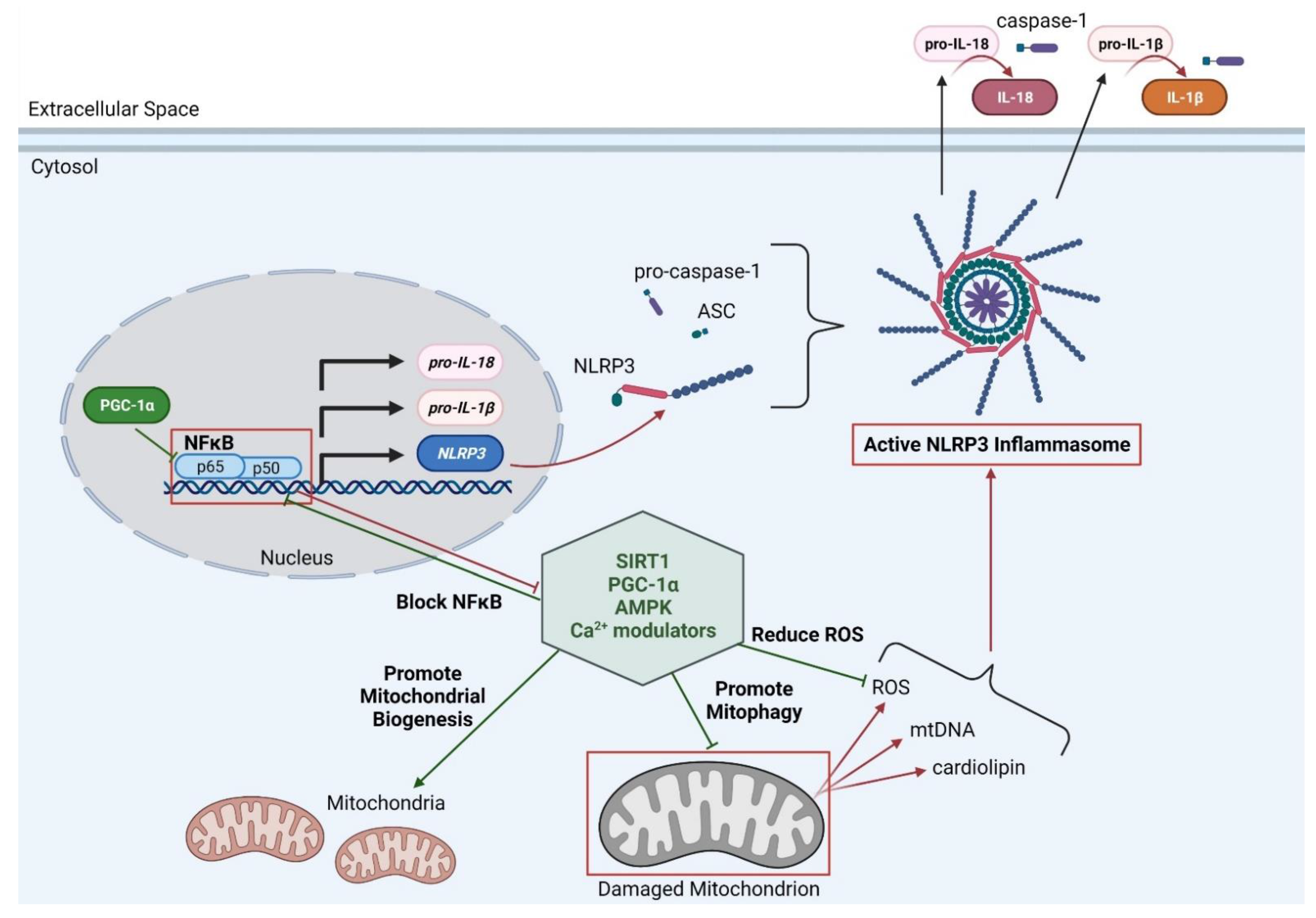
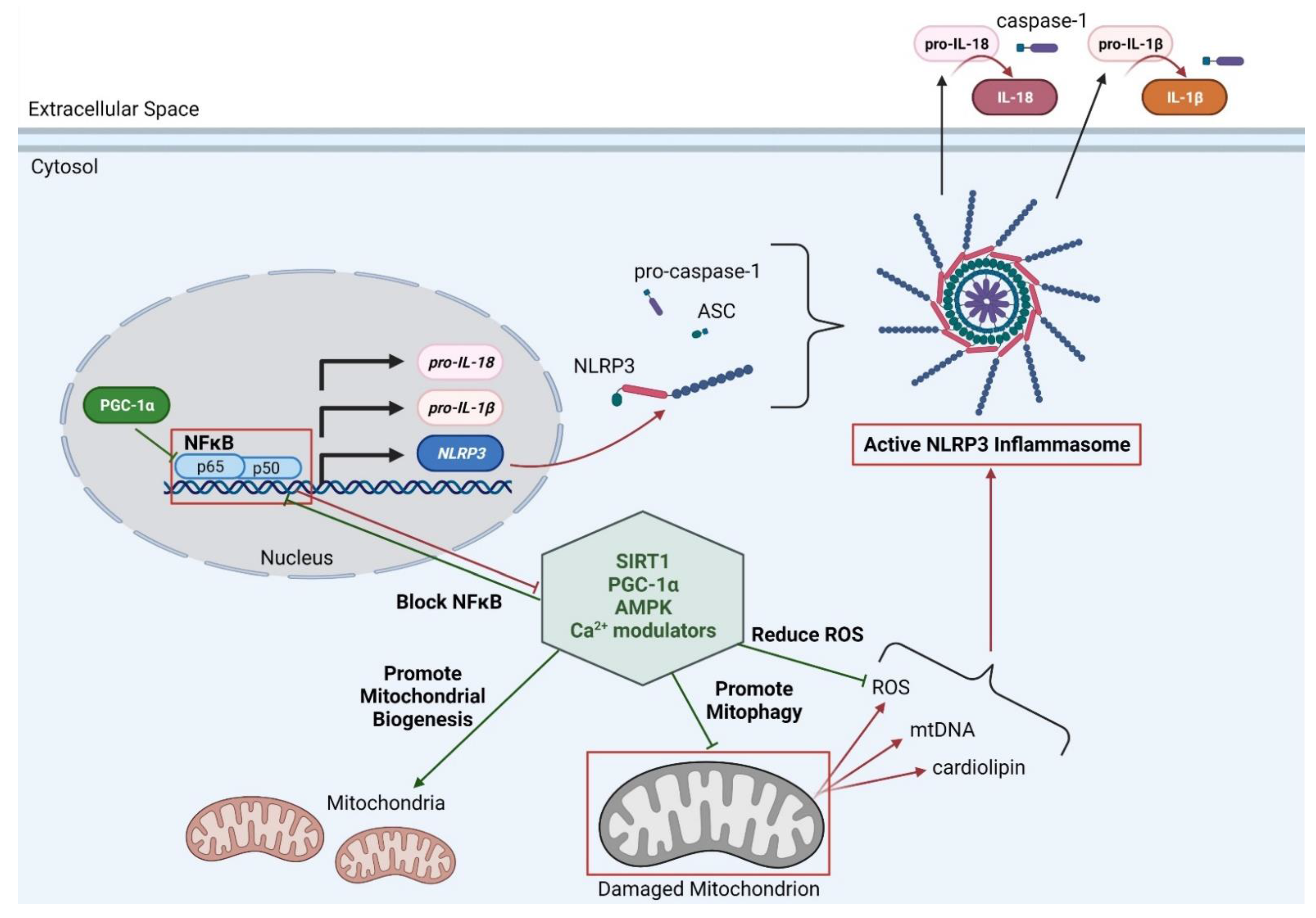
1.4. Mitochondria and Inflammation
1.5. Possible Therapeutic Targets for Improving Mitophagy and Inflammation in DMD
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendell, J.R.; Lloyd-Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013, 48, 21–26. [Google Scholar] [CrossRef]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Monaco, A.P.; Neve, R.L.; Colletti-Feener, C.; Bertelson, C.J.; Kurnit, D.M.; Kunkel, L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Fischbeck, K.H.; Brown, R.H.; Johnson, M.; Medori, R.; Loike, J.D.; Harris, J.B.; Waterston, R.; Brooke, M.; Specht, L.; et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N. Engl. J. Med. 1988, 318, 1363–1368. [Google Scholar] [CrossRef]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic strategies for duchenne muscular dystrophy: An update. Genes 2020, 11, 837. [Google Scholar] [CrossRef]
- Duan, D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther. 2018. [Google Scholar] [CrossRef] [Green Version]
- Duan, D. Micro-dystrophin gene therapy goes systemic in duchenne muscular dystrophy patients. Hum. Gene Ther. 2018, 29, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, S.J.; Nicolau, S.; Connolly, A.M.; Mendell, J.R. Therapeutic approaches for duchenne muscular dystrophy: Old and new. Semin. Pediatr. Neurol. 2021, 37, 100877. [Google Scholar] [CrossRef]
- Chemello, F.; Wang, Z.; Li, H.; McAnally, J.R.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Degenerative and regenerative pathways underlying Duchenne muscular dystrophy revealed by single-nucleus RNA sequencing. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Franke, V.; Brandt, B.; Lowenstein, E.D.; Schöwel, V.; Spuler, S.; Akalin, A.; Birchmeier, C. Single-nucleus transcriptomics reveals functional compartmentalization in syncytial skeletal muscle cells. Nat. Commun. 2020, 11, 6375. [Google Scholar] [CrossRef]
- Rahimov, F.; Kunkel, L.M. Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [Green Version]
- Constantin, B.; Sebille, S.; Cognard, C. New insights in the regulation of calcium transfers by muscle dystrophin-based cytoskeleton: Implications in DMD. J. Muscle Res. Cell Motil. 2006, 27, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Campbell, K.P. Dystrophin-glycoprotein complex: Its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve 1994, 17, 2–15. [Google Scholar] [CrossRef]
- Chen, Y.W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rosenberg, A.S.; Bluestone, J.A. The immune system in Duchenne muscular dystrophy: Friend or foe. Rare Dis. 2015, 3, e1010966. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Manzur, A.Y.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2008, 1. [Google Scholar] [CrossRef]
- Ricotti, V.; Ridout, D.A.; Scott, E.; Quinlivan, R.; Robb, S.A.; Manzur, A.Y.; Muntoni, F. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 2013, 84, 698–705. [Google Scholar] [CrossRef]
- Griggs, R.C.; Miller, J.P.; Greenberg, C.R.; Fehlings, D.L.; Pestronk, A.; Mendell, J.R.; Moxley, R.T.; King, W.; Kissel, J.T.; Cwik, V.; et al. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology 2016, 87, 2123–2131. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, S.J.; Chakkalakal, J.V.; Kolodziejczyk, S.M.; Knudson, J.C.; Jasmin, B.J.; Megeney, L.A. Glucocorticoid treatment alleviates dystrophic myofiber pathology by activation of the calcineurin/NF-AT pathway. FASEB J. 2004, 18, 1937–1939. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Shah, S.N.; Lu, P.; Richardson, S.M.; Bollinger, L.E.; Blaeser, A.; Madden, K.L.; Sun, Y.; Luckie, T.M.; Cox, M.D.; et al. Glucocorticoid steroid and alendronate treatment alleviates dystrophic phenotype with enhanced functional glycosylation of α-dystroglycan in mouse model of limb-girdle muscular dystrophy with FKRPP448L mutation. Am. J. Pathol. 2016, 186, 1635–1648. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Tolmeijer, S.; Oskam, J.M.; Tonkens, T.; Meijer, A.H.; Schaaf, M.J.M. Glucocorticoids inhibit macrophage differentiation towards a pro-inflammatory phenotype upon wounding without affecting their migration. Dis. Models Mech. 2019, dmm.037887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabtree, N.J.; Adams, J.E.; Padidela, R.; Shaw, N.J.; Högler, W.; Roper, H.; Hughes, I.; Daniel, A.; Mughal, M.Z. Growth, bone health & ambulatory status of boys with DMD treated with daily vs. intermittent oral glucocorticoid regimen. Bone 2018, 116, 181–186. [Google Scholar] [CrossRef]
- Rutter, M.M.; Collins, J.; Rose, S.R.; Woo, J.G.; Sucharew, H.; Sawnani, H.; Hor, K.N.; Cripe, L.H.; Wong, B.L. Growth hormone treatment in boys with Duchenne muscular dystrophy and glucocorticoid-induced growth failure. Neuromuscul. Disord. 2012, 22, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Torrisi, J. Anti-TNFα (Remicade®) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 2004, 18, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.L.; Carathers, M.; Li, Z.-W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-κB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogra, C.; Changotra, H.; Wergedal, J.E.; Kumar, A. Regulation of phosphatidylinositol 3-kinase (PI3K)/Akt and nuclear factor-kappa B signaling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J. Cell. Physiol. 2006, 208, 575–585. [Google Scholar] [CrossRef]
- Kumar, A.; Boriek, A.M. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. 2003, 17, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Hightower, R.M.; Reid, A.L.; Gibbs, D.E.; Wang, Y.; Widrick, J.J.; Kunkel, L.M.; Kastenschmidt, J.M.; Villalta, S.A.; van Groen, T.; Chang, H. The SINE Compound KPT-350 Blocks Dystrophic Pathologies in DMD Zebrafish and Mice. Mol. Ther. 2020, 28, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Tang, Y.; Li, J.; Dzuricky, A.T.; Pu, C.; Fu, F.; Wang, B. Genetic ablation of P65 subunit of NF-κB in mdx mice to improve muscle physiological function. Muscle Nerve 2017, 56, 759–767. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small molecule NF-κB pathway inhibitors in clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 inflammasome by adiponectin rescues Duchenne muscular dystrophy. BMC Biol. 2018, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef] [PubMed]
- Luan, P.; D’Amico, D.; Andreux, P.A.; Laurila, P.P.; Wohlwend, M.; Li, H.; Imamura de Lima, T.; Place, N.; Rinsch, C.; Zanou, N.; et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; Van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteom. 2013, 12, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.R.; von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [Green Version]
- Kuno, A.; Hosoda, R.; Sebori, R.; Hayashi, T.; Sakuragi, H.; Tanabe, M.; Horio, Y. Resveratrol ameliorates mitophagy disturbance and improves cardiac pathophysiology of dystrophin-deficient mdx mice. Sci. Rep. 2018, 8, 15555. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebori, R.; Kuno, A.; Hosoda, R.; Hayashi, T.; Horio, Y. Resveratrol decreases oxidative stress by restoring mitophagy and improves the pathophysiology of dystrophin-deficient mdx mice. Oxid. Med. Cell. Longev. 2018, 2018, 9179270. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, N.P.; Yeung, E.W.; Allen, D.G. Muscle damage in mdx (dystrophic) mice: Role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006, 33, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padman, B.S.; Nguyen, T.N.; Uoselis, L.; Skulsuppaisarn, M.; Nguyen, L.K.; Lazarou, M. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun. 2019, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Badr, M.A.; Kyrychenko, V.; Eskelinen, E.L.; Shirokova, N. Deficit in PINK1/PARKIN-mediated mitochondrial autophagy at late stages of dystrophic cardiomyopathy. Cardiovasc. Res. 2018, 114, 90–102. [Google Scholar] [CrossRef]
- Moulin, M.; Ferreiro, A. Muscle redox disturbances and oxidative stress as pathomechanisms and therapeutic targets in early-onset myopathies. Semin. Cell Dev. Biol. 2017, 64, 213–223. [Google Scholar] [CrossRef] [PubMed]
- da Silva, H.N.M.; Covatti, C.; da Rocha, G.L.; Mizobuti, D.S.; Mâncio, R.D.; Hermes, T.A.; Kido, L.A.; Cagnon, V.H.A.; Pereira, E.C.L.; Minatel, E. Oxidative stress, inflammation, and activators of mitochondrial biogenesis: Tempol targets in the diaphragm muscle of exercise trained-mdx mice. Front. Physiol. 2021, 12, 649793. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kwak, H.B.; Thompson, L.V.; Lawler, J.M. Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abou-Samra, M.; Lecompte, S.; Schakman, O.; Noel, L.; Many, M.C.; Gailly, P.; Brichard, S.M. Involvement of adiponectin in the pathogenesis of dystrophinopathy. Skelet Muscle 2015, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angebault, C.; Panel, M.; Lacôte, M.; Rieusset, J.; Lacampagne, A.; Fauconnier, J. Metformin Reverses the Enhanced Myocardial SR/ER-Mitochondria Interaction and Impaired Complex I-Driven Respiration in Dystrophin-Deficient Mice. Front. Cell Dev. Biol. 2020, 8, 609493. [Google Scholar] [CrossRef]
- Pauly, M.; Daussin, F.; Burelle, Y.; Li, T.; Godin, R.; Fauconnier, J.; Koechlin-Ramonatxo, C.; Hugon, G.; Lacampagne, A.; Coisy-Quivy, M.; et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 2012, 181, 583–592. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Igarashi, M.; Nasamu, A.S.; Knezevic, J.; Guarente, L. Muscle-specific SIRT1 gain-of-function increases slow-twitch fibers and ameliorates pathophysiology in a mouse model of duchenne muscular dystrophy. PLoS Genet. 2014, 10, e1004490. [Google Scholar] [CrossRef]
- Ryu, D.; Zhang, H.; Ropelle, E.R.; Sorrentino, V.; Mázala, D.A.; Mouchiroud, L.; Marshall, P.L.; Campbell, M.D.; Ali, A.S.; Knowels, G.M.; et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016, 8, 361ra139. [Google Scholar] [CrossRef] [Green Version]
- Handschin, C.; Kobayashi, Y.M.; Chin, S.; Seale, P.; Campbell, K.P.; Spiegelman, B.M. PGC-1α regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007, 21, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suntar, I.; Sureda, A.; Belwal, T.; Sanches Silva, A.; Vacca, R.A.; Tewari, D.; Sobarzo-Sánchez, E.; Nabavi, S.F.; Shirooie, S.; Dehpour, A.R.; et al. Natural products, PGC-1 α and Duchenne muscular dystrophy. Acta Pharm. Sin. B 2020, 10, 734–745. [Google Scholar] [CrossRef]
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP(3) receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3685–3695. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Mounier, R.; Théret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisele, P.S.; Salatino, S.; Sobek, J.; Hottiger, M.O.; Handschin, C. The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J. Biol. Chem. 2013, 288, 2246–2260. [Google Scholar] [CrossRef] [Green Version]
- Eisele, P.S.; Furrer, R.; Beer, M.; Handschin, C. The PGC-1 coactivators promote an anti-inflammatory environment in skeletal muscle in vivo. Biochem. Biophys. Res. Commun. 2015, 464, 692–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remels, A.H.; Gosker, H.R.; Bakker, J.; Guttridge, D.C.; Schols, A.M.; Langen, R.C. Regulation of skeletal muscle oxidative phenotype by classical NF-κB signalling. Biochim. Biophys. Acta 2013, 1832, 1313–1325. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal 2013, 25, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Laguna, J.C.; Vázquez-Carrera, M. The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res. 2010, 87, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubicic, V.; Miura, P.; Burt, M.; Boudreault, L.; Khogali, S.; Lunde, J.A.; Renaud, J.M.; Jasmin, B.J. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum. Mol. Genet. 2011, 20, 3478–3493. [Google Scholar] [CrossRef] [Green Version]
- Ljubicic, V.; Jasmin, B.J. Metformin increases peroxisome proliferator-activated receptor γ Co-activator-1α and utrophin a expression in dystrophic skeletal muscle. Muscle Nerve 2015, 52, 139–142. [Google Scholar] [CrossRef]
- Ljubicic, V.; Burt, M.; Lunde, J.A.; Jasmin, B.J. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1-PGC-1α axis. Am. J. Physiol. Cell Physiol. 2014, 307, C66–C82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Gordon, B.S.; Delgado Díaz, D.C.; Kostek, M.C. Resveratrol decreases inflammation and increases utrophin gene expression in the mdx mouse model of Duchenne muscular dystrophy. Clin. Nutr. 2013, 32, 104–111. [Google Scholar] [CrossRef]
- Woodman, K.G.; Coles, C.A.; Lamandé, S.R.; White, J.D. Resveratrol Promotes Hypertrophy in Wildtype Skeletal Muscle and Reduces Muscle Necrosis and Gene Expression of Inflammatory Markers in Mdx Mice. Molecules 2021, 26, 853. [Google Scholar] [CrossRef]
- Capogrosso, R.F.; Cozzoli, A.; Mantuano, P.; Camerino, G.M.; Massari, A.M.; Sblendorio, V.T.; De Bellis, M.; Tamma, R.; Giustino, A.; Nico, B.; et al. Assessment of resveratrol, apocynin and taurine on mechanical-metabolic uncoupling and oxidative stress in a mouse model of duchenne muscular dystrophy: A comparison with the gold standard, α-methyl prednisolone. Pharmacol. Res. 2016, 106, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Tonon, E.; Ferretti, R.; Shiratori, J.H.; Santo Neto, H.; Marques, M.J.; Minatel, E. Ascorbic acid protects the diaphragm muscle against myonecrosis in mdx mice. Nutrition 2012, 28, 686–690. [Google Scholar] [CrossRef]
- de Senzi Moraes Pinto, R.; Ferretti, R.; Moraes, L.H.; Neto, H.S.; Marques, M.J.; Minatel, E. N-acetylcysteine treatment reduces TNF-α levels and myonecrosis in diaphragm muscle of mdx mice. Clin. Nutr. 2013, 32, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Mâncio, R.D.; Hermes, T.A.; Macedo, A.B.; Mizobuti, D.S.; Valduga, A.H.; Rupcic, I.F.; Minatel, E. Vitamin E treatment decreases muscle injury in mdx mice. Nutrition 2017, 43-44, 39–46. [Google Scholar] [CrossRef]
- Woodman, K.G.; Coles, C.A.; Lamandé, S.R.; White, J.D. Nutraceuticals and their potential to treat duchenne muscular dystrophy: Separating the credible from the conjecture. Nutrients 2016, 8, 713. [Google Scholar] [CrossRef]
- Ballmann, C.; Denney, T.S.; Beyers, R.J.; Quindry, T.; Romero, M.; Amin, R.; Selsby, J.T.; Quindry, J.C. Lifelong quercetin enrichment and cardioprotection in Mdx/Utrn+/− mice. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H128–H140. [Google Scholar] [CrossRef] [Green Version]
- Burns, D.P.; Ali, I.; Rieux, C.; Healy, J.; Jasionek, G.; O’Halloran, K.D. Tempol supplementation restores diaphragm force and metabolic enzyme activities in mdx mice. Antioxidants 2017, 6, 101. [Google Scholar] [CrossRef] [Green Version]
- Hermes, T.A.; Mâncio, R.D.; Macedo, A.B.; Mizobuti, D.S.; Rocha, G.L.D.; Cagnon, V.H.A.; Minatel, E. Tempol treatment shows phenotype improvement in mdx mice. PLoS ONE 2019, 14, e0215590. [Google Scholar] [CrossRef] [PubMed]
- Hermes, T.A.; Mizobuti, D.S.; da Rocha, G.L.; da Silva, H.N.M.; Covatti, C.; Pereira, E.C.L.; Ferretti, R.; Minatel, E. Tempol improves redox status in mdx dystrophic diaphragm muscle. Int. J. Exp. Pathol. 2020, 101, 289–297. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The effect of deflazacort treatment on the functioning of skeletal muscle mitochondria in duchenne muscular dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Reay, D.P.; Niizawa, G.A.; Watchko, J.F.; Daood, M.; Reay, J.C.; Raggi, E.; Clemens, P.R. Effect of nuclear factor κB inhibition on serotype 9 adeno-associated viral (AAV9) minidystrophin gene transfer to the mdx mouse. Mol. Med. 2012, 18, 466–476. [Google Scholar] [CrossRef]
- Boccanegra, B.; Verhaart, I.E.C.; Cappellari, O.; Vroom, E.; De Luca, A. Safety issues and harmful pharmacological interactions of nutritional supplements in Duchenne muscular dystrophy: Considerations for standard of care and emerging virus outbreaks. Pharmacol. Res. 2020, 158, 104917. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reid, A.L.; Alexander, M.S. The Interplay of Mitophagy and Inflammation in Duchenne Muscular Dystrophy. Life 2021, 11, 648. https://doi.org/10.3390/life11070648
Reid AL, Alexander MS. The Interplay of Mitophagy and Inflammation in Duchenne Muscular Dystrophy. Life. 2021; 11(7):648. https://doi.org/10.3390/life11070648
Chicago/Turabian StyleReid, Andrea L., and Matthew S. Alexander. 2021. "The Interplay of Mitophagy and Inflammation in Duchenne Muscular Dystrophy" Life 11, no. 7: 648. https://doi.org/10.3390/life11070648
APA StyleReid, A. L., & Alexander, M. S. (2021). The Interplay of Mitophagy and Inflammation in Duchenne Muscular Dystrophy. Life, 11(7), 648. https://doi.org/10.3390/life11070648