Cardiotoxicity of Novel Targeted Hematological Therapies


Abstract
1. Introduction
2. TKIs
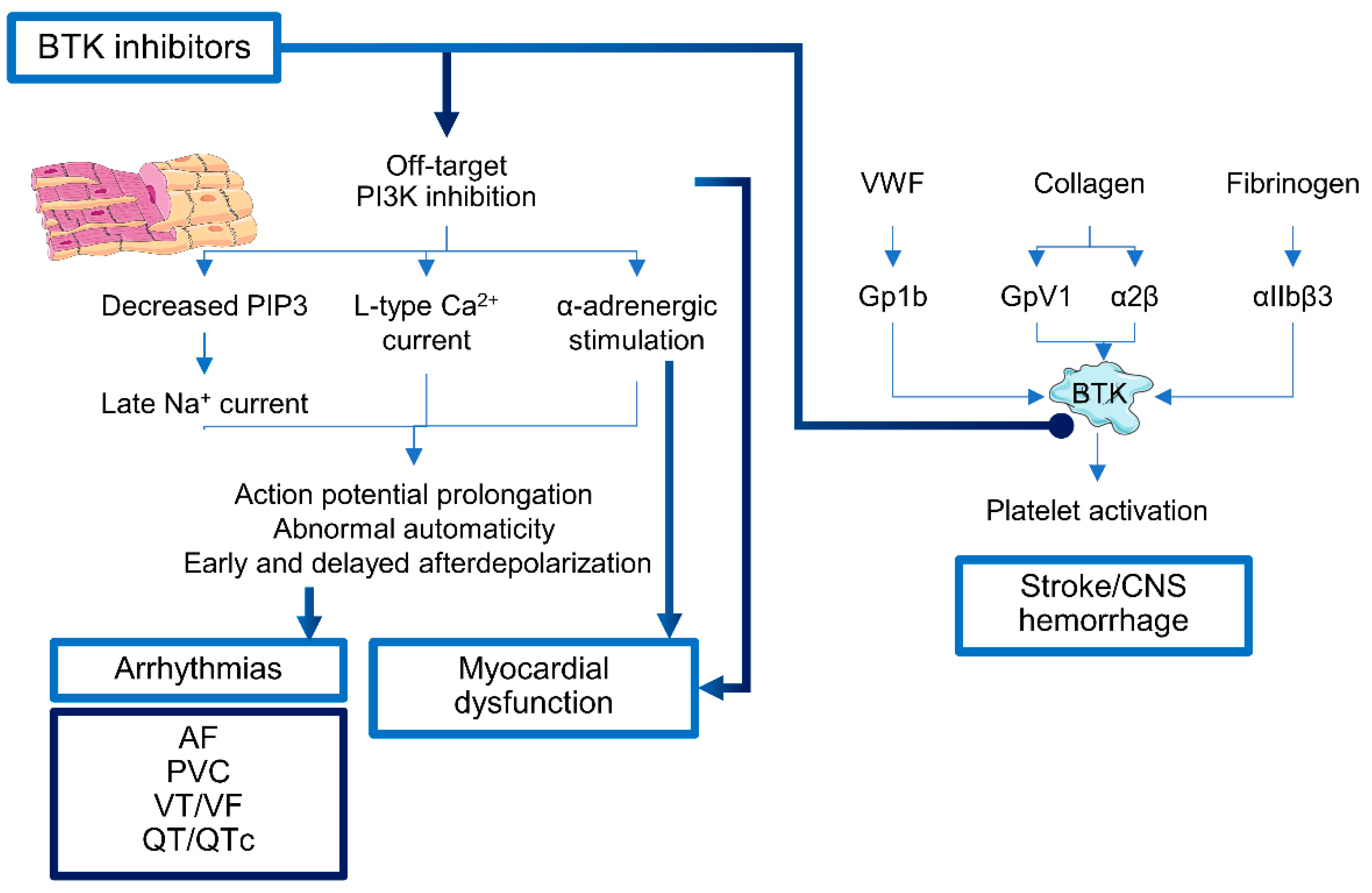
2.1. Bruton’s Tyrosine Kinase Inhibitors
2.2. PI3K Inhibitors
2.3. Isocitrate Dehydrogenase (IDH) Inhibitors
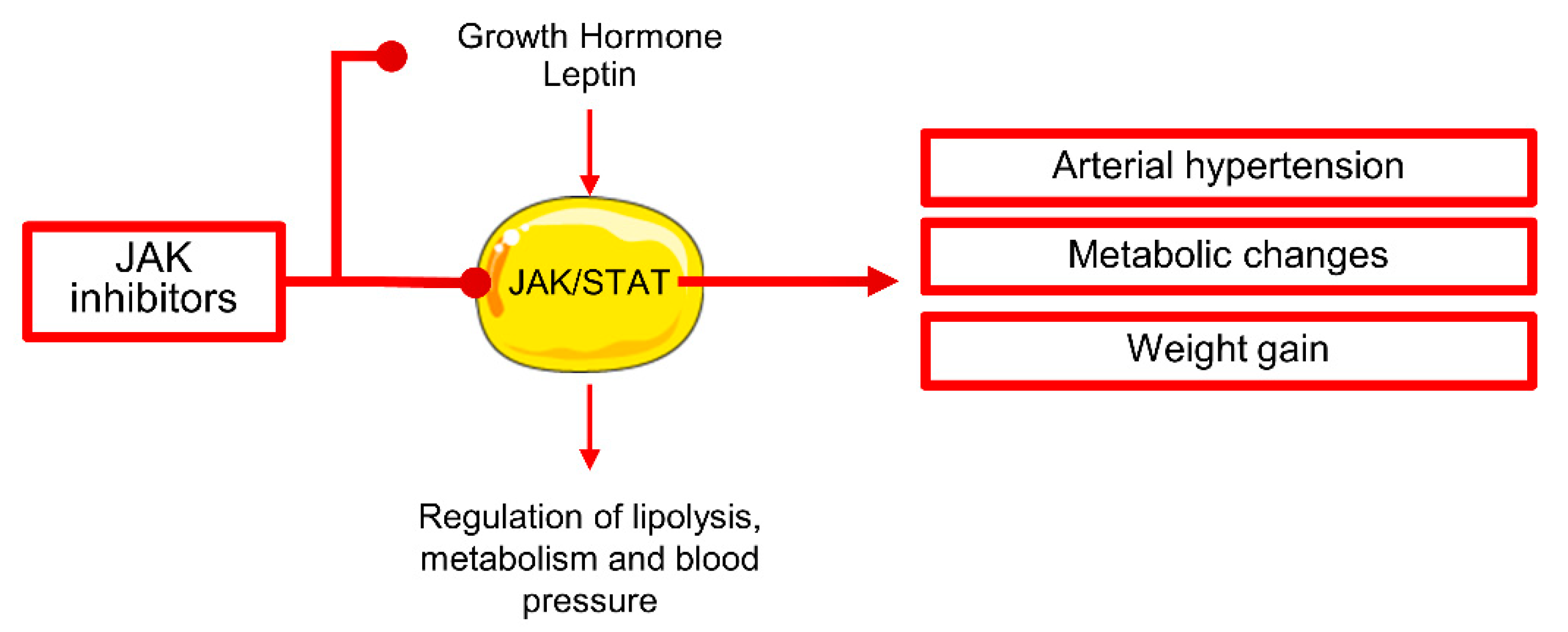
2.4. Janus Kinase Inhibitor
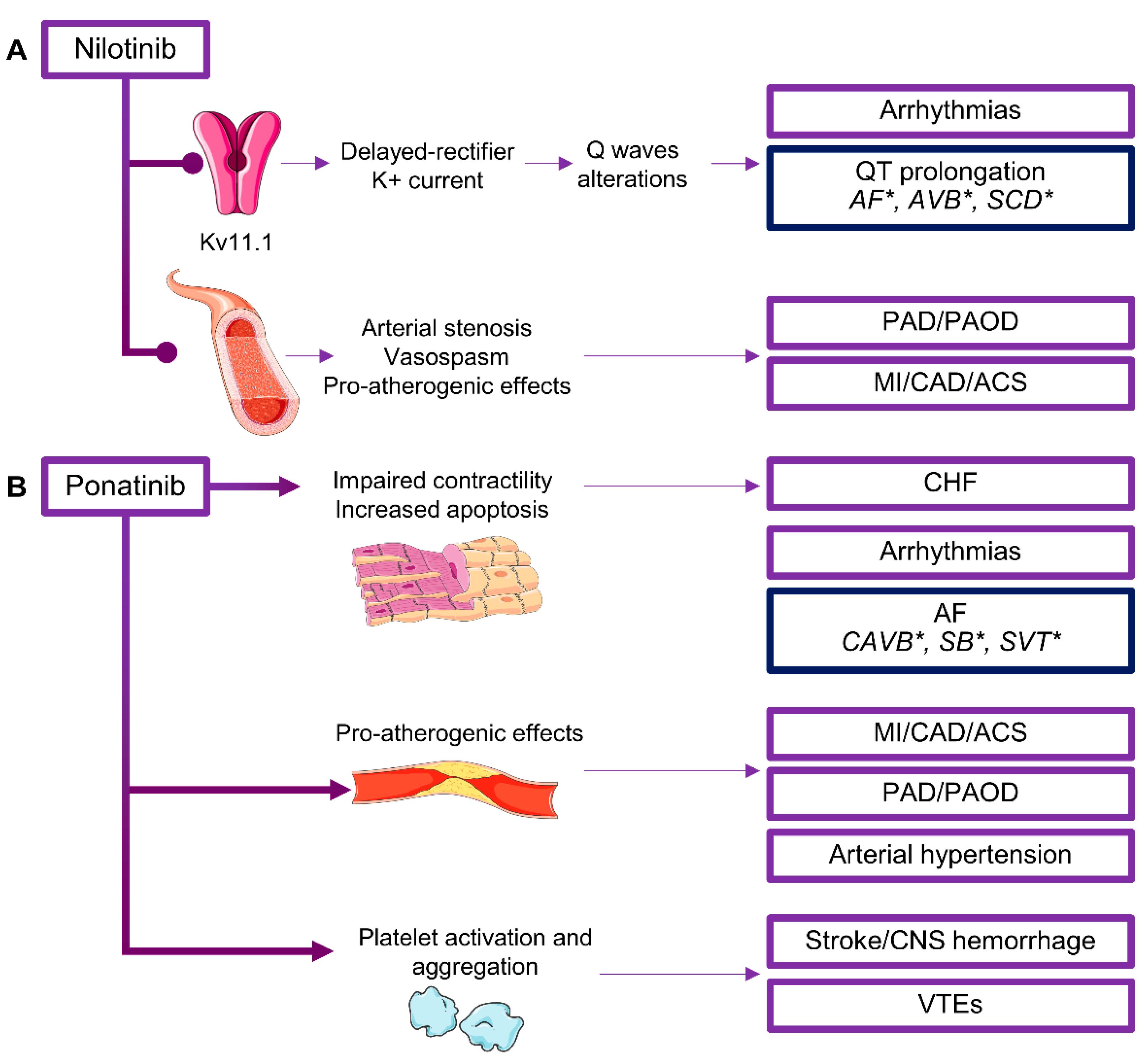
2.5. BCR/ABL Inhibitors
2.6. Other TKIs
3. Proteasome Inhibitors
4. IMiDs
5. Demethylating Agents
6. Monoclonal Antibodies
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef]
- Feijen, E.A.M.L.; Font-Gonzalez, A.; Van der Pal, H.J.H.; Kok, W.E.M.; Geskus, R.B.; Ronckers, C.M.; Bresters, D.; van Dalen, E.C.; van Dulmen-den Broeder, E.; van den Berg, M.H.; et al. Risk and Temporal Changes of Heart Failure Among 5-Year Childhood Cancer Survivors: A DCOG-LATER Study. J. Am. Heart Assoc. 2019, 8, e009122. [Google Scholar] [CrossRef]
- Lenneman, C.G.; Sawyer, D.B. Cardio-Oncology: An Update on Cardiotoxicity of Cancer-Related Treatment. Circ. Res. 2016, 118, 1008–1020. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhou, Y.; Liu, W. Precision cardio-oncology: Understanding the cardiotoxicity of cancer therapy. NPJ Precis. Oncol. 2017, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Galderisi, M.; Santoro, C.; Bossone, E.; Mancusi, C. Rationale and proposal for cardio-oncology services in Italy. J. Cardiovasc. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Esposito, R.; Lembo, M.; Sorrentino, R.; De Santo, I.; Luciano, F.; Casciano, O.; Giuliano, M.; De Placido, S.; Trimarco, B.; et al. Strain-oriented strategy for guiding cardioprotection initiation of breast cancer patients experiencing cardiac dysfunction. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 1345–1352. [Google Scholar] [CrossRef]
- Linschoten, M.; Teske, A.J.; Cramer, M.J.; van der Wall, E.; Asselbergs, F.W. Chemotherapy-Related Cardiac Dysfunction: A Systematic Review of Genetic Variants Modulating Individual Risk. Circ. Genom. Precis. Med. 2018, 11, e001753. [Google Scholar] [CrossRef]
- Michel, L.; Mincu, R.I.; Mahabadi, A.A.; Settelmeier, S.; Al-Rashid, F.; Rassaf, T.; Totzeck, M. Troponins and brain natriuretic peptides for the prediction of cardiotoxicity in cancer patients: A meta-analysis. Eur. J. Heart Fail. 2020, 22, 350–361. [Google Scholar] [CrossRef]
- Morandi, P.; Ruffini, P.A.; Benvenuto, G.M.; Raimondi, R.; Fosser, V. Cardiac toxicity of high-dose chemotherapy. Bone Marrow Transplant. 2005, 35, 323–334. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr. A selective history of the therapy of Hodgkin’s disease. Br. J. Haematol. 2003, 122, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, B.; Griffin, J.D. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene 2002, 21, 3314–3333. [Google Scholar] [CrossRef] [PubMed]
- Giudice, V.; Mensitieri, F.; Izzo, V.; Filippelli, A.; Selleri, C. Aptamers and Antisense Oligonucleotides for Diagnosis and Treatment of Hematological Diseases. Int. J. Mol. Sci. 2020, 21, 3252. [Google Scholar] [CrossRef]
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Anderson, J.S.; Teutsch, M.; Dong, Z.; Wortis, H.H. An essential role for Bruton’s [corrected] tyrosine kinase in the regulation of B-cell apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 10966–10971. [Google Scholar] [CrossRef]
- Glassford, J.; Soeiro, I.; Skarell, S.M.; Banerji, L.; Holman, M.; Klaus, G.G.; Kadowaki, T.; Koyasu, S.; Lam, E.W. BCR targets cyclin D2 via Btk and the p85alpha subunit of PI3-K to induce cell cycle progression in primary mouse B cells. Oncogene 2003, 22, 2248–2259. [Google Scholar] [CrossRef]
- Spaargaren, M.; Beuling, E.A.; Rurup, M.L.; Meijer, H.P.; Klok, M.D.; Middendorp, S.; Hendriks, R.W.; Pals, S.T. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J. Exp. Med. 2003, 198, 1539–1550. [Google Scholar] [CrossRef]
- Brachtl, G.; Piñón Hofbauer, J.; Greil, R.; Hartmann, T.N. The pathogenic relevance of the prognostic markers CD38 and CD49d in chronic lymphocytic leukemia. Ann. Hematol. 2014, 93, 361–374. [Google Scholar] [CrossRef]
- Elices, M.J.; Osborn, L.; Takada, Y.; Crouse, C.; Luhowskyj, S.; Hemler, M.E.; Lobb, R.R. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell 1990, 60, 577–584. [Google Scholar] [CrossRef]
- Osborn, L.; Hession, C.; Tizard, R.; Vassallo, C.; Luhowskyj, S.; Chi-Rosso, G.; Lobb, R. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell 1989, 59, 1203–1211. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Geyer, S.M.; Kay, N.E. Prognosis at diagnosis: Integrating molecular biologic insights into clinical practice for patients with CLL. Blood 2004, 103, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Moslehi, J.J.; Roden, D.M. Proarrhythmic effects of ibrutinib, a clinically approved inhibitor of Bruton’s tyrosine kinase (BTK) used in cancer therapy (abstr). Circulation 2015, 132, A14587. [Google Scholar]
- McMullen, J.R.; Boey, E.J.; Ooi, J.Y.; Seymour, J.F.; Keating, M.J.; Tam, C.S. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood 2014, 124, 3829–3830. [Google Scholar] [CrossRef]
- Zhabyeyev, P.; Chen, X.; Vanhaesebroeck, B.; Oudit, G.Y. PI3Kα in cardioprotection: Cytoskeleton, late Na+ current, and mechanism of arrhythmias. Channels 2019, 13, 520–532. [Google Scholar] [CrossRef]
- Ghigo, A.; Li, M. Phosphoinositide 3-kinase: Friend and foe in cardiovascular disease. Front. Pharmacol. 2015, 6, 169. [Google Scholar] [CrossRef]
- Chong, E.; Chang, S.L.; Hsiao, Y.W.; Singhal, R.; Liu, S.H.; Leha, T.; Lin, W.Y.; Hsu, C.P.; Chen, Y.C.; Chen, Y.J.; et al. Resveratrol, a red wine antioxidant, reduces atrial fibrillation susceptibility in the failing heart by PI3K/AKT/eNOS signaling pathway activation. Heart Rhythm. 2015, 12, 1046–1056. [Google Scholar] [CrossRef]
- Li, M.; Sala, V.; De Santis, M.C.; Cimino, J.; Cappello, P.; Pianca, N.; Di Bona, A.; Margaria, J.P.; Martini, M.; Lazzarini, E.; et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects From Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 2018, 138, 696–711. [Google Scholar] [CrossRef]
- Jiang, L.; Li, L.; Ruan, Y.; Zuo, S.; Wu, X.; Zhao, Q.; Xing, Y.; Zhao, X.; Xia, S.; Bai, R.; et al. Ibrutinib promotes atrial fibrillation by inducing structural remodeling and calcium dysregulation in the atrium. Heart Rhythm. 2019, 16, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Fradley, M.G.; Gliksman, M.; Emole, J.; Viganego, F.; Rhea, I.; Welter-Frost, A.; Armanious, M.; Lee, D.H.; Walko, C.; Shah, B.; et al. Rates and Risk of Atrial Arrhythmias in Patients Treated With Ibrutinib Compared With Cytotoxic Chemotherapy. Am. J. Cardiol. 2019, 124, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Ganatra, S.; Sharma, A.; Shah, S.; Chaudhry, G.M.; Martin, D.T.; Neilan, T.G.; Mahmood, S.S.; Barac, A.; Groarke, J.D.; Hayek, S.S.; et al. Ibrutinib-Associated Atrial Fibrillation. JACC Clin. Electrophysiol. 2018, 4, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Psaty, B.M.; Manolio, T.A.; Kuller, L.H.; Kronmal, R.A.; Cushman, M.; Fried, L.P.; White, R.; Furberg, C.D.; Rautaharju, P.M. Incidence of and risk factors for atrial fibrillation in older adults. Circulation 1997, 96, 2455–2461. [Google Scholar] [CrossRef]
- Brown, J.R.; Moslehi, J.; O’Brien, S.; Ghia, P.; Hillmen, P.; Cymbalista, F.; Shanafelt, T.D.; Fraser, G.; Rule, S.; Kipps, T.J.; et al. Characterization of atrial fibrillation adverse events reported in ibrutinib randomized controlled registration trials. Haematologica 2017, 102, 1796–1805. [Google Scholar] [CrossRef]
- Pineda-Gayoso, R.; Alomar, M.; Lee, D.H.; Fradley, M.G. Cardiovascular Toxicities of Bruton’s Tyrosine Kinase Inhibitors. Curr. Treat. Options Oncol. 2020, 21, 67. [Google Scholar] [CrossRef]
- Lampson, B.L.; Yu, L.; Glynn, R.J.; Barrientos, J.C.; Jacobsen, E.D.; Banerji, V.; Jones, J.A.; Walewska, R.; Savage, K.J.; Michaud, G.F.; et al. Ventricular arrhythmias and sudden death in patients taking ibrutinib. Blood 2017, 129, 2581–2584. [Google Scholar] [CrossRef]
- Bernardeschi, P.; Pirrotta, M.T.; Del Rosso, A.; Fontanelli, G.; Milandri, C. Sudden ventricular fibrillation and death during ibrutinib therapy-A case report. Eur. J. Haematol. 2019, 103, 442–443. [Google Scholar] [CrossRef]
- Fradley, M.G.; Welter-Frost, A.; Gliksman, M.; Emole, J.; Viganego, F.; Lee, D.H.; Shah, B.; Chavez, J.C.; Pinilla-Ibarz, J.; Schabath, M.B. Electrocardiographic Changes Associated With Ibrutinib Exposure. Cancer Control 2020, 27, 1073274820931808. [Google Scholar] [CrossRef]
- Salem, J.E.; Manouchehri, A.; Bretagne, M.; Lebrun-Vignes, B.; Groarke, J.D.; Johnson, D.B.; Yang, T.; Reddy, N.M.; Funck-Brentano, C.; Brown, J.R.; et al. Cardiovascular Toxicities Associated With Ibrutinib. J. Am. Coll. Cardiol. 2019, 74, 1667–1678. [Google Scholar] [CrossRef]
- Ahn, I.E. Cardiovascular adverse events of ibrutinib. Blood 2019, 134, 1881–1882. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, T.; Wiczer, T.; Waller, A.; Philippon, J.; Porter, K.; Haddad, D.; Guha, A.; Rogers, K.A.; Bhat, S.; Byrd, J.C.; et al. Hypertension and incident cardiovascular events following ibrutinib initiation. Blood 2019, 134, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, D.; Alves, D.; Costa, J.; Ferreira, J.J.; Pinto, F.J. Ibrutinib increases the risk of hypertension and atrial fibrillation: Systematic review and meta-analysis. PLoS ONE 2019, 14, e0211228. [Google Scholar] [CrossRef] [PubMed]
- Roeker, L.E.; Sarraf Yazdy, M.; Rhodes, J.; Goodfriend, J.; Narkhede, M.; Carver, J.; Mato, A. Hypertension in Patients Treated With Ibrutinib for Chronic Lymphocytic Leukemia. JAMA Netw. Open 2019, 2, e1916326. [Google Scholar] [CrossRef] [PubMed]
- Busygina, K.; Jamasbi, J.; Seiler, T.; Deckmyn, H.; Weber, C.; Brandl, R.; Lorenz, R.; Siess, W. Oral Bruton tyrosine kinase inhibitors selectively block atherosclerotic plaque-triggered thrombus formation in humans. Blood 2018, 131, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, J.C. Idelalisib for the treatment of indolent non-Hodgkin lymphoma: A review of its clinical potential. Onco Targets Ther. 2016, 9, 2945–2953. [Google Scholar] [CrossRef] [PubMed]
- Sapon-Cousineau, V.; Sapon-Cousineau, S.; Assouline, S. PI3K Inhibitors and Their Role as Novel Agents for Targeted Therapy in Lymphoma. Curr. Treat. Options Oncol. 2020, 21, 51. [Google Scholar] [CrossRef]
- de Weerdt, I.; Koopmans, S.M.; Kater, A.P.; van Gelder, M. Incidence and management of toxicity associated with ibrutinib and idelalisib: A practical approach. Haematologica 2017, 102, 1629–1639. [Google Scholar] [CrossRef]
- Nair, K.S.; Cheson, B. The role of idelalisib in the treatment of relapsed and refractory chronic lymphocytic leukemia. Ther. Adv. Hematol. 2016, 7, 69–84. [Google Scholar] [CrossRef]
- Mahida, H.; Gharia, B.; Ugoeke, N.; Maludum, O.; Asif, A.; Calderon, D. Abstract 11835: Evaluation of Cardiovascular Adverse Events Associated With Ibrutinib; Venetoclax and Idelalisib Used in Treatment of Chronic Lymphocytic Leukemia. Circulation 2018, 138, A11835. [Google Scholar]
- Dreyling, M.; Morschhauser, F.; Bouabdallah, K.; Bron, D.; Cunningham, D.; Assouline, S.E.; Verhoef, G.; Linton, K.; Thieblemont, C.; Vitolo, U.; et al. Phase II study of copanlisib; a PI3K inhibitor; in relapsed or refractory; indolent or aggressive lymphoma. Ann. Oncol. 2017, 28, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; O’Brien, S.; Ewer, M.S.; Goncalves, M.D.; Farooki, A.; Lenz, G.; Yu, A.; Fisher, R.I.; Zinzani, P.L.; Dreyling, M. Optimal Management of Adverse Events From Copanlisib in the Treatment of Patients With Non-Hodgkin Lymphomas. Clin. Lymphoma Myeloma Leuk. 2019, 19, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Appleman, L.J.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Weiss, G.J.; Sachdev, J.C.; Chadha, M.; Fulk, M.; et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. 2016, 27, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell. 2018, 34, 186–195. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Hernandez Burgos, P.; Patel, J.; Chen, A. Ivosidenib induction therapy complicated by myopericarditis and cardiogenic shock: A case report and literature review. J. Oncol. Pharm. Pract. 2020, 26, 754–757. [Google Scholar] [CrossRef]
- Galkin, M.; Jonas, B.A. Enasidenib in the treatment of relapsed/refractory acute myeloid leukemia: An evidence-based review of its place in therapy. Core Evid. 2019, 14, 3–17. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Tallman, M.S.; de Botton, S.; Kantarjian, H.M.; Collins, R.; Stein, A.S.; Frattini, M.G.; Xu, Q.; Tosolini, A.; See, W.L.; et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 2019, 33, 2575–2584. [Google Scholar] [CrossRef]
- Giudice, V.; Pagliano, P.; Vatrella, A.; Masullo, A.; Poto, S.; Polverino, B.M.; Gammaldi, R.; Maglio, A.; Sellitto, C.; Vitale, C.; et al. Combination of Ruxolitinib and Eculizumab for Treatment of Severe SARS-CoV-2-Related Acute Respiratory Distress Syndrome: A Controlled Study. Front. Pharmacol. 2020, 11, 857. [Google Scholar] [CrossRef]
- Anand, S.; Stedham, F.; Gudgin, E.; Campbell, P.; Beer, P.; Green, A.R.; Huntly, B.J. Increased basal intracellular signaling patterns do not correlate with JAK2 genotype in human myeloproliferative neoplasms. Blood 2011, 118, 1610–1621. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, G.P.; Young, N.S. The Bethesda Handbook of Clinical Hematology, 4th ed.; Lippincott Williams & Wilkins (LWW): Philadelphia, PA, USA, 2018; ISBN 978-1-49-635400-6. [Google Scholar]
- Pardanani, A.; Tefferi, A. How I treat myelofibrosis after failure of JAK inhibitors. Blood 2018, 132, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; Di Persio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef]
- von Bubnoff, N.; Ihorst, G.; Grishina, O.; Röthling, N.; Bertz, H.; Duyster, J.; Finke, J.; Zeiser, R. Ruxolitinib in GvHD (RIG) study: A multicenter, randomized phase 2 trial to determine the response rate of Ruxolitinib and best available treatment (BAT) versus BAT in steroid-refractory acute graft-versus-host disease (aGvHD) (NCT02396628). BMC Cancer 2018, 18, 1132. [Google Scholar] [CrossRef]
- Sapre, M.; Tremblay, D.; Wilck, E.; James, A.; Leiter, A.; Coltoff, A.; Koshy, A.G.; Kremyanskaya, M.; Hoffman, R.; Mascarenhas, J.O.; et al. Metabolic Effects of JAK1/2 Inhibition in Patients with Myeloproliferative Neoplasms. Sci. Rep. 2019, 9, 16609. [Google Scholar] [CrossRef]
- Punwani, N.; Yeleswaram, S.; Chen, X.; Bowman, J.; Soloviev, M.; Williams, W. Evaluation of the effect of ruxolitinib on cardiac repolarization: A thorough QT study. Clin. Pharmacol. Drug Dev. 2014, 3, 207–214. [Google Scholar] [CrossRef]
- Miyawaki, H.; Kioka, H.; Sato, K.; Kurashige, M.; Ozawa, T.; Shibayama, H.; Hikoso, S.; Morii, E.; Yamauchi-Takihara, K.; Sakata, Y. Long-term Effects of the Janus Kinase 1/2 Inhibitor Ruxolitinib on Pulmonary Hypertension and the Cardiac Function in a Patient with Myelofibrosis. Intern. Med. 2020, 59, 229–233. [Google Scholar] [CrossRef]
- Talpaz, M.; Kiladjian, J.J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2020, 2020, 1–17. [Google Scholar] [CrossRef]
- Mullally, A.; Hood, J.; Harrison, C.; Mesa, R. Fedratinib in myelofibrosis. Blood Adv. 2020, 4, 1792–1800. [Google Scholar] [CrossRef]
- Schieber, M.; Crispino, J.D.; Stein, B. Myelofibrosis in 2019: Moving beyond JAK2 inhibition. Blood Cancer J. 2019, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Bewersdorf, J.P.; Jaszczur, S.M.; Afifi, S.; Zhao, J.C.; Zeidan, A.M. Beyond Ruxolitinib: Fedratinib and Other Emergent Treatment Options for Myelofibrosis. Cancer Manag. Res. 2019, 11, 10777–10790. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and Efficacy of Fedratinib in Patients With Primary or Secondary Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Marcucci, G.; Perrotti, D.; Caligiuri, M.A. Understanding the molecular basis of imatinib mesylate therapy in chronic myelogenous leukemia and the related mechanisms of resistance. Commentary re: A. N. Mohamed et al., The effect of imatinib mesylate on patients with Philadelphia chromosome-positive chronic myeloid leukemia with secondary chromosomal aberrations. Clin. Cancer Res. 2003, 9, 1248–1252. [Google Scholar]
- Kerkelä, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef]
- Distler, J.H.; Distler, O. Cardiotoxicity of imatinib mesylate: An extremely rare phenomenon or a major side effect? Ann. Rheum Dis. 2007, 66, 836. [Google Scholar] [CrossRef]
- Ribeiro, A.L.; Marcolino, M.S.; Bittencourt, H.N.; Barbosa, M.M.; Nunes Mdo, C.; Xavier, V.F.; Clementino, N.C. An evaluation of the cardiotoxicity of imatinib mesylate. Leuk. Res. 2008, 32, 1809–1814. [Google Scholar] [CrossRef]
- Francisco, A.R.G.; Alves, D.; David, C.; Guerra, L.; Pinto, F.J.; Almeida, A.G. Cardiotoxicity in Hematological Diseases: Are the Tyrosine Kinase Inhibitors Imatinib and Nilotinib Safe? Cardiovasc. Toxicol. 2018, 18, 431–435. [Google Scholar] [CrossRef]
- Estabragh, Z.R.; Knight, K.; Watmough, S.J.; Lane, S.; Vinjamuri, S.; Hart, G.; Clark, R.E. A prospective evaluation of cardiac function in patients with chronic myeloid leukaemia treated with imatinib. Leuk. Res. 2011, 35, 49–51. [Google Scholar] [CrossRef]
- Orphanos, G.S.; Ioannidis, G.N.; Ardavanis, A.G. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol. 2009, 48, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Atallah, E.; Durand, J.B.; Kantarjian, H.; Cortes, J. Congestive heart failure is a rare event in patients receiving imatinib therapy. Blood 2007, 110, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Maharsy, W.; Aries, A.; Mansour, O.; Komati, H.; Nemer, M. Ageing is a risk factor in imatinib mesylate cardiotoxicity. Eur. J. Heart Fail. 2014, 16, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Cang, S.; Yang, T.; Liu, D. Cardiotoxicity of tyrosine kinase inhibitors in chronic myelogenous leukemia therapy. Hematol. Rev. 2009, 1, e4. [Google Scholar] [CrossRef]
- Magdy, T.; Burmeister, B.T.; Burridge, P.W. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther. 2016, 168, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Brave, M.; Goodman, V.; Kaminskas, E.; Farrell, A.; Timmer, W.; Pope, S.; Harapanhalli, R.; Saber, H.; Morse, D.; Bullock, J.; et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin. Cancer Res. 2008, 14, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Krauth, M.T.; Herndlhofer, S.; Schmook, M.T.; Mitterbauer-Hohendanner, G.; Schlögl, E.; Valent, P. Extensive pleural and pericardial effusion in chronic myeloid leukemia during treatment with dasatinib at 100 mg or 50 mg daily. Haematologica 2011, 96, 163–166. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Kantarjian, H.; O’brien, S.; Borthakur, G.; Bruzzi, J.; Munden, R.; Cortes, J. Pleural effusion in patients with chronic myelogenous leukemia treated with dasatinib after imatinib failure. J. Clin. Oncol. 2007, 25, 3908–3914. [Google Scholar] [CrossRef]
- Cortes, J.E.; Jimenez, C.A.; Mauro, M.J.; Geyer, A.; Pinilla-Ibarz, J.; Smith, B.D. Pleural Effusion in Dasatinib-Treated Patients With Chronic Myeloid Leukemia in Chronic Phase: Identification and Management. Clin. Lymphoma Myeloma Leuk. 2017, 17, 78–82. [Google Scholar] [CrossRef]
- Porkka, K.; Khoury, H.J.; Paquette, R.L.; Matloub, Y.; Sinha, R.; Cortes, J.E. Dasatinib 100 mg once daily minimizes the occurrence of pleural effusion in patients with chronic myeloid leukemia in chronic phase and efficacy is unaffected in patients who develop pleural effusion. Cancer 2010, 116, 377–386. [Google Scholar] [CrossRef]
- Visani, G.; Breccia, M.; Montefusco, E.; Morra, E.; Santini, V.; Isidori, A. The incidence of pleural and pericardial effusion is not higher in patients receiving dasatinib at low doses. (Reply). Haematologica 2011, 96, e23–e24, author reply e25. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Giles, F.; Wunderle, L.; Bhalla, K.; O’Brien, S.; Wassmann, B.; Tanaka, C.; Manley, P.; Rae, P.; Mietlowski, W.; et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N. Engl. J. Med. 2006, 354, 2542–2551. [Google Scholar] [CrossRef] [PubMed]
- Shopp, G.M.; Helson, L.; Bouchard, A.; Salvail, D.; Majeed, M. Liposomes ameliorate Crizotinib- and Nilotinib-induced inhibition of the cardiac IKr channel and QTc prolongation. Anticancer Res. 2014, 34, 4733–4740. [Google Scholar] [PubMed]
- Valent, P.; Hadzijusufovic, E.; Schernthaner, G.H.; Wolf, D.; Rea, D.; le Coutre, P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood 2015, 125, 901–906. [Google Scholar] [CrossRef]
- Doherty, K.R.; Wappel, R.L.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Kramer, J.W.; Brown, A.M.; Shell, S.A.; Bacus, S. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol. Appl. Pharmacol. 2013, 272, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; le Coutre, P.; Schwarz, M.; Grille, P.; Levitin, M.; Fateh-Moghadam, S.; Giles, F.J.; Dörken, B.; Haverkamp, W.; Köhncke, C. Clinical cardiac safety profile of nilotinib. Haematologica 2012, 97, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Aichberger, K.J.; Herndlhofer, S.; Schernthaner, G.H.; Schillinger, M.; Mitterbauer-Hohendanner, G.; Sillaber, C.; Valent, P. Progressive peripheral arterial occlusive disease and other vascular events during nilotinib therapy in CML. Am. J. Hematol. 2011, 86, 533–539. [Google Scholar] [CrossRef]
- Cortes, J.E.; Jean Khoury, H.; Kantarjian, H.; Brümmendorf, T.H.; Mauro, M.J.; Matczak, E.; Pavlov, D.; Aguiar, J.M.; Fly, K.D.; Dimitrov, S.; et al. Long-term evaluation of cardiac and vascular toxicity in patients with Philadelphia chromosome-positive leukemias treated with bosutinib. Am. J. Hematol. 2016, 91, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Mahida, H.; Gharia, B.; Ugoeke, N.; Maludum, O.; Asif, A.; Calderon, D. Abstract 11853: Cardiovascular Adverse Events Associated With First, Second and Third Generation Tyrosine Kinase Inhibitors Used in Treatment of Chronic Myeloid Leukemia. Circulation 2018, 138, A11853. [Google Scholar]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef]
- Tamai, M.; Inukai, T.; Kojika, S.; Abe, M.; Kagami, K.; Harama, D.; Shinohara, T.; Watanabe, A.; Oshiro, H.; Akahane, K.; et al. T315I mutation of BCR-ABL1 into human Philadelphia chromosome-positive leukemia cell lines by homologous recombination using the CRISPR/Cas9 system. Sci. Rep. 2018, 8, 9966. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Umbarkar, P.; Tousif, S.; Lal, H. Cardiotoxicity of the BCR-ABL1 tyrosine kinase inhibitors: Emphasis on ponatinib. Int. J. Cardiol. 2020, 316, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Xu, Z.; Yan, H.; He, Q.; Yang, X.; Luo, P. A Comprehensive Review of Clinical Cardiotoxicity Incidence of FDA-Approved Small-Molecule Kinase Inhibitors. Front. Pharmacol. 2020, 11, 891. [Google Scholar] [CrossRef] [PubMed]
- Dorer, D.J.; Knickerbocker, R.K.; Baccarani, M.; Cortes, J.E.; Hochhaus, A.; Talpaz, M.; Haluska, F.G. Impact of dose intensity of ponatinib on selected adverse events: Multivariate analyses from a pooled population of clinical trial patients. Leuk. Res. 2016, 48, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.D.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: Final 5-year results of the phase 2 PACE trial. Blood 2018, 132, 393–404. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef]
- Caocci, G.; Mulas, O.; Bonifacio, M.; Abruzzese, E.; Galimberti, S.; Orlandi, E.M.; Iurlo, A.; Annunziata, M.; Luciano, L.; Castagnetti, F.; et al. Recurrent arterial occlusive events in patients with chronic myeloid leukemia treated with second- and third-generation tyrosine kinase inhibitors and role of secondary prevention. Int. J. Cardiol. 2019, 288, 124–127. [Google Scholar] [CrossRef]
- Caocci, G.; Mulas, O.; Abruzzese, E.; Luciano, L.; Iurlo, A.; Attolico, I.; Castagnetti, F.; Galimberti, S.; Sgherza, N.; Bonifacio, M.; et al. Arterial occlusive events in chronic myeloid leukemia patients treated with ponatinib in the real-life practice are predicted by the Systematic Coronary Risk Evaluation (SCORE) chart. Hematol. Oncol. 2019, 37, 296–302. [Google Scholar] [CrossRef]
- Tzogani, K.; Røshol, H.; Olsen, H.H.; Aas, I.B.; Dalhus, M.L.; Håkonsen, G.D.; Nilssen, L.S.; Lindberg, V.; Økvist, M.; Bolstad, B.; et al. The European Medicines Agency Review of Gilteritinib (Xospata) for the Treatment of Adult Patients with Relapsed or Refractory Acute Myeloid Leukemia with an FLT3 Mutation. Oncologist 2020, 25, e1070–e1076. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Fostvedt, L.; Shaik, N.; Martinelli, G.; Wagner, A.; Ruiz-Garcia, A. Abstract 3889: Population pharmacokinetic/pharmacodynamic evaluation of the effect of glasdegib exposure on cardiac repolarization (QT interval) in cancer patients. Cancer Res. 2019, 79, 3889. [Google Scholar]
- del Corral, A.; Dutreix, C.; Huntsman-Labed, A.; Lorenzo, S.; Morganroth, J.; Harrell, R.; Wang, Y. Midostaurin does not prolong cardiac repolarization defined in a thorough electrocardiogram trial in healthy volunteers. Cancer Chemother. Pharmacol. 2012, 69, 1255–1263. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gallogly, M.M.; Lazarus, H.M. Midostaurin: An emerging treatment for acute myeloid leukemia patients. J. Blood Med. 2016, 7, 73–83. [Google Scholar] [PubMed]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef]
- Vaidya, P.; Khedro, T.; Yaghmour, B.; Yaghmour, G. Midostaurin-Related Interstitial Lung Injury in FLT3+ Acute Myeloid Leukemia Post-Allogeneic Transplant. World J. Oncol. 2019, 10, 237–239. [Google Scholar] [CrossRef]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef]
- Plummer, C.; Driessen, C.; Szabo, Z.; Mateos, M.V. Management of cardiovascular risk in patients with multiple myeloma. Blood Cancer J. 2019, 9, 26. [Google Scholar] [CrossRef]
- Nowis, D.; Maczewski, M.; Mackiewicz, U.; Kujawa, M.; Ratajska, A.; Wieckowski, M.R.; Wilczyński, G.M.; Malinowska, M.; Bil, J.; Salwa, P.; et al. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am. J. Pathol. 2010, 176, 2658–2668. [Google Scholar] [CrossRef]
- Xiao, Y.; Yin, J.; Wei, J.; Shang, Z. Incidence and risk of cardiotoxicity associated with bortezomib in the treatment of cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e87671. [Google Scholar] [CrossRef]
- Jaramillo Restrepo, V.; Shah, V.S.; Jain, T.; Larsen, J.T.; Narayanasamy, H.; Arsanjani, R.; Hardaway, B.W.; Mookadam, F.; Larsen, C. Cardiovascular outcomes following bortezomib reinitiation after congestive heart failure diagnosis in multiple myeloma. J. Am. Coll. Cardiol. 2020, 75, 723. [Google Scholar] [CrossRef]
- Reneau, J.; Asante, D.; Van Houten, H.; Buadi, F.; Lerman, A.; Herrmann, J. Cardiotoxicity Risk with Bortezomib Versus Lenalidomide for Treatment of Multiple Myeloma: A Propensity-Matched Study of 1128 Patients. Blood 2015, 126, 3046. [Google Scholar] [CrossRef]
- Land, J.; Afifi, S.; Adel, N.G.; Devlin, S.; Arora, A.; Lendvai, N.; Landgren, O. Incidence and Management of Proteasome Inhibitor-Related Cardiotoxicity in Multiple Myeloma Patients at Memorial Sloan Kettering Cancer Center. Blood 2015, 126, 4265. [Google Scholar] [CrossRef]
- Wu, P.; Oren, O.; Gertz, M.A.; Yang, E.H. Proteasome Inhibitor-Related Cardiotoxicity: Mechanisms, Diagnosis, and Management. Curr. Oncol. Rep. 2020, 22, 66. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, M.; Chakravarty, S.; Schulman, P.; Bockorny, B.; Bona, R. Severe heart failure after bortezomib treatment in a patient with multiple myeloma: A case report and review of the literature. Acta Haematol. 2012, 128, 244–247. [Google Scholar] [CrossRef]
- Jerkins, J.H.; Suciu, A.; Mazimba, S.; Calvo, A. Bortezomib-induced Severe Congestive Heart Failure. Cardiol. Res. 2010, 1, 20–23. [Google Scholar] [CrossRef][Green Version]
- Diwadkar, S.; Patel, A.A.; Fradley, M.G. Bortezomib-Induced Complete Heart Block and Myocardial Scar: The Potential Role of Cardiac Biomarkers in Monitoring Cardiotoxicity. Case Rep. Cardiol. 2016, 2016, 3456287. [Google Scholar] [CrossRef]
- Buza, V.; Rajagopalan, B.; Curtis, A.B. Cancer Treatment-Induced Arrhythmias: Focus on Chemotherapy and Targeted Therapies. Circ. Arrhythm. Electrophysiol. 2017, 10, e005443. [Google Scholar] [CrossRef]
- Chen, J.H.; Lenihan, D.J.; Phillips, S.E.; Harrell, S.L.; Cornell, R.F. Cardiac events during treatment with proteasome inhibitor therapy for multiple myeloma. Cardiooncology 2017, 3, 4. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; DeCara, J.M.; Mezzi, K. Cardiovascular events during carfilzomib therapy for relapsed myeloma: Practical management aspects from two case studies. Hematology 2017, 22, 585–591. [Google Scholar] [CrossRef]
- Siegel, D.; Martin, T.; Nooka, A.; Harvey, R.D.; Vij, R.; Niesvizky, R.; Badros, A.Z.; Jagannath, S.; McCulloch, L.; Rajangam, K.; et al. Integrated safety profile of single-agent carfilzomib: Experience from 526 patients enrolled in 4 phase II clinical studies. Haematologica 2013, 98, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Bringhen, S.; Milan, A.; Ferri, C.; Wäsch, R.; Gay, F.; Larocca, A.; Salvini, M.; Terpos, E.; Goldschmidt, H.; Cavo, M.; et al. Cardiovascular adverse events in modern myeloma therapy—Incidence and risks. A review from the European Myeloma Network (EMN) and Italian Society of Arterial Hypertension (SIIA). Haematologica 2018, 103, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef]
- Di Lullo, G.; Marcatti, M.; Heltai, S.; Tresoldi, C.; Paganoni, A.M.; Bordignon, C.; Ciceri, F.; Protti, M.P. Immunomodulatory Drugs in the Context of Autologous Hematopoietic Stem Cell Transplantation Associate With Reduced Pro-tumor T Cell Subsets in Multiple Myeloma. Front. Immunol. 2019, 9, 3171. [Google Scholar] [CrossRef]
- Delforge, M.; Ludwig, H. How I manage the toxicities of myeloma drugs. Blood 2017, 129, 2359–2367. [Google Scholar] [CrossRef]
- Li, W.; Cornell, R.F.; Lenihan, D.; Slosky, D.; Jagasia, M.; Piazza, G.; Moslehi, J. Cardiovascular Complications of Novel Multiple Myeloma Treatments. Circulation 2016, 133, 908–912. [Google Scholar] [CrossRef]
- Reneau, J.C.; Asante, D.; van Houten, H.; Sangaralingham, L.R.; Buadi, F.K.; Lerman, A.; Herrmann, J. Cardiotoxicity risk with bortezomib versus lenalidomide for treatment of multiple myeloma: A propensity matched study of 1790 patients. Am. J. Hematol. 2017, 92, 15–17. [Google Scholar] [CrossRef]
- Li, W.; Garcia, D.; Cornell, R.F.; Gailani, D.; Laubach, J.; Maglio, M.E.; Richardson, P.G.; Moslehi, J. Cardiovascular and Thrombotic Complications of Novel Multiple Myeloma Therapies: A Review. JAMA Oncol. 2017, 3, 980–988. [Google Scholar] [CrossRef]
- Fradley, M.G.; Groarke, J.D.; Laubach, J.; Alsina, M.; Lenihan, D.J.; Cornell, R.F.; Maglio, M.; Shain, K.H.; Richardson, P.G.; Moslehi, J. Recurrent cardiotoxicity potentiated by the interaction of proteasome inhibitor and immunomodulatory therapy for the treatment of multiple myeloma. Br. J. Haematol. 2018, 180, 271–275. [Google Scholar] [CrossRef]
- El Accaoui, R.N.; Shamseddeen, W.A.; Taher, A.T. Thalidomide and thrombosis. A meta-analysis. Thromb. Haemost. 2007, 97, 1031–1036. [Google Scholar] [PubMed]
- Dimopoulos, M.A.; Chen, C.; Spencer, A.; Niesvizky, R.; Attal, M.; Stadtmauer, E.A.; Petrucci, M.T.; Yu, Z.; Olesnyckyj, M.; Zeldis, J.B.; et al. Long-term follow-up on overall survival from the MM-009 and MM-010 phase III trials of lenalidomide plus dexamethasone in patients with relapsed or refractory multiple myeloma. Leukemia 2009, 23, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Issa, J.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948. [Google Scholar] [CrossRef] [PubMed]
- Kambara, Y.; Yamamoto, A.; Masunari, T.; Sezaki, N.; Sugiura, H.; Ikegawa, S.; Meguri, Y.; Kiguchi, T.; Maeda, Y. A Risk-Scoring System to Predict Heart Failure Onset during Treatment of Myelodysplastic Syndrome with Azacitidine. Ann. Hematol. Oncol. 2019, 6, 1248. [Google Scholar]
- Newman, M.; Malla, M.; Gojo, I. Azacitidine-Induced Pericarditis: A Case Series. Pharmacotherapy 2016, 36, 443–448. [Google Scholar] [CrossRef]
- Agasthi, P.; Narayanasamy, H.; Sorajja, D.; Slack, J.; Mookadam, F. Decitabine Induced Delayed Cardiomyopathy in Hematologic Malignancy. Case Rep. Cardiol. 2018, 2018, 3953579. [Google Scholar] [CrossRef]
- Cuesta-Mateos, C.; Alcaraz-Serna, A.; Somovilla-Crespo, B.; Muñoz-Calleja, C. Monoclonal Antibody Therapies for Hematological Malignancies: Not Just Lineage-Specific Targets. Front. Immunol. 2018, 8, 1936. [Google Scholar] [CrossRef]
- Weiner, G.J. Monoclonal antibody mechanisms of action in cancer. Immunol. Res. 2007, 39, 271–278. [Google Scholar] [CrossRef]
- Craig, F.E.; Foon, K.A. Flow cytometric immunophenotyping for hematologic neoplasms. Blood 2008, 111, 3941–3967. [Google Scholar] [CrossRef]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun. Highlights 2017, 8, 12. [Google Scholar] [CrossRef]
- Alduaij, W.; Illidge, T.M. The future of anti-CD20 monoclonal antibodies: Are we making progress? Blood 2011, 117, 2993–3001. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.C.J.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Jelínek, T.; Mihályová, J.; Hájek, R. CD38 targeted treatment for multiple myeloma. Vnitr. Lek. 2018, 64, 939–948. [Google Scholar] [PubMed]
- Trudel, S.; Moreau, P.; Touzeau, C. Update on elotuzumab for the treatment of relapsed/refractory multiple myeloma: Patients’ selection and perspective. Onco Targets Ther. 2019, 12, 5813–5822. [Google Scholar] [CrossRef]
- Richardson, P.G.; Lee, H.C.; Abdallah, A.O.; Cohen, A.D.; Kapoor, P.; Voorhees, P.M.; Hoos, A.; Wang, K.; Baron, J.; Piontek, T.; et al. Single-agent belantamab mafodotin for relapsed/refractory multiple myeloma: Analysis of the lyophilised presentation cohort from the pivotal DREAMM-2 study. Blood Cancer J. 2020, 10, 106. [Google Scholar] [CrossRef]
- Scott, L.J. Brentuximab Vedotin: A Review in CD30-Positive Hodgkin Lymphoma. Drugs 2017, 77, 435–445. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Dhimolea, E. Brentuximab vedotin. MAbs 2012, 4, 458–465. [Google Scholar] [CrossRef]
- De Re, V.; Caggiari, L.; Repetto, O.; Mussolin, L.; Mascarin, M. Classical Hodgkin’s Lymphoma in the Era of Immune Checkpoint Inhibition. J. Clin. Med. 2019, 8, 1596. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; González Barca, E.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Douglas, M. Polatuzumab Vedotin for the Treatment of Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Transplant-Ineligible Patients. J. Adv. Pract. Oncol. 2020, 11, 521–528. [Google Scholar]
- Trapani, D.; Zagami, P.; Nicolò, E.; Pravettoni, G.; Curigliano, G. Management of Cardiac Toxicity Induced by Chemotherapy. J. Clin. Med. 2020, 9, 2885. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.S.; Ayub-Ferreira, S.M.; de Barros Wanderley, M.R., Jr.; das Dores Cruz, F.; Gonçalves Brandão, S.M.; Rigaud, V.O.C.; Higuchi-Dos-Santos, M.H.; Hajjar, L.A.; Kalil Filho, R.; Hoff, P.M.; et al. Carvedilol for Prevention of Chemotherapy-Related Cardiotoxicity: The CECCY Trial. J. Am. Coll. Cardiol. 2018, 71, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, R.; Ramirez, M.C.; Nes, K.; Schuster, A.; Aguayo, R.; Morales, M.; Ramos, C.; Hasson, D.; Sotomayor, C.G.; Henriquez, P.; et al. Prevention of doxorubicin-induced Cardiotoxicity by pharmacological non-hypoxic myocardial preconditioning based on Docosahexaenoic Acid (DHA) and carvedilol direct antioxidant effects: Study protocol for a pilot, randomized, double-blind, controlled trial (CarDHA trial). Trials 2020, 21, 137. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| MoAb | Target | Indications | Cardiotoxicity |
|---|---|---|---|
| Rituximab | CD20 | NHL CLL | IRRs |
| Hypertension (6–12%) | |||
| Transient hypotension (10%) | |||
| SVT, AF | |||
| Takotsubo * | |||
| CAVB *, ST *, SB *, PVC *, VT *, QTc/TdP *, SCD * | |||
| Obinutuzumab | CD20 | CLL/FL | IRRs, SCD *, HF * |
| Ofatumumab | CD20 | CLL | IRRs |
| Hypertension/hypotension | |||
| Tachycardia | |||
| AF *, HF *, MI *, pericarditis * | |||
| Daratumumab | CD38 | MM | IRRs |
| Hypertension | |||
| AF | |||
| SCD * | |||
| Isatuximab | CD38 | MM | IRRs |
| AF | |||
| Elotuzumab | SLAMF7 | MM | IRRs |
| DVT | |||
| Brentuximab vedotin | CD30 | HL | ST (6%) |
| Pericardial effusion (3%) | |||
| CHF *, MI * | |||
| Nivolumab | PD-1 | HL | Myocarditis |
| Pericardial diseases | |||
| Stress cardiomyopathy | |||
| VT | |||
| CAVB *, SCD * | |||
| Pembrolizumab | PD-1 | HL | Myocarditis |
| Pericardial diseases | |||
| Stress cardiomyopathy | |||
| ST *, AF *, PVC *, VT *, SCD * | |||
| Gemtuzumab ozogamicin | CD33 | AML | VOD (2%) |
| Tachycardia, ST, SVT (13%) | |||
| Hypertension (17.3%) | |||
| Blinatumomab | CD19/CD3 | R/R Ph-ALL | Tachycardia |
| HF * | |||
| Belantamab mafodotin | BCMA | MM | Not reported |
| Inotuzumab ozogamicin | CD22 | Ph+ ALL | VOD |
| QT/QTc | |||
| Moxetumomab pasudotox | CD22 | R/R HCL | HUS * |
| Pericardial/pleural effusion * | |||
| Hypertension/hypotension * | |||
| Tachycardia * | |||
| Tafasitamab | CD19 | DLBCL | Pulmonary embolism (4%) |
| AF (2%) | |||
| CHF (2%) | |||
| Polatuzumab vedotin | CD79a | DLBCL | Hypotension |
| Ravulizumab | C5a | PNH HUS | Hypertension/hypotension * |
| Eculizumab | C5a | PNH HUS | Hypertension |
| Tachycardia/Palpitation | |||
| Cardiomyopathy * | |||
| Hypotension * | |||
| Emapalumab | IFNγ | R/R HLH | Hypertension (41%) |
| Tachycardia (12%) | |||
| Bradycardia * | |||
| Siltuximab | IL-6 | Castleman disease | Hypertension |
| Peripheral edema (26%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giudice, V.; Vecchione, C.; Selleri, C. Cardiotoxicity of Novel Targeted Hematological Therapies. Life 2020, 10, 344. https://doi.org/10.3390/life10120344
Giudice V, Vecchione C, Selleri C. Cardiotoxicity of Novel Targeted Hematological Therapies. Life. 2020; 10(12):344. https://doi.org/10.3390/life10120344
Chicago/Turabian StyleGiudice, Valentina, Carmine Vecchione, and Carmine Selleri. 2020. "Cardiotoxicity of Novel Targeted Hematological Therapies" Life 10, no. 12: 344. https://doi.org/10.3390/life10120344
APA StyleGiudice, V., Vecchione, C., & Selleri, C. (2020). Cardiotoxicity of Novel Targeted Hematological Therapies. Life, 10(12), 344. https://doi.org/10.3390/life10120344