
The Structure, Activity, and Function of the SETD3 Protein Histidine Methyltransferase


Abstract
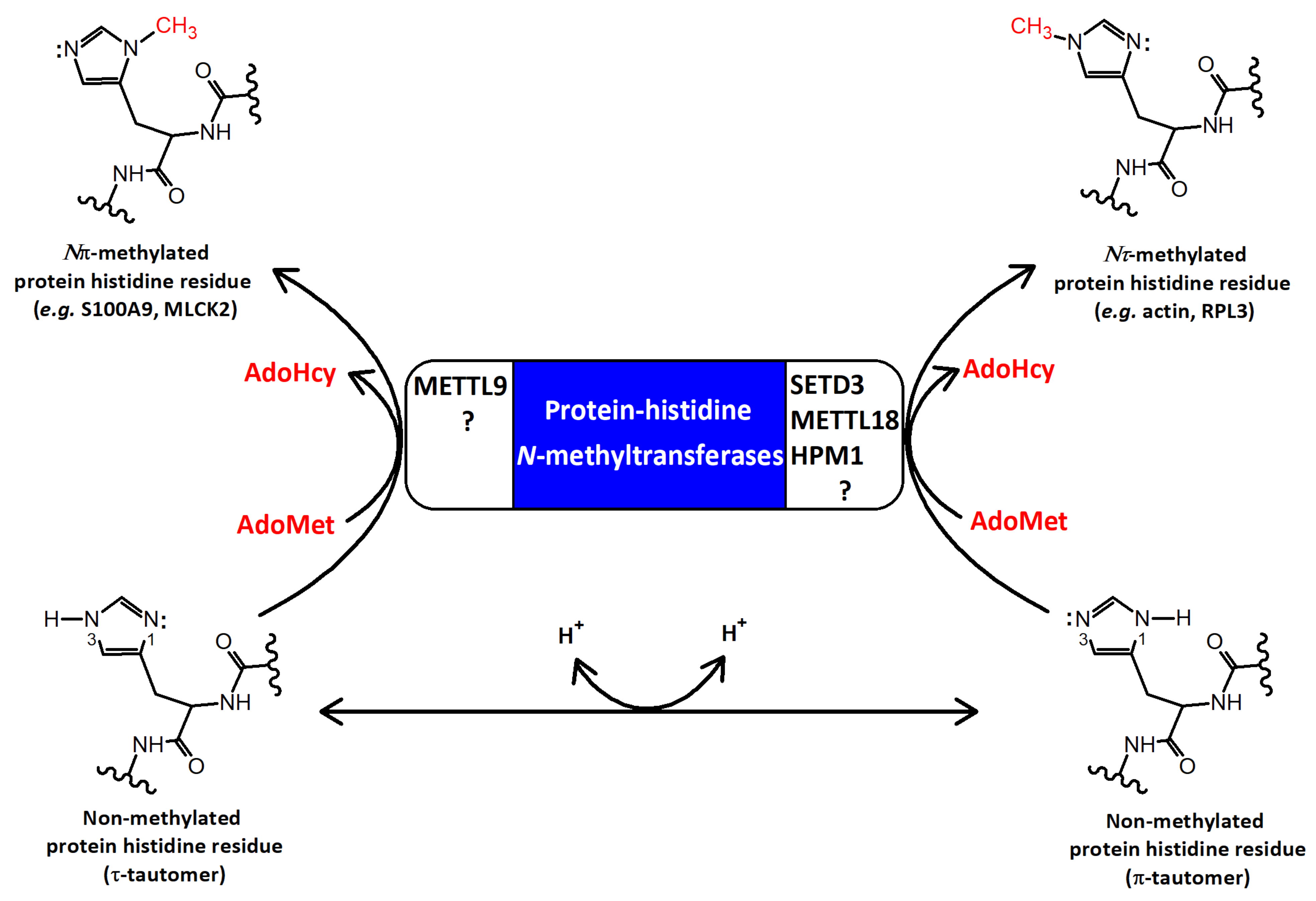
1. Introduction
2. The Structural Features of SETD3
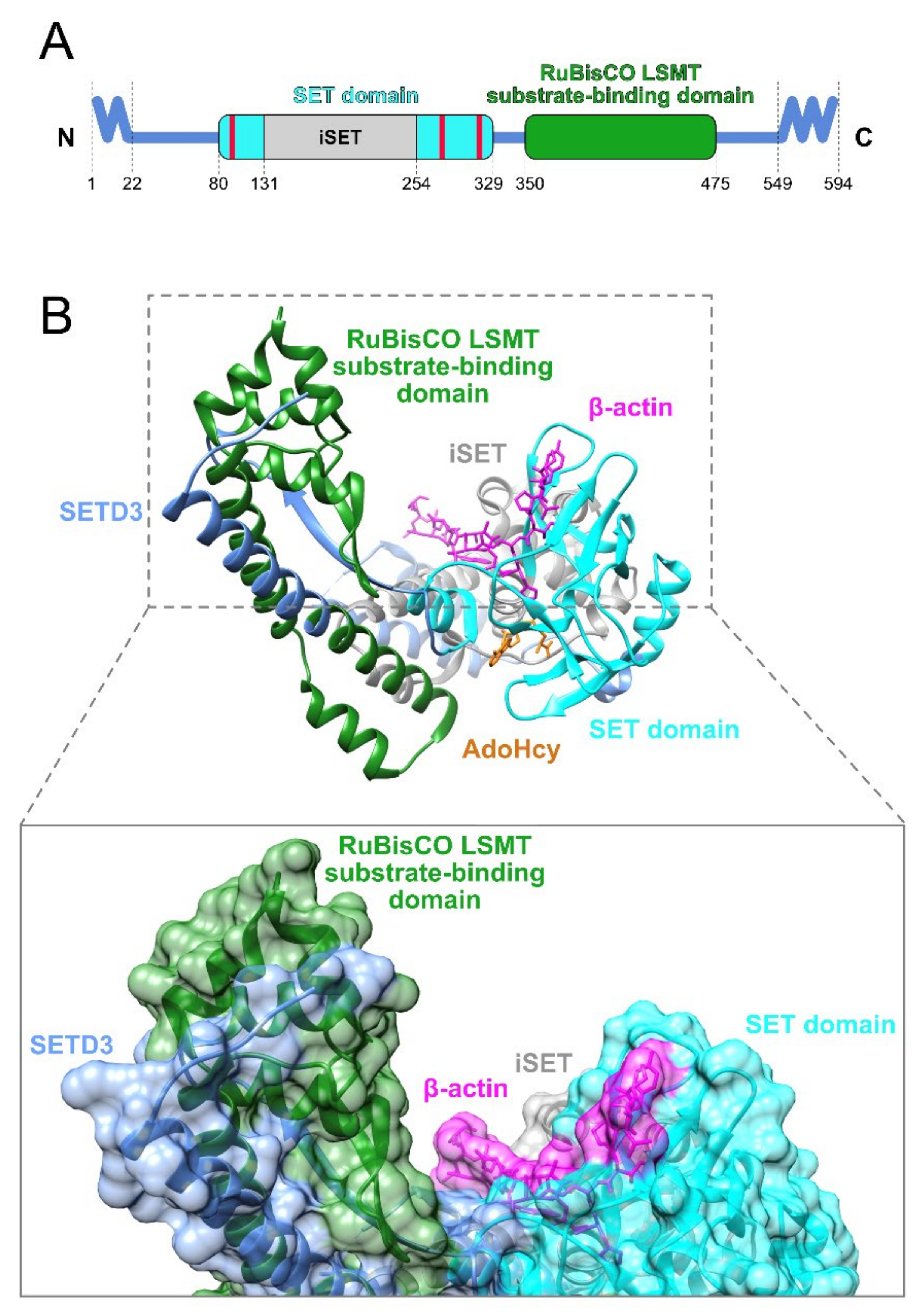
2.1. Domain Architecture
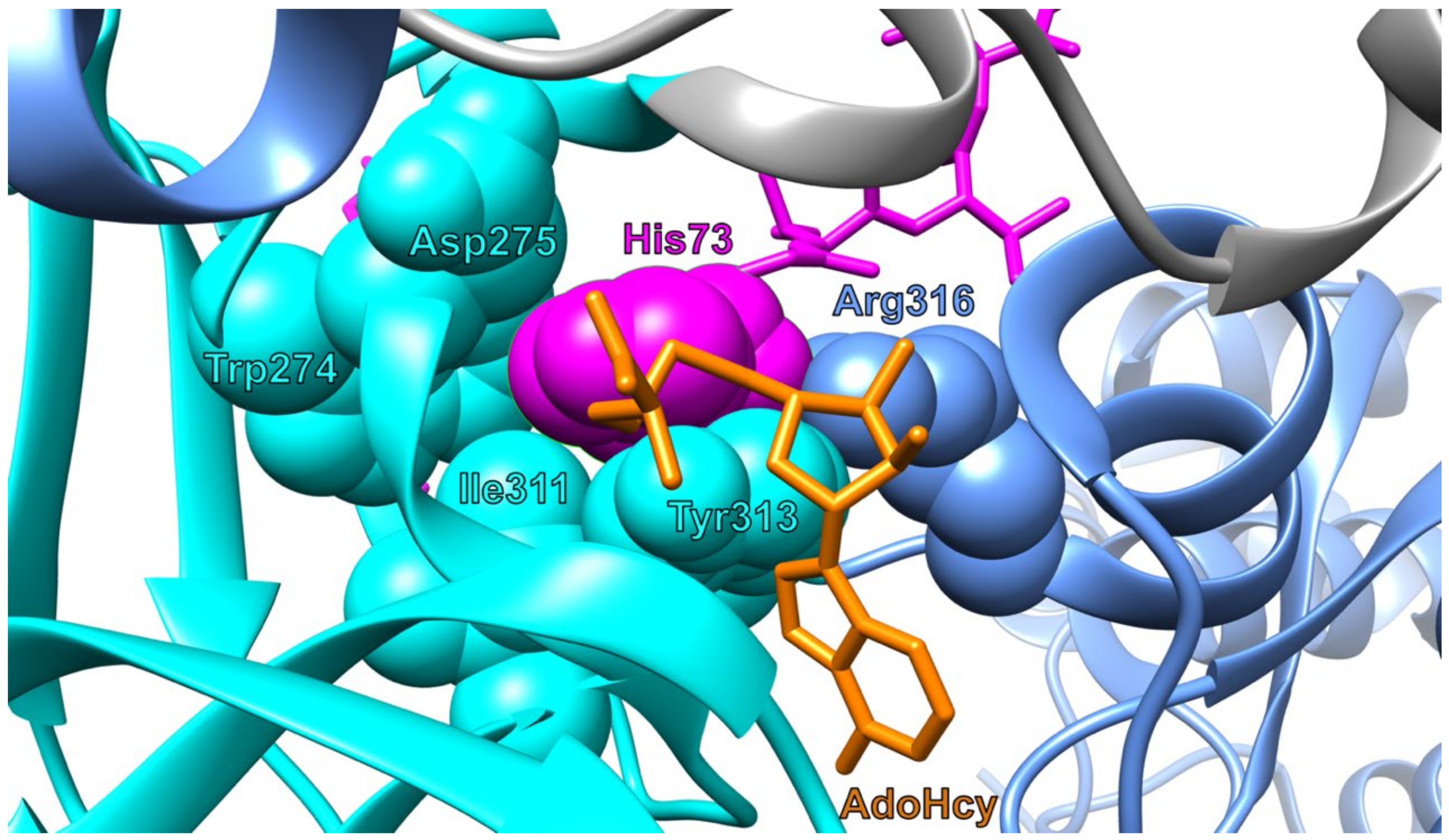
2.2. Structure
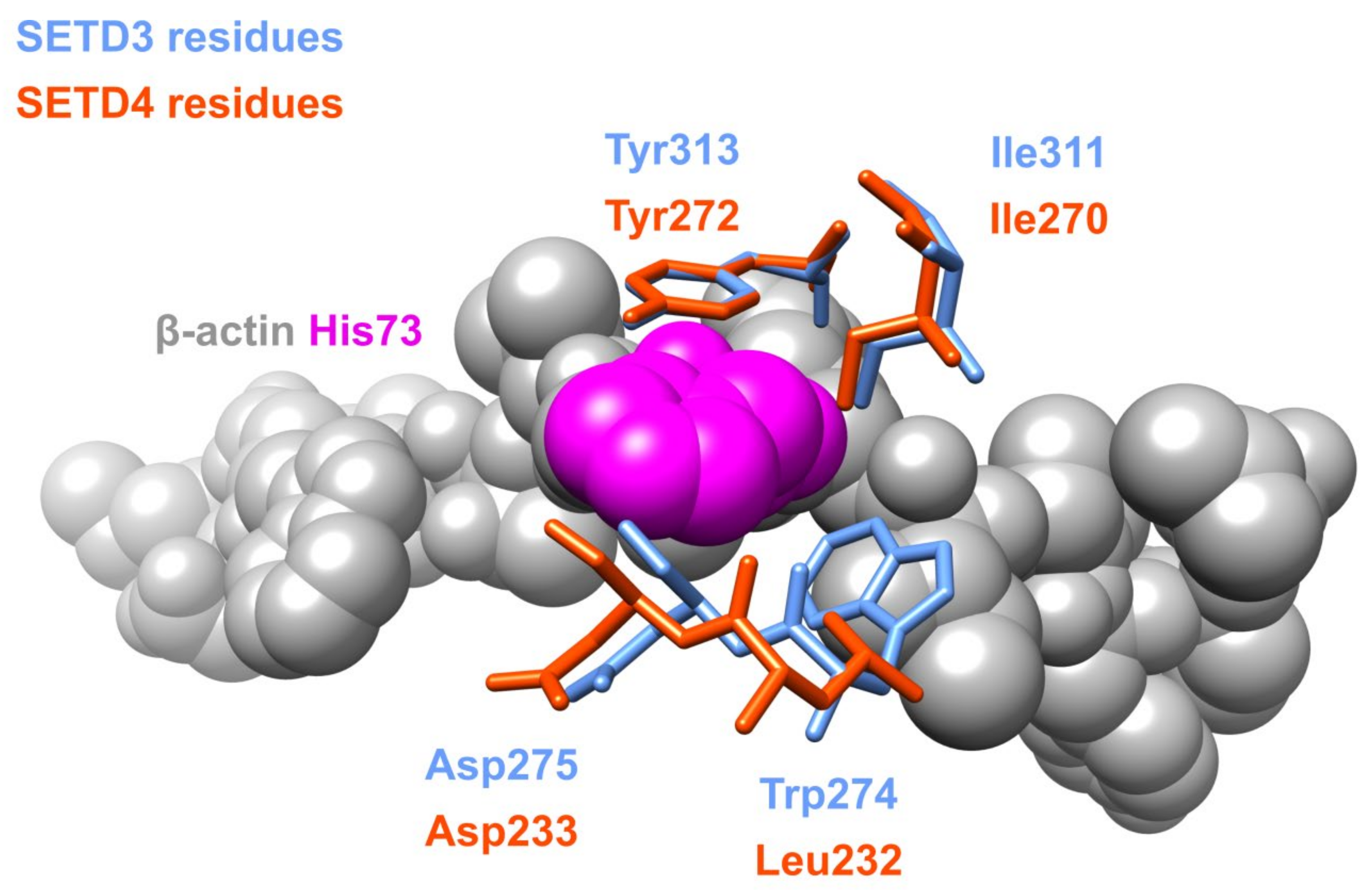
2.3. Paralogs
3. The Biochemical Features of SETD3
3.1. Substrate Specificity
3.1.1. Actin
3.1.2. Other Substrates
3.1.3. Inhibitors
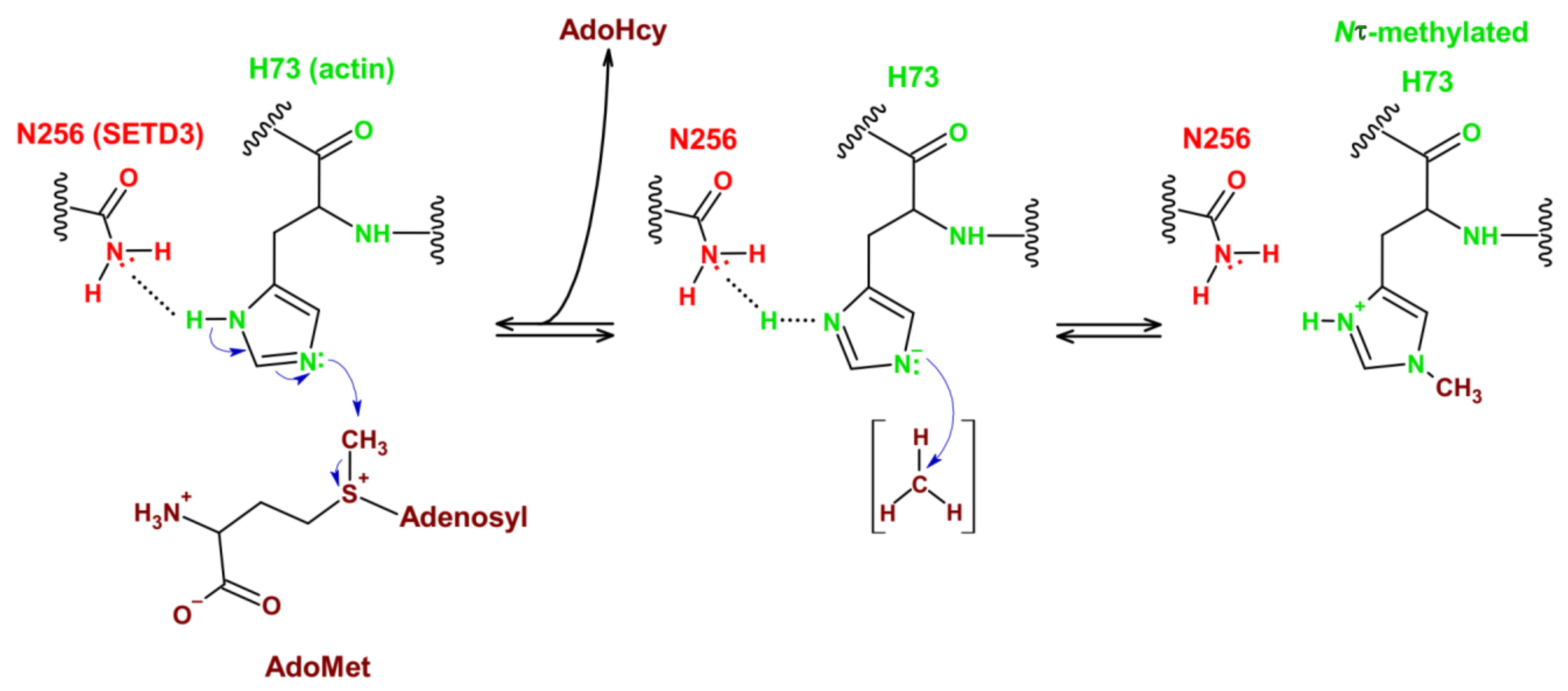
3.2. Reaction Mechanism
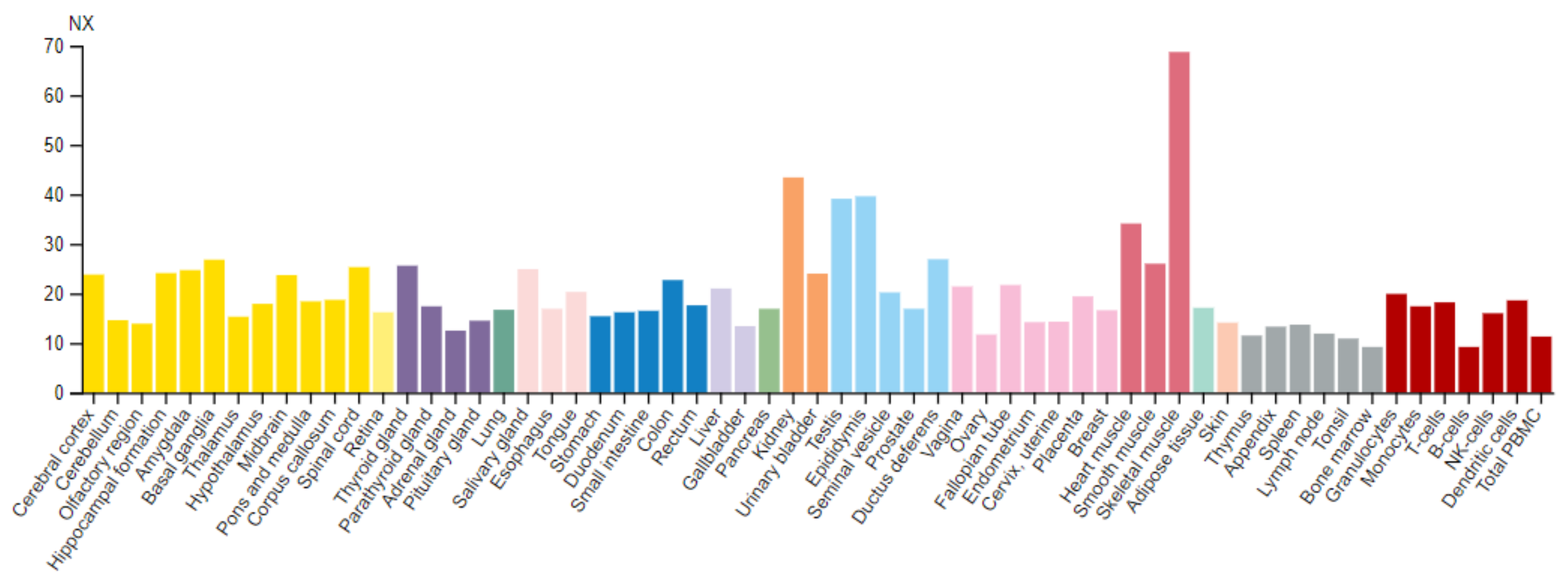
3.3. Tissue Distribution and Intracellular Localization
4. The Cellular Features of SETD3
4.1. Biological Effect of Actin Methylation by SETD3
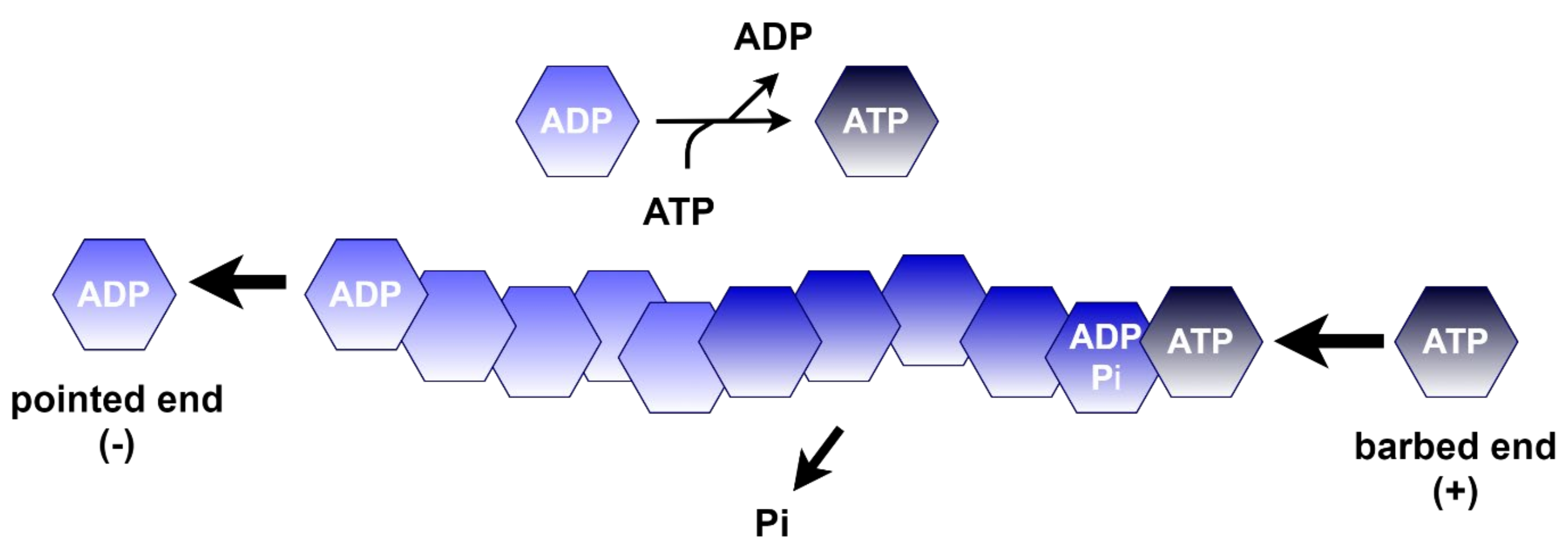
4.1.1. Polymerization of Actin
4.1.2. Effect of Actin H73 Methylation
4.2. The Cellular Roles of SETD3 and Association with Signaling Pathways
The Functions of Cytosolic SETD3
4.3. Other Postulated Functions of the SETD3 Protein
5. The Role of SETD3 in Diseases
5.1. Cancer
5.2. Other Diseases
6. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Clarke, S.G. Protein methylation at the surface and buried deep: Thinking outside the histone box. Trends Biochem. Sci. 2013, 38, 243–252. [Google Scholar] [CrossRef]
- Nyman, T.; Schüler, H.; Korenbaum, E.; Schutt, C.E.; Karlsson, R.; Lindberg, U. The role of MeH73 in actin polymerization and ATP hydrolysis. J. Mol. Biol. 2002, 317, 577–589. [Google Scholar] [CrossRef]
- Raftery, M.J.; Harrison, C.A.; Alewood, P.; Jones, A.; Geczy, C.L. Isolation of the murine S100 protein MRP14 (14 kDa migration-inhibitory-factor-related protein) from activated spleen cells: Characterization of posttranslational modifications and zinc binding. Biochem. J. 1996, 316, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, M.; Collins, J.H. Amino acid sequence of a myosin fragment that contains SH-1, SH-2, and Ntau-methylhistidine. Proc. Natl. Acad. Sci. USA 1977, 74, 4281–4284. [Google Scholar] [CrossRef]
- Meyer, H.E.; Mayr, G.W. N pi-methylhistidine in myosin-light-chain kinase. Biol. Chem. Hoppe-Seyler 1987, 368, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Webb, K.J.; Zurita-Lopez, C.I.; Al-Hadid, Q.; Laganowsky, A.; Young, B.D.; Lipson, R.S.; Souda, P.; Faull, K.F.; Whitelegge, J.P.; Clarke, S.G. A novel 3-methylhistidine modification of yeast ribosomal protein Rpl3 is dependent upon the YIL110W methyltransferase. J. Biol. Chem. 2010, 285, 37598–37606. [Google Scholar] [CrossRef]
- MacTaggart, B.; Kashina, A. Posttranslational modifications of the cytoskeleton. Cytoskeleton 2021, 78, 142–173. [Google Scholar] [CrossRef]
- Johnson, P.; Harris, C.I.; Perry, S.V. 3-methylhistidine in actin and other muscle proteins. Biochem. J. 1967, 105, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Asatoor, A.M.; Armstrong, M.D. 3-methylhistidine, a component of actin. Biochem. Biophys. Res. Commun. 1967, 26, 168–174. [Google Scholar] [CrossRef]
- Elzinga, M. Amino acid sequence aroung 3-methylhistidine in rabbit skeletal muscle actin. Biochemistry 1971, 10, 224–229. [Google Scholar] [CrossRef]
- Johnson, P.; Perry, S.V. Biological activity and the 3-methylhistidine content of actin and myosin. Biochem. J. 1970, 119, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, M.; Collins, J.H.; Kuehl, W.M.; Adelstein, R.S. Complete amino-acid sequence of actin of rabbit skeletal muscle. Proc. Natl. Acad. Sci. USA 1973, 70, 2687–2691. [Google Scholar] [CrossRef]
- Vijayasarathy, C.; Rao, B.S. Partial purification and characterisation of S-adenosylmethionine:protein-histidine N-methyltransferase from rabbit skeletal muscle. Biochim. Biophys. Acta 1987, 923, 156–165. [Google Scholar] [CrossRef]
- Raghavan, M.; Lindberg, U.; Schutt, C. The use of alternative substrates in the characterization of actin-methylating and carnosine-methylating enzymes. Eur. J. Biochem. 1992, 210, 311–318. [Google Scholar] [CrossRef]
- Drozak, J.; Piecuch, M.; Poleszak, O.; Kozlowski, P.; Chrobok, L.; Baelde, H.J.; de Heer, E. UPF0586 Protein C9orf41 Homolog Is Anserine-producing Methyltransferase. J. Biol. Chem. 2015, 290, 17190–17205. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Seliga, A.K.; Vertommen, D.; Terreri, M.; Ishikawa, T.; Grabowska, I.; Tiebe, M.; Teleman, A.A.; Jagielski, A.K.; Veiga-da-Cunha, M.; et al. SETD3 protein is the actin-specific histidine N-methyltransferase. eLife 2018, 7, e37921. [Google Scholar] [CrossRef]
- Wilkinson, A.W.; Diep, J.; Dai, S.; Liu, S.; Ooi, Y.S.; Song, D.; Li, T.M.; Horton, J.R.; Zhang, X.; Liu, C.; et al. SETD3 is an actin histidine methyltransferase that prevents primary dystocia. Nature 2019, 565, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Davydova, E.; Shimazu, T.; Schuhmacher, M.K.; Jakobsson, M.E.; Willemen, H.L.D.M.; Liu, T.; Moen, A.; Ho, A.Y.Y.; Małecki, J.; Schroer, L.; et al. The methyltransferase METTL9 mediates pervasive 1-methylhistidine modification in mammalian proteomes. Nat. Commun. 2021, 12, 891. [Google Scholar] [CrossRef]
- Lv, M.; Cao, D.; Zhang, L.; Hu, C.; Li, S.; Zhang, P.; Zhu, L.; Yi, X.; Li, C.; Yang, A.; et al. METTL9 mediated N1-histidine methylation of zinc transporters is required for tumor growth. Protein Cell 2021, 1–6. [Google Scholar] [CrossRef]
- Kapell, S.; Jakobsson, M.E. Large-scale identification of protein histidine methylation in human cells. NAR Genom. Bioinform. 2021, 3, lqab045. [Google Scholar] [CrossRef] [PubMed]
- Małecki, J.M.; Odonohue, M.F.; Kim, Y.; Jakobsson, M.E.; Gessa, L.; Pinto, R.; Wu, J.; Davydova, E.; Moen, A.; Olsen, J.V.; et al. Human METTL18 is a histidine-specific methyltransferase that targets RPL3 and affects ribosome biogenesis and function. Nucleic Acids Res. 2021, 49, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Matsuura-Suzuki, E.; Shimazu, T.; Takahashi, M.; Kotoshiba, K.; Suzuki, T.; Kashiwagi, K.; Sohtome, Y.; Akakabe, M.; Sodeoka, M.; Dohmae, N.; et al. METTL18-mediated histidine methylation on RPL3 modulates translation elongation for proteostasis maintenance. bioRxiv 2021, 454307. [Google Scholar] [CrossRef]
- Al-Hadid, Q.; Roy, K.; Chanfreau, G.; Clarke, S.G. Methylation of yeast ribosomal protein Rpl3 promotes translational elongation fidelity. RNA 2016, 22, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Kim, K.B.; Kim, J.Y.; Seo, S.B. Characterization of a novel histone H3K36 methyltransferase setd3 in zebrafish. Biosci. Biotechnol. Biochem. 2011, 75, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Kim, K.B.; Kim, J.H.; Kim, J.Y.; Kim, J.R.; Kee, H.J.; Kim, D.W.; Choe, N.; Park, H.J.; Son, H.J.; et al. Histone methyltransferase SETD3 regulates muscle differentiation. J. Biol. Chem. 2011, 286, 34733–34742. [Google Scholar] [CrossRef]
- Dai, S.; Holt, M.V.; Horton, J.R.; Woodcock, C.B.; Patel, A.; Zhang, X.; Young, N.L.; Wilkinson, A.W.; Cheng, X. Characterization of SETD3 methyltransferase-mediated protein methionine methylation. J. Biol. Chem. 2020, 295, 10901–10910. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M. Driver mutations in histone H3.3 and chromatin remodeling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef]
- Guo, Q.; Liao, S.; Kwiatkowski, S.; Tomaka, W.; Yu, H.; Wu, G.; Tu, X.; Min, J.; Drozak, J.; Xu, C. Structural insights into SETD3-mediated histidine methylation on β-actin. eLife 2019, 8, e43676. [Google Scholar] [CrossRef]
- Dai, S.; Horton, J.R.; Woodcock, C.B.; Wilkinson, A.W.; Zhang, X.; Gozani, O.; Cheng, X. Structural basis for the target specificity of actin histidine methyltransferase SETD3. Nat. Commun. 2019, 10, 3541. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, X.; Li, H. Molecular basis for histidine N3-specific methylation of actin H73 by SETD3. Cell Discovery 2020, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Trievel, R.C.; Beach, B.M.; Dirk, L.M.; Houtz, R.L.; Hurley, J.H. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell 2002, 111, 91–103. [Google Scholar] [CrossRef]
- Trievel, R.C.; Flynn, E.M.; Houtz, R.L.; Hurley, J.H. Mechanism of multiple lysine methylation by the SET domain enzyme Rubisco LSMT. Nat. Struct. Biol. 2003, 10, 545–552. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005, 6, 227. [Google Scholar] [CrossRef]
- Raunser, S.; Magnani, R.; Huang, Z.; Houtz, R.L.; Trievel, R.C.; Penczek, P.A.; Walz, T. Rubisco in complex with Rubisco large subunit methyltransferase. Proc. Nat. Acad. Sci. USA 2009, 106, 3160–3165. [Google Scholar] [CrossRef]
- Chang, Y.; Levy, D.; Horton, J.R.; Peng, J.; Zhang, X.; Gozani, O.; Cheng, X. Structural basis of SETD6-mediated regulation of the NF-kB network via methyl-lysine signaling. Nucleic Acids Res. 2011, 39, 6380–6389. [Google Scholar] [CrossRef]
- Ye, S.; Ding, Y.F.; Jia, W.H.; Liu, X.L.; Feng, J.Y.; Zhu, Q.; Cai, S.L.; Yang, Y.S.; Lu, Q.Y.; Huang, X.T.; et al. SET Domain-Containing Protein 4 Epigenetically Controls Breast Cancer Stem Cell Quiescence. Cancer Res. 2019, 79, 4729–4743. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Herz, H.M.; Garruss, A.; Shilatifard, A. SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639. [Google Scholar] [CrossRef]
- Lukinović, V.; Casanova, A.G.; Roth, G.S.; Chuffart, F.; Reynoird, N. Lysine Methyltransferases Signaling: Histones are Just the Tip of the Iceberg. Curr. Protein Pept. Sci. 2020, 21, 655–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yan, C.T.; Dou, Y.; Viboolsittiseri, S.S.; Wang, J.H. The role of a newly identified SET domain-containing protein, SETD3, in oncogenesis. Haematologica 2013, 98, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Kudryashov, D.S.; Reisler, E. ATP and ADP actin states. Biopolymers 2013, 99, 245–256. [Google Scholar] [CrossRef]
- Stemp, M.J.; Guha, S.; Hartl, F.U.; Barral, J.M. Efficient production of native actin upon translation in a bacterial lysate supplemented with the eukaryotic chaperonin TRiC. Biol. Chem. 2005, 386, 753–757. [Google Scholar] [CrossRef]
- Frankel, A.; Brown, J.I. Evaluation of kinetic data: What the numbers tell us about PRMTs. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Horton, J.R.; Wilkinson, A.W.; Gozani, O.; Zhang, X.; Cheng, X. An engineered variant of SETD3 methyltransferase alters target specificity from histidine to lysine methylation. J. Biol. Chem. 2020, 295, 2582–2589. [Google Scholar] [CrossRef]
- Cohn, O.; Feldman, M.; Weil, L.; Kublanovsky, M.; Levy, D. Chromatin associated SETD3 negatively regulates VEGF expression. Sci. Rep. 2016, 6, 37115. [Google Scholar] [CrossRef]
- Hintzen, J.C.J.; Moesgaard, L.; Kwiatkowski, S.; Drozak, J.; Kongsted, J.; Mecinović, J. β-Actin Peptide-Based Inhibitors of Histidine Methyltransferase SETD3. Chem. Med. Chem. 2021, 16, 2695–2702. [Google Scholar] [CrossRef]
- Bachovchin, W.W.; Roberts, J.D. Nitrogen-15 nuclear magnetic resonance spectroscopy. The state of histidine in the catalytic triad of .alpha.-lytic protease. Implications for the charge-relay mechanism of peptide-bond cleavage by serine proteases. J. Am. Chem. Soc. 1978, 100, 8041–8047. [Google Scholar] [CrossRef]
- Grimsley, G.R.; Scholtz, J.M.; Pace, C.N. A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 2009, 18, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000183576-SETD3/cell (accessed on 29 July 2021).
- Sanger, J.W.; Wang, J.; Fan, Y.; White, J.; Mi-Mi, L.; Dube, D.K.; Sanger, J.M.; Pruyne, D. Assembly and Maintenance of Myofibrils in Striated Muscle. Handb. Exp. Pharmacol. 2017, 235, 39–75. [Google Scholar] [CrossRef]
- Su, W.; Mruk, D.D.; Cheng, C.Y. Regulation of actin dynamics and protein trafficking during spermatogenesis—Insights into a complex process. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 153–172. [Google Scholar] [CrossRef]
- Sever, S.; Schiffer, M. Actin dynamics at focal adhesions: A common endpoint and putative therapeutic target for proteinuric kidney diseases. Kidney Int. 2018, 93, 1298–1307. [Google Scholar] [CrossRef]
- Cheng, X.; Hao, Y.; Shu, W.; Zhao, M.; Zhao, C.; Wu, Y.; Peng, X.; Yao, P.; Xiao, D.; Qing, G.; et al. Cell cycle-dependent degradation of the methyltransferase SETD3 attenuates cell proliferation and liver tumorigenesis. J. Biol. Chem. 2017, 292, 9022–9033. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef]
- DiNubile, M.J. Nucleation and elongation of actin filaments in the presence of high speed supernate from neutrophil lysates: Modulating effects of Ca2+ and phosphatidylinositol-4,5-bisphosphate. Biochim. Biophys. Acta 1998, 1405, 85–98. [Google Scholar] [CrossRef][Green Version]
- Choua, S.Z.; Pollarda, T.D. Mechanism of actin polymerization revealed by cryo-EM structures of actin filaments with three different bound nucleotides. Proc. Natl. Acad. Sci. USA 2019, 116, 4265–4274. [Google Scholar] [CrossRef]
- Solomon, L.R.; Rubenstein, P.A. Studies on the role of actin’s N tau-methylhistidine using oligodeoxynucleotide-directed site-specific mutagenesis. J. Biol. Chem. 1987, 262, 11382–11388. [Google Scholar] [CrossRef]
- Diep, J.; Ooi, Y.S.; Wilkinson, A.W.; Peters, C.E.; Foy, E.; Johnson, J.R.; Zengel, J.; Ding, S.; Weng, K.F.; Laufman, O.; et al. Enterovirus pathogenesis requires the host methyltransferase SETD3. Nat. Microbiol. 2019, 4, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, O.H.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytönen, V.P.; Flodström-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267. [Google Scholar] [CrossRef]
- Visser, L.J.; Langereis, M.A.; Rabouw, H.H.; Wahedi, M.; Muntjewerff, E.M.; de Groot, R.J.; van Kuppeveld, F.J.M. Essential role of enterovirus 2A protease in counteracting stress granule formation and the induction of type I interferon. J. Virol. 2019, 93, e00222-19. [Google Scholar] [CrossRef]
- Pires-Luís, A.S.; Vieira-Coimbra, M.; Vieira, F.Q.; Costa-Pinheiro, P.; Silva-Santos, R.; Dias, P.C.; Antunes, L.; Lobo, F.; Oliveira, J.; Gonçalves, C.S.; et al. Expression of histone methyltransferases as novel biomarkers for renal cell tumor diagnosis and prognostication. Epigenetics 2015, 10, 1033–1043. [Google Scholar] [CrossRef]
- Xu, L.; Wang, P.; Feng, X.; Tang, J.; Li, L.; Zheng, X.; Zhang, J.; Hu, Y.; Lan, T.; Yuan, K.; et al. SETD3 is regulated by a couple of microRNAs and plays opposing roles in proliferation and metastasis of hepatocellular carcinoma. Clin. Sci. 2019, 133, 2085–2105. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.; Rutsch, N.; Győrffy, B.; Espinoza-Sánchez, N.A.; Götte, M. SETD3 acts as a prognostic marker in breast cancer patients and modulates the viability and invasion of breast cancer cells. Sci. Rep. 2020, 10, 2262. [Google Scholar] [CrossRef]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; et al. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Feng, X.; Hao, X.; Wang, P.; Zhang, Y.; Zheng, X.; Li, L.; Ren, S.; Zhang, M.; Xu, M. CircSETD3 (Hsa_circ_0000567) acts as a sponge for microRNA-421 inhibiting hepatocellular carcinoma growth. J. Exp. Clin. Cancer Res. 2019, 38, 98. [Google Scholar] [CrossRef]
- Tang, L.; Xiong, W.; Zhang, L.; Wang, D.; Wang, Y.; Wu, Y.; Wei, F.; Mo, Y.; Hou, X.; Shi, L.; et al. CircSETD3 regulates MAPRE1 through miR-615-5p and miR-1538 sponges to promote migration and invasion in nasopharyngeal carcinoma. Oncogene 2021, 40, 307–321. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Jiang, Q. SETD3 reduces KLC4 expression to improve the sensitization of cervical cancer cell to radiotherapy. Biochem. Biophys. Res. Commun. 2019, 516, 619–625. [Google Scholar] [CrossRef]
- Baek, J.H.; Lee, J.; Yun, H.S.; Lee, C.W.; Song, J.Y.; Um, H.D.; Park, J.K.; Park, I.C.; Kim, J.S.; Kim, E.H.; et al. Kinesin light chain-4 depletion induces apoptosis of radioresistant cancer cells by mitochondrial dysfunction via calcium ion influx. Cell Death Dis. 2018, 9, 496. [Google Scholar] [CrossRef]
- Abaev-Schneiderman, E.; Admoni-Elisha, L.; Levy, D. SETD3 is a positive regulator of DNA-damage-induced apoptosis. Cell Death Dis. 2019, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Engqvist, H.; Parris, T.Z.; Kovács, A.; Rönnerman, E.W.; Sundfeldt, K.; Karlsson, P.; Helou, K. Validation of Novel Prognostic Biomarkers for Early-Stage Clear-Cell, Endometrioid and Mucinous Ovarian Carcinomas Using Immunohistochemistry. Front. Oncol. 2020, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, T.; Sun, J.; Liu, J.; Wu, H. SETD3 negatively regulates VEGF expression during hypoxic pulmonary hypertension in rats. Hypertens. Res. 2018, 41, 691–698. [Google Scholar] [CrossRef]
- Liao, J.; Luo, S.; Yang, M.; Lu, Q. Overexpression of CXCR5 in CD4+ T cells of SLE patients caused by excessive SETD3. Clin. Immunol. 2020, 214, 108406. [Google Scholar] [CrossRef]
- Xu, X.; Cui, Y.; Li, C.; Wang, Y.; Cheng, J.; Chen, S.; Sun, J.; Ren, J.; Yao, X.; Gao, J.; et al. SETD3 Downregulation Mediates PTEN Upregulation-Induced Ischemic Neuronal Death Through Suppression of Actin Polymerization and Mitochondrial Function. Mol. Neurobiol. 2021, 15. [Google Scholar] [CrossRef]
- Garza-Rodríguez, M.L.; Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Sánchez-Domínguez, C.N.; Berlanga-Garza, A.; Antonio-Macedo, M.; Valdés-Chapa, L.D.; Vidal-Torres, D.; Vidal-Gutiérrez, O.; Pérez-Ibave, D.C.; et al. Analysis of HPV Integrations in Mexican Pre-Tumoral Cervical Lesions Reveal Centromere-Enriched Breakpoints and Abundant Unspecific HPV Regions. Int. J. Mol. Sci. 2021, 22, 3242. [Google Scholar] [CrossRef]
- Gershey, E.L.; Haslett, G.W.; Vidali, G.; Allfrey, V.G. Chemical studies of histone methylation. Evidence for the occurrence of 3-methylhistidine in avian erythrocyte histone fractions. J. Biol. Chem. 1969, 244, 4871–4877. [Google Scholar] [CrossRef]
- Woodcock, C.B.; Yu, D.; Zhang, X.; Cheng, X. Human HemK2/KMT9/N6AMT1 is an active protein methyltransferase, but does not act on DNA in vitro, in the presence of Trm112. Cell Discov. 2019, 5, 50. [Google Scholar] [CrossRef]
- Gao, J.; Wang, B.; Yu, H.; Wu, G.; Wan, C.; Liu, W.; Liao, S.; Cheng, L.; Zhu, Z. Structural insight into HEMK2-TRMT112-mediated glutamine methylation. Biochem. J. 2020, 477, 3833–3838. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | SETD3 Activity | Proposed Functions | References |
|---|---|---|---|
| Actin polymerization | Methylation of H73 in β-actin | Stabilization of actin monomers | [17,18] |
| Promoting actin polymerization | |||
| Enterovirus pathogenesis | Unknown | Supporting viral genome replication | [63] |
| Interaction with viral protease 2A | |||
| Epigenetic regulation of chromatin | Methylation of core histones, plausibly H3 | Regulation of gene expression | [25,26,44] |
| Response to hypoxia conditions | Methylation of FoxM1 | Increasing expression of VEGF and promotion of angiogenesis | [49] |
| Carcinogenesis | Unknown | Regulation of cancer development and progression | [44,49,57,66,67,68,70,71,72,73,74,75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witecka, A.; Kwiatkowski, S.; Ishikawa, T.; Drozak, J. The Structure, Activity, and Function of the SETD3 Protein Histidine Methyltransferase. Life 2021, 11, 1040. https://doi.org/10.3390/life11101040
Witecka A, Kwiatkowski S, Ishikawa T, Drozak J. The Structure, Activity, and Function of the SETD3 Protein Histidine Methyltransferase. Life. 2021; 11(10):1040. https://doi.org/10.3390/life11101040
Chicago/Turabian StyleWitecka, Apolonia, Sebastian Kwiatkowski, Takao Ishikawa, and Jakub Drozak. 2021. "The Structure, Activity, and Function of the SETD3 Protein Histidine Methyltransferase" Life 11, no. 10: 1040. https://doi.org/10.3390/life11101040
APA StyleWitecka, A., Kwiatkowski, S., Ishikawa, T., & Drozak, J. (2021). The Structure, Activity, and Function of the SETD3 Protein Histidine Methyltransferase. Life, 11(10), 1040. https://doi.org/10.3390/life11101040