Geochemical Characterization of Iron and Steel Slag and Its Potential to Remove Phosphate and Neutralize Acid

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description and Sampling Strategy
2.2. Mineralogical and Geochemical Investigations
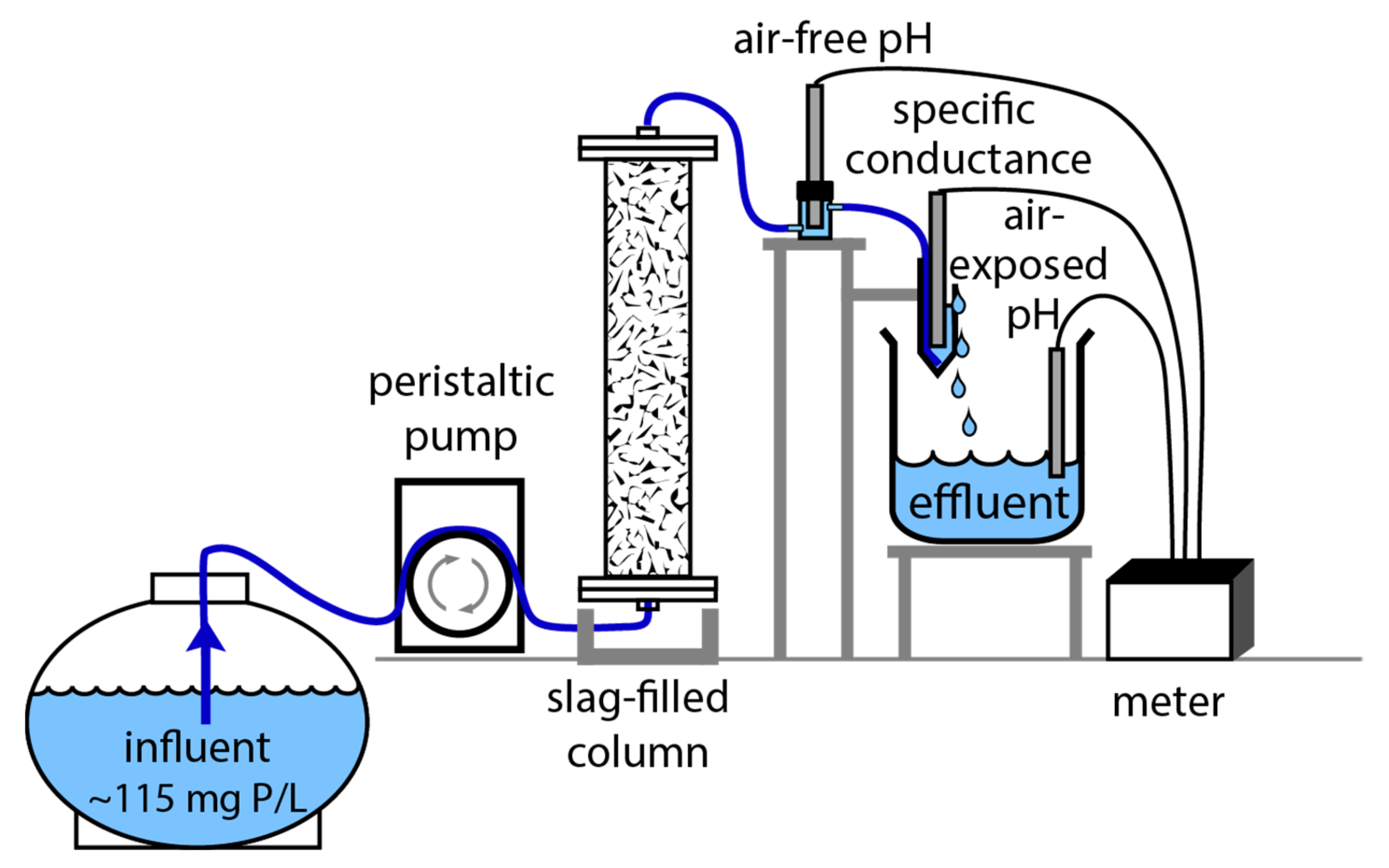
2.3. Phosphate Removal Experiments
3. Results
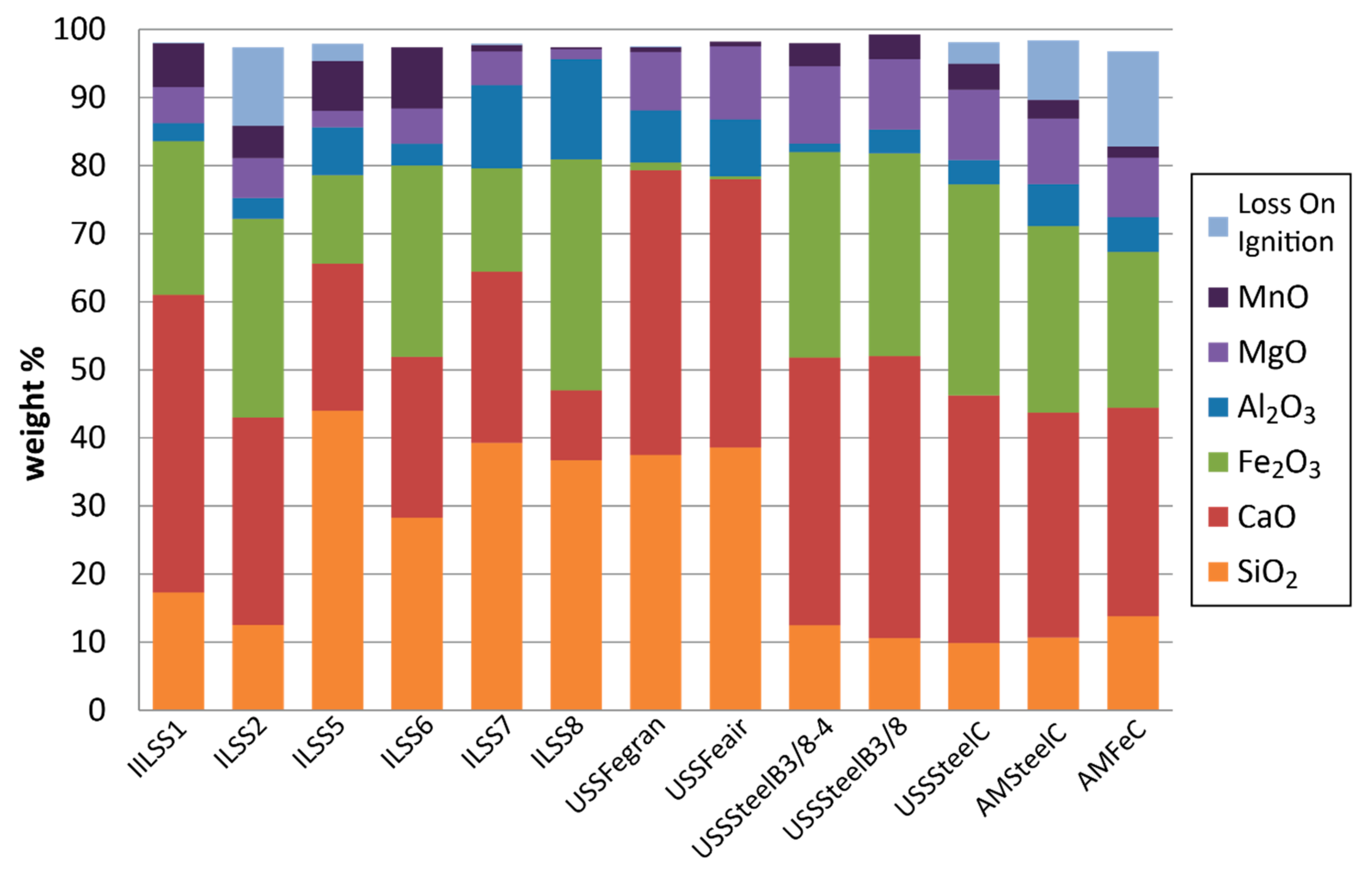
3.1. Geochemical Characteristics
3.2. Mineralogy
3.3. Synthetic Precipitation Leaching Procedure
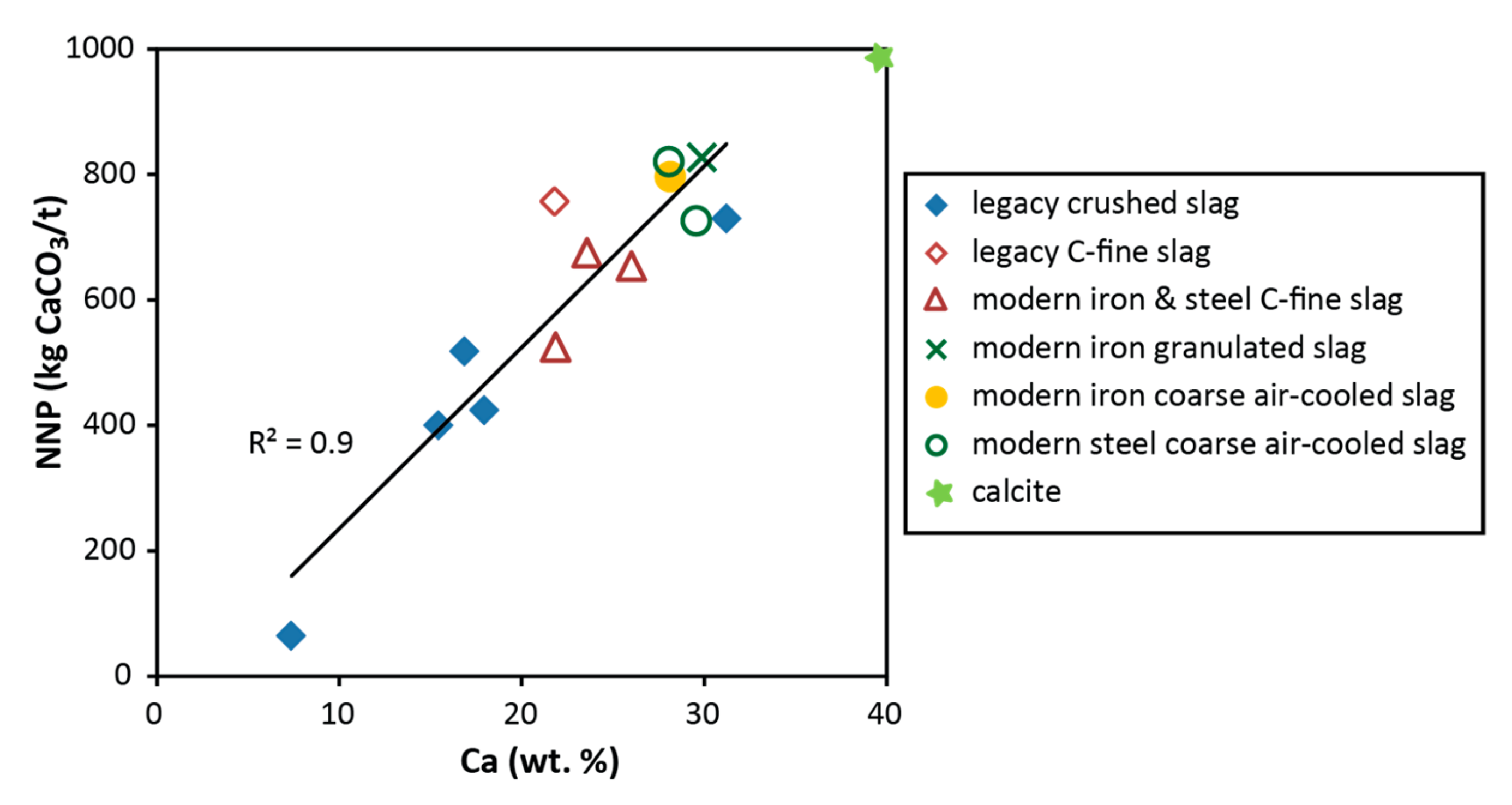
3.4. Neutralization Potential
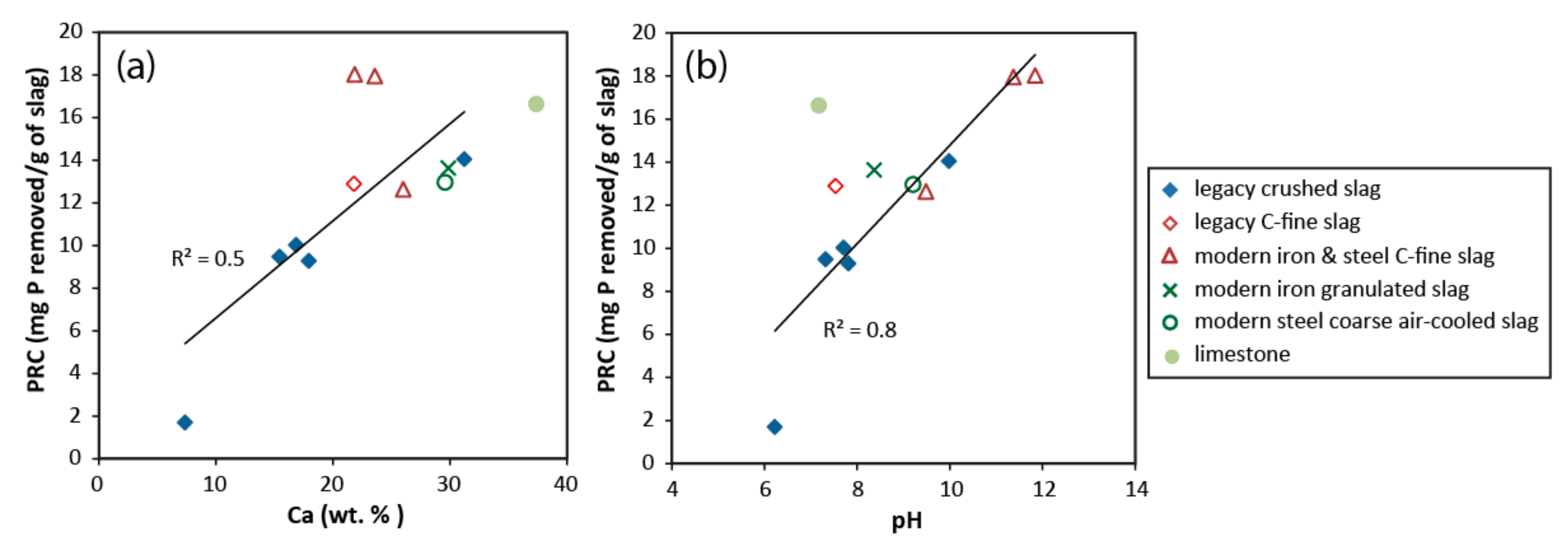
3.5. Phosphate Removal Capacity
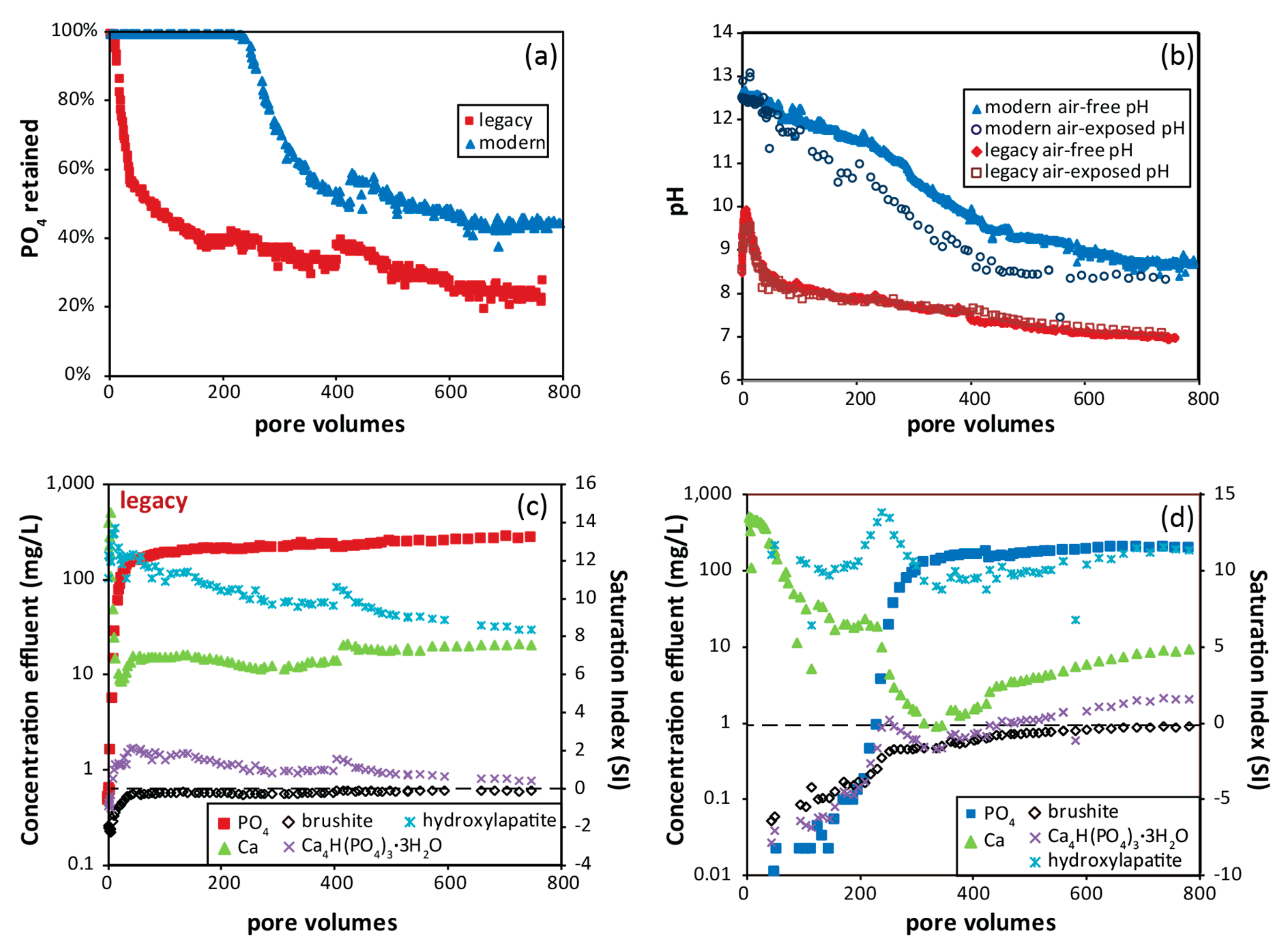
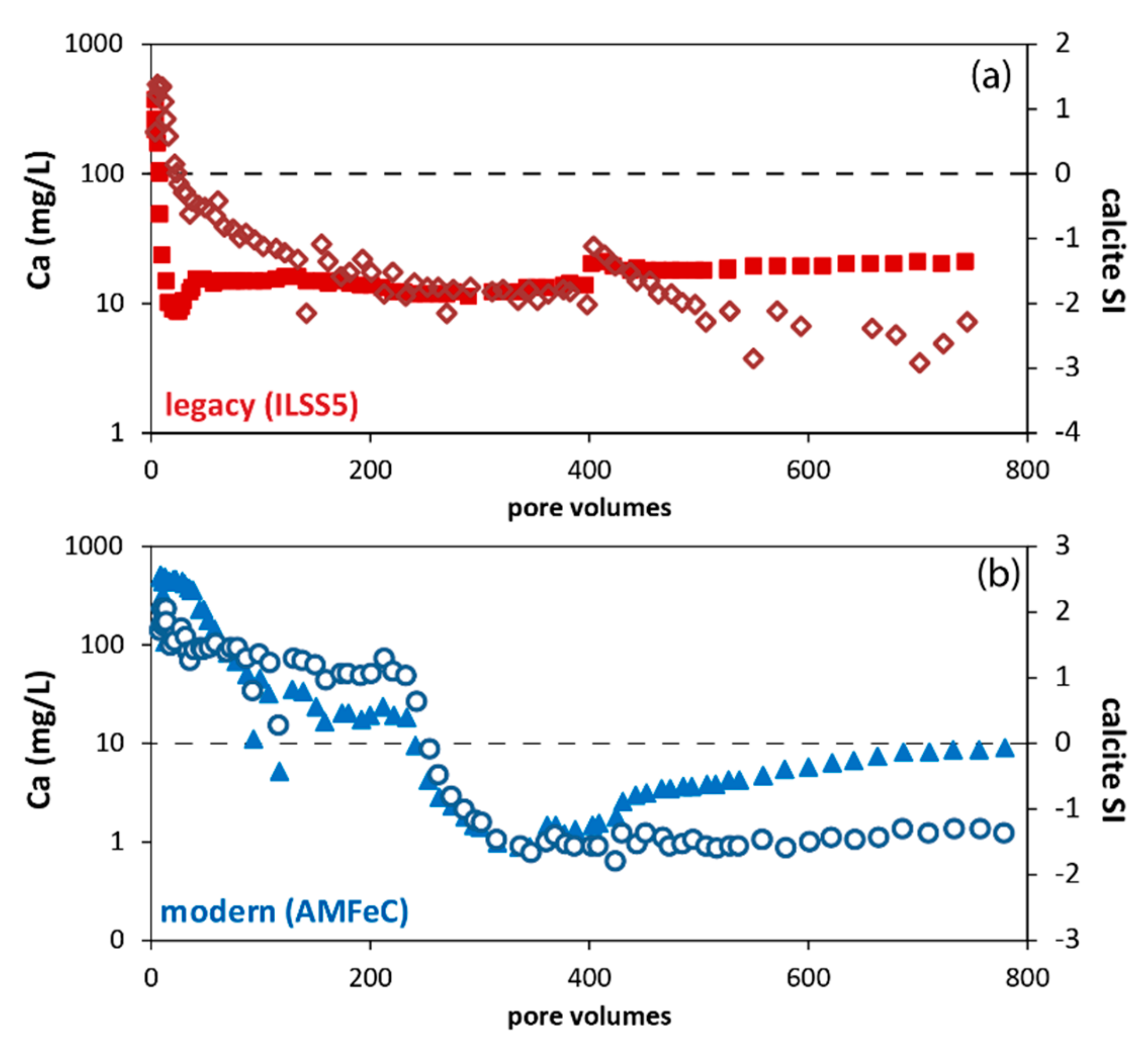
3.6. Phosphate Removal Column Experiments
4. Discussion
4.1. Environmental Characteristics of Slag Samples
4.2. Acid Neutralizatino and Potential for Application in Water Treatment
4.3. Phosphate Removal Mechanisms and Chemistry of Reacted Solutions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Piatak, N.M.; Parsons, M.B.; Seal, R.R., II. Characteristics and Environmental Aspects of Slag: A Review. Appl. Geochem. 2015, 57, 236–266. [Google Scholar] [CrossRef]
- Van Oss, H.G. Slag, iron and steel. U.S. Geol. Surv. Miner. Yearb. 2018, 1, 69.1–69.8. [Google Scholar]
- Baker, M.J.; Blowes, D.W.; Ptacek, C.J. Laboratory development of permeable reactive mixtures for the removal of phosphorus from onsite wastewater disposal systems. Environ. Sci. Technol. 1998, 32, 2308–2316. [Google Scholar] [CrossRef]
- Drizo, A.; Comeau, Y.; Forget, C.; Chapuis, R.P. Phosphorus saturation potential: A parameter for estimating the longevity of constructed wetland systems. Environ. Sci. Technol. 2002, 36, 4642–4648. [Google Scholar] [CrossRef] [PubMed]
- Chazarenc, F.; Kacem, M.; Gérente, C.; Andrès, Y. ‘Active’ filters: A mini-review on the use of industrial by-products for upgrading phosphorus removal from treatment wetlands. In Proceedings of the 11th International Conference on Wetland Systems for Water Pollution Control, Indore, India, 1–7 November 2008; pp. 727–737. [Google Scholar]
- Bowden, L.I.; Jarvis, A.P.; Younger, P.L.; Johnson, K.L. Phosphorus removal from waste waters using basic oxygen steel slag. Environ. Sci. Technol. 2009, 43, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Pratt, C.; Shilton, A.; Haverkamp, R.G.; Pratt, S. Assessment of physical techniques to regenerate active slag filters removing phosphorus from wastewater. Water Res. 2009, 43, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Pratt, C.; Shilton, A. Active slag filters-simple and sustainable phosphorus removal from wastewater using steel industry byproduct. Water Sci. Technol. 2010, 62, 1713–1718. [Google Scholar] [CrossRef][Green Version]
- Barca, C.; Gérente, C.; Meyer, D.; Chazarenc, F.; Andrès, Y. Phosphate removal from synthetic and real wastewater using steel slags produced in Europe. Water Res. 2012, 46, 2376–2384. [Google Scholar] [CrossRef]
- Barca, C.; Meyer, D.; Liira, M.; Drissen, P.; Comeau, Y.; Andrès, Y.; Chazarenc, F. Steel slag filters to upgrade phosphorus removal in constructed wetlands: Two years of field experiments. Environ. Sci. Technol. 2013, 47, 549–556. [Google Scholar] [CrossRef]
- Barca, C.; Meyer, D.; Liira, M.; Drissen, P.; Comeau, Y.; Andrès, Y.; Chazarenc, F. Steel slag filters to upgrade phosphorus removal in small wastewater treatment plants: Removal mechanisms and performance. Ecol. Eng. 2014, 68, 214–222. [Google Scholar] [CrossRef]
- Claveau-Mallet, D.; Wallace, S.; Comeau, Y. Removal of phosphorus, fluoride and metals from a gypsum mining leachate using steel slag filters. Water Res. 2013, 47, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Claveau-Mallet, D.; Courcelles, B.; Comeau, Y. Phosphorus removal by steel slag filters: Modeling dissolution and precipitation kinetics to predict longevity. Water Sci. Technol. 2014, 48, 7486–7493. [Google Scholar] [CrossRef] [PubMed]
- Claveau-Mallet, D.; Boutet, É.; Comeau, Y. Steel slag filter design criteria for phosphorus removal from wastewater in decentralized applications. Water Res. 2018, 143, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Sellner, B.M.; Hua, G.; Ahiablame, L.M. Fixed bed column evaluation of phosphate adsorption and recovery from aqueous solutions using recycled steel byproducts. J. Environ. Manag. 2019, 233, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Ziemkiewicz, P.; Skousen, J. Steel slag in acid mine drainage treatment and control. In Proceedings of the Annual National Meeting of the Society for Surface Mining and Reclamation, Scottsdale, AZ, USA, 13–19 August 1999; Volume 16, pp. 651–656. [Google Scholar]
- Simmons, J.; Ziemkiewicz, P.; Black, D.C. Use of steel slag leach beds for the treatment of acid mine drainage. Mine Water Environ. 2002, 21, 91–99. [Google Scholar] [CrossRef]
- Cravotta, C.A., III. Assessment of Characteristics and Remedial Alternatives for Abandoned Mine Drainage: Case Study at Staple Bend Tunnel Unit of Allegheny Portage Railroad National Historic Site, Cambria Country, Pennsylvania, 2004; U.S. Geological Survey Open-File Report 2005-1283; United States Geological Survey: Reston, VA, USA, 2005; 52p.
- Gahan, C.S.; Cunha, M.L.; Sandström, Å. Comparative study on different steel slags as neutralising agent in bioleaching. Hydrometallurgy 2009, 95, 190–197. [Google Scholar] [CrossRef]
- Kruse, N.A.; Mackey, A.L.; Bowman, J.R.; Brewster, K.; Riefler, R.G. Alkalinity production as an indicator of failure in steel slag leach beds treating acid mine drainage. Environ. Earth Sci. 2012, 67, 1389–1395. [Google Scholar] [CrossRef]
- Goetz, E.R.; Riefler, R.G. Performance of steel slag leach beds in acid mine drainage treatment. Chem. Eng. J. 2014, 240, 579–588. [Google Scholar] [CrossRef]
- Name, T.; Sheridan, C. Remediation of acid mine drainage using metallurgical slags. Miner. Eng. 2014, 64, 15–22. [Google Scholar] [CrossRef]
- Bayless, E.R.; Greeman, T.K.; Harvey, C.C. Hydrology and Geochemistry of a Slag-Affected Aquifer and Chemical Characteristics of Slag-Affected Ground Water, Northwestern Indiana and Northeastern Illinois; U.S. Geological Survey Water-Resources Investigations Report 97-4198; United States Geological Survey: Reston, VA, USA, 1998; 67p.
- Bayless, E.R.; Schulz, M.S. Mineral precipitation and dissolution at two slag-disposal sites in northwestern Indiana, USA. Environ. Geol. 2003, 45, 252–261. [Google Scholar] [CrossRef]
- Roadcap, G.S.; Kelly, W.R.; Bethke, C.M. Geochemistry of extremely alkaline (pH > 12) ground water in slag-fill aquifers. Ground Water 2005, 43, 806–816. [Google Scholar] [CrossRef]
- Gahala, A.M.; Seal, R.R., II; Piatak, N.M. Preliminary Evaluation of Hydrologic and Chemical-Mass Balance, and Geochemical Modeling at Hyper-Alkaline Ponds of Big March, Chicago, Illinois. U.S. Geological Survey Scientific Investigations Report 2018; United States Geological Survey: Reston, VA, USA, in press.
- Carmichael, W.W.; Boyer, G.L. Health impacts from cyanobacteria harmful algae blooms: Implications for the North American Great Lakes. Harmful Algae 2016, 54, 194–212. [Google Scholar] [CrossRef]
- Powell, J.D. Origin and influence of coal mine drainage on streams of the United States. Environ. Geol. Water Sci. 1988, 11, 141–152. [Google Scholar] [CrossRef]
- Piatak, N.M.; Seal, R.R., II; Hoppe, D.A.; Green, C.G.; Buszka, P.M. Slag: What is it good for? Utilization of steelmaking slag to remove phosphate and neutralize acid. In Proceedings of the 13th International Mine Water Association Congress—Mine Water & Circular Economy, Lappeenranta, Finland, 25–30 June 2017; pp. 869–876. Available online: http://www.imwa.info/docs/imwa_2017/IMWA2017_Piatak_869.pdf (accessed on 23 July 2019).
- International Centre for Diffraction Data. JCPDS PDF-4+ 2018 Powder Diffraction Database; ICDD: Newton Square, PA, USA, 2018. [Google Scholar]
- Hoppe, D.A.; Piatak, N.M.; Seal, R.R., II; Green, C.J. Geochemical Characterization, Acid Neutralization Potential, and Phosphate Removal Capacity of Modern and Legacy Iron and Steel Slag from the Chicago-Gary Area of Illinois and Indiana, USA. U.S. Geological Survey Data Release. Available online: https://doi:10.5066/P9X7SPIK (accessed on 31 July 2019).
- MEND. Acid Rock Drainage Prediction Manual. Project 1.16.1b. 1991, Report Prepared by Coastech Research Inc., Ottawa, MEND, Natural Resources Canada. Available online: http://mend-nedem.org/wp-content/uploads/2013/01/1.16.1b.pdf (accessed on 23 July 2019).
- U.S. Environmental Protection Agency (USEPA). Method 1312; Synthetic Precipitation Leaching Procedure, part of Test Methods for Evaluating Solid Waste, Physical/Chemical Methods. 1994. Available online: https://www.epa.gov/sites/production/files/2015-12/documents/1312.pdf (accessed on 23 July 2019).
- Toride, N.; Leij, F.J.; Van Genuchten, M.T. The CXTFIT Code for Estimating Transport Parameters from Laboratory or Field; Version 2.1. Research Report No. 137; U.S. Department of Agriculture: Riverside, CA, USA, 1999.
- Parker, J.C.; van Genuchten, M.T. Determining Transport Parameters from Laboratory and Field Tracer Experiments; Bulletin 84-3; Virginia Polytechnic Institute and State University: Blacksburg, VA, USA, 1984; pp. 1–97. [Google Scholar]
- Alizadeh, A.; Naseri, P.S.Z. An Investigation on the Influence of EAF Slag Particle Size on AMD Neutralization Behavior in Static and Dynamic Slag Leaching Systems. Mine Water Environ. 2015, 34, 204–212. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency (USEPA). Regional Screening Levels (RSLs)–Generic Tables. 2019. Available online: https://www.epa.gov/risk/regional-screening-levels-rsls-generic-tables (accessed on 23 July 2019).
- Canadian Council of Ministries of the Environment (CCME). Soil Quality Guidelines for the Protection of Environmental and Human Health. 2019. Available online: http://st-ts.ccme.ca/en/index.html (accessed on 23 July 2019).
- Smith, D.B.; Cannon, W.F.; Woodruff, L.G.; Solano, F.; Kilburn, J.E.; Fey, D.L. Geochemical and Mineralogical Data for Soils of the Conterminous United States. U.S. Geological Survey Data Series 801. 2013; 19p. Available online: https://pubs.usgs.gov/ds/801/pdf/ds801.pdf (accessed on 23 July 2019).
- U.S. Environmental Protection Agency (USPEA). 2018 Edition of the Drinking Water Standards and Health Advisories National Drinking Water Regulations. EPA 822-F-18-001. 2018. Available online: https://www.epa.gov/sites/production/files/2018-03/documents/dwtable2018.pdf (accessed on 23 July 2019).
- U.S. Environmental Protection Agency (USEPA). National Recommended Water Quality Criteria—Aquatic Life Criteria Table. 2019. Available online: https://www.epa.gov/wqc/national-recommended-water-quality-criteria-aquatic-life-criteria-table (accessed on 23 July 2019).
- World Health Organization (WHO). Guidelines for Drinking-water Quality, 4th ed.; Incorporating the 1st Addendum. 2017. Available online: https://www.who.int/water_sanitation_health/publications/drinking-water-quality-guidelines-4-including-1st-addendum/en/ (accessed on 23 July 2019).
- Haynes, W.M. CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data, 97th ed.; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Spanka, M.; Mansfeldt, T.; Bialucha, R. Sequential extraction of chromium, molybdenum, and vanadium in basic oxygen furnace slags. Environ. Sci. Pollut. Res. Int. 2018, 25, 23082–23090. [Google Scholar] [CrossRef]
- Kasina, M.; Kowalski, P.R.; Michalik, M. Mineral carbonation of metallurgical slags. Mineralogia 2015, 45, 27–45. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Prog Biomatter 2016, 5, 9–70. [Google Scholar] [CrossRef]
- Sellner, B.M.; Hua, G.; Ahiablame, L.M.; Trooien, T.P.; Hay, C.H.; Kjaersgaard, J. Evaluation of industrial by-products and natural minerals for phosphate adsorption from subsurface drainage. Environ. Technol. 2019, 40, 756–767. [Google Scholar] [CrossRef]
- Almeelbi, T.; Bezbaruah, A. Aqueous phosphate removal using nanoscale zero-valent iron. J. Nanopart. Res. 2012, 14. [Google Scholar] [CrossRef]
- Klimeski, A.; Chardon, W.J.; Turtola, E.; Uusitalo, R. Potential and limitations of phosphate retention media in water protection: A process-based review of laboratory and field scale tests. Agric. Food Sci. 2012, 21, 206–223. [Google Scholar] [CrossRef]
- Penn, C.; Bowen, J.; McGrath, J.; Nairn, R.; Fox, G.; Brown, G.; Wilson, S.; Gill, C. Evaluation of a universal flow-through model for predicting and designing phosphorus removal structures. Chemosphere 2016, 151, 345–355. [Google Scholar] [CrossRef]
- Shilton, A.; Pratt, S.; Drizo, A.; Mahmood, B.; Banker, S.; Billings, L.; Glenny, S.; Luo, D. “Active” filters for upgrading phosphorus removal from pond systems. Water Sci. Technol. 2005, 51, 111–116. [Google Scholar] [CrossRef]
- Korkusuz, E.A.; Beklioǧlu, M.; Demirer, G.N. Comparison of the treatment performances of blast furnace slag-based and gravel-based vertical flow wetlands operated identically for domestic wastewater treatment in turkey. Ecol. Eng. 2005, 24, 185–198. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Site | Location | Size Fraction | Furnace |
|---|---|---|---|---|
| ILSS1 | legacy | 41°41′12.05″ N 87°33′57.82″ W | crushed, <9.5 mm | unknown |
| ILSS2 | legacy | 41°39′44.46″ N 87°32′38.04″ W | C-fines, <9.5 mm | unknown |
| ILSS5 * | legacy | 41°45′06.95″ N 87°32′33.97″ W | crushed, <9.5 mm | unknown |
| ILSS6 | legacy | 41°39′41.00″ N 87°29′45.20″ W | crushed, <9.5 mm | unknown |
| ILSS7 | legacy | 41°39′44.93″ N 87°30‘59.82″ W | crushed, <9.5 mm | unknown |
| ILSS8 | legacy | 41°39′02.02″ N 87°31′22.66″ W | crushed, <9.5 mm | unknown |
| AMFeC * | modern | Arcelor Mittal Indiana Harbor | C-fines, <9.5 mm | blast |
| AMSteelC | modern | Arcelor Mittal Indiana Harbor | C-fines, <9.5 mm | steel (BOF) |
| USSFeair | modern | U.S. Steel Corp. Gary Works | 25–75 mm (1–3 in.) | blast |
| USSFegran | modern | U.S. Steel Corp. Gary Works | 0.075–4.7 mm (0.003–3/16 in.) | blast |
| USSSteelB_3/8 | modern | U.S. Steel Corp. Gary Works | 9.5 mm (3/8 in.) | steel (BOF) |
| USSSteelB_3/8-4 | modern | U.S. Steel Corp. Gary Works | 9.5–100 mm (3/8–4 in.) | steel (BOF) |
| USSSteelC | modern | U.S. Steel Corp. Gary Works | C-fines, <9.5 mm | steel (BOF) |
| Sample | Paste pH | Total S | HCl Extractable S | Sulfide S * | AP | NP | NNP | NPR |
|---|---|---|---|---|---|---|---|---|
| pH Units | wt. % | wt. % | wt. % | kg CaCO3/t | kg CaCO3/t | kg CaCO3/t | - | |
| ILSS1 | 12.5 | 0.09 | 0.02 | 0.07 | 2.2 | 733 | 730 | 333.0 |
| ILSS2 | 11.4 | 0.13 | 0.07 | 0.06 | 1.9 | 759 | 757 | 399.4 |
| ILSS5 | 10.6 | 0.56 | 0.11 | 0.45 | 14.1 | 414 | 400 | 29.3 |
| ILSS6 | 12.0 | 0.11 | 0.03 | 0.08 | 2.5 | 520 | 518 | 208.0 |
| ILSS6duplicate | 12.1 | 0.13 | 0.03 | 0.10 | 3.1 | 503 | 499 | 162.1 |
| ILSS7 | 10.6 | 0.42 | 0.04 | 0.38 | 11.9 | 436 | 424 | 36.7 |
| ILSS8 | 9.91 | 0.10 | 0.04 | 0.06 | 1.9 | 66.5 | 64.6 | 35.0 |
| USSFegran | 10.1 | 1.00 | 0.03 | 0.97 | 30.3 | 858 | 827 | 28.3 |
| USSFeair | 10.7 | 1.08 | 0.04 | 1.04 | 32.5 | 829 | 796 | 25.5 |
| USSSteel B 3/8-4 | 12.5 | 0.04 | 0.12 | <0.02 | <0.6 | 820 | 820 | - |
| USSSteel B 3/8 | 12.5 | 0.05 | 0.40 | <0.02 | <0.6 | 726 | 726 | - |
| USSSteelC | 12.5 | 0.06 | 0.10 | <0.02 | <0.6 | 654 | 654 | - |
| AMSteelC | 12.5 | 0.22 | 0.13 | 0.09 | 2.8 | 678 | 675 | 242.0 |
| AMFeC | 12.4 | 0.58 | 0.45 | 0.13 | 4.1 | 529 | 525 | 129.0 |
| detection limits | - | 0.02 | 0.01 | 0.02 | 0.6 | 0.1 | 0.1 | - |
| Constituent | ILSS5 >500 | AMFeC >500 |
|---|---|---|
| wt. % | wt. % | |
| Al2O3 | 6.62 | 7.21 |
| CaO | 20.3 | 34.4 |
| Cr2O3 | 0.03 | 0.27 |
| Fe2O3 | 11.9 | 28.6 |
| MgO | 2.31 | 9.28 |
| MnO | 8.04 | 3.16 |
| SiO2 | 45.3 | 11.1 |
| K2O | 0.23 | 0.03 |
| Na2O | 0.12 | 0.02 |
| P2O5 | 0.23 | 0.58 |
| S | 0.58 | 0.169 |
| C | 1.27 | 1.64 |
| Carbonate C | 0.21 | 0.66 |
| LOI | 1.48 | 5.01 |
| Sample | Initial Leaching Solution (mg P/L) | PRC (mg P/g) | |||||
|---|---|---|---|---|---|---|---|
| 10 | 54 | 108 | 256 | 501 | 1085 | ||
| Legacy | |||||||
| ILSS1 | 99.8% | 100.0% | 100.0% | 99.9% | 88.4% | 64.7% | 14.1 |
| ILSS2 | 99.5% | 97.2% | 74.3% | 75.5% | 71.8% | 59.4% | 12.9 |
| ILSS5 | 99.6% | - | - | - | - | 43.7% | 9.5 |
| ILSS6 | 99.6% | - | - | - | - | 46.2% | 10.0 |
| ILSS7 | 99.2% | - | - | - | - | 42.8% | 9.3 |
| ILSS8 | 74.2% | - | - | - | - | 7.8% | 1.7 |
| Modern | |||||||
| AM Fe C | 99.9% | 100.0% | 100.0% | 100.0% | 100.0% | 71.8% | 18.0 |
| AM Steel C | 99.9% | 100.0% | 100.0% | 100.0% | 100.0% | 82.7% | 18.0 |
| USSFeair | 97.9% | - | - | - | - | - | - |
| USSFegran | 98.9% | - | - | - | - | 62.8% | 13.6 |
| USSSteelB_3/8 | 100.0% | - | - | - | - | 59.7% | 13.0 |
| USSSteelB_3/8-4 | 99.7% | - | - | - | - | - | - |
| USSSteelC | 99.9% | - | - | - | - | 58.2% | 12.6 |
| Other | |||||||
| Limestone | - | - | 93.7% | 89.8% | 83.4% | 76.7% | 16.6 |
| Sand | - | 1.8% | −5.4% | - | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piatak, N.M.; Seal, R.R., II; Hoppe, D.A.; Green, C.J.; Buszka, P.M. Geochemical Characterization of Iron and Steel Slag and Its Potential to Remove Phosphate and Neutralize Acid. Minerals 2019, 9, 468. https://doi.org/10.3390/min9080468
Piatak NM, Seal RR II, Hoppe DA, Green CJ, Buszka PM. Geochemical Characterization of Iron and Steel Slag and Its Potential to Remove Phosphate and Neutralize Acid. Minerals. 2019; 9(8):468. https://doi.org/10.3390/min9080468
Chicago/Turabian StylePiatak, Nadine M., Robert R. Seal, II, Darryl A. Hoppe, Carlin J. Green, and Paul M. Buszka. 2019. "Geochemical Characterization of Iron and Steel Slag and Its Potential to Remove Phosphate and Neutralize Acid" Minerals 9, no. 8: 468. https://doi.org/10.3390/min9080468
APA StylePiatak, N. M., Seal, R. R., II, Hoppe, D. A., Green, C. J., & Buszka, P. M. (2019). Geochemical Characterization of Iron and Steel Slag and Its Potential to Remove Phosphate and Neutralize Acid. Minerals, 9(8), 468. https://doi.org/10.3390/min9080468