X-ray Photoelectron Spectroscopy (XPS) Study of the Products Formed on Sulfide Minerals Upon the Interaction with Aqueous Platinum (IV) Chloride Complexes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
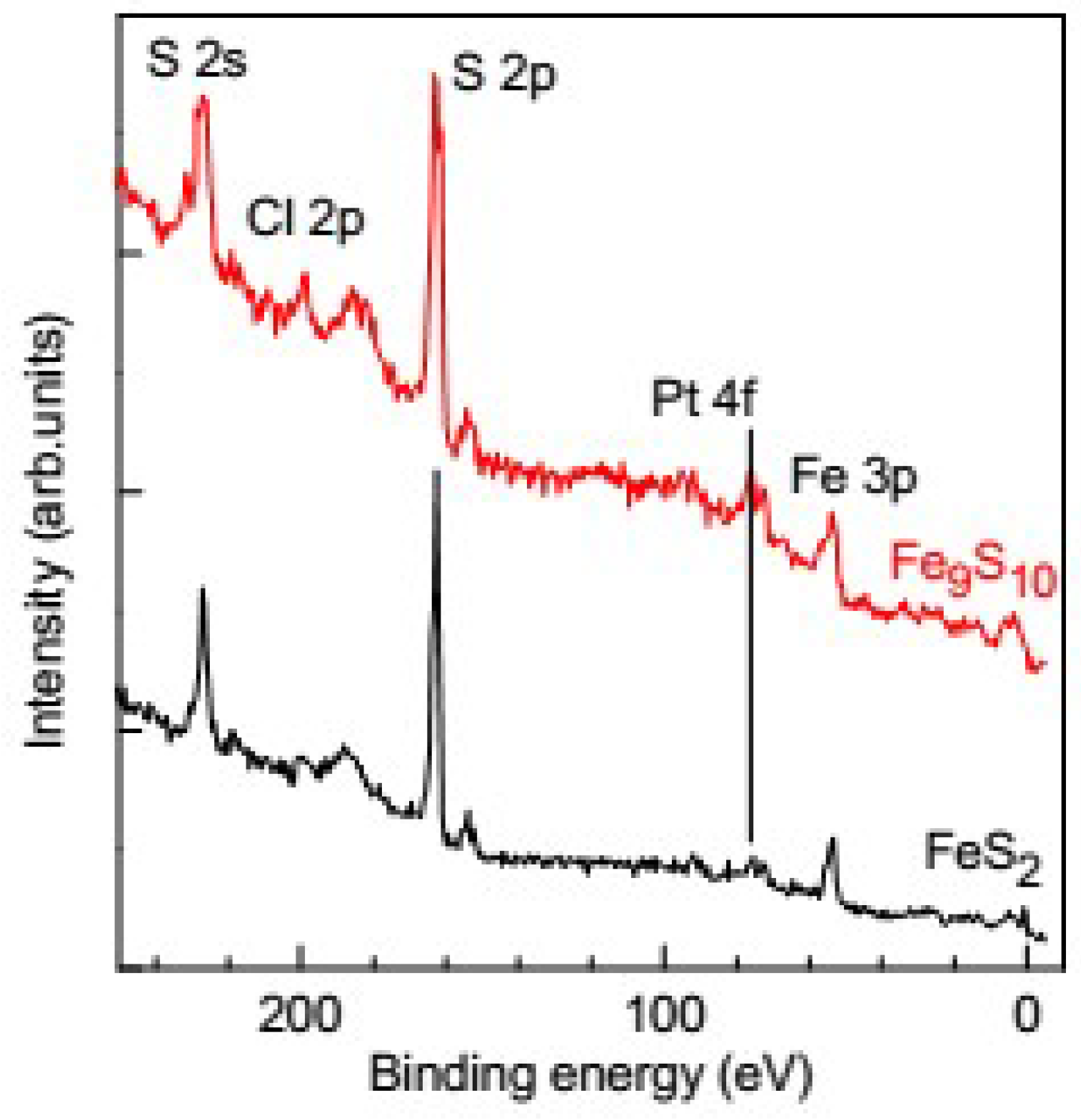
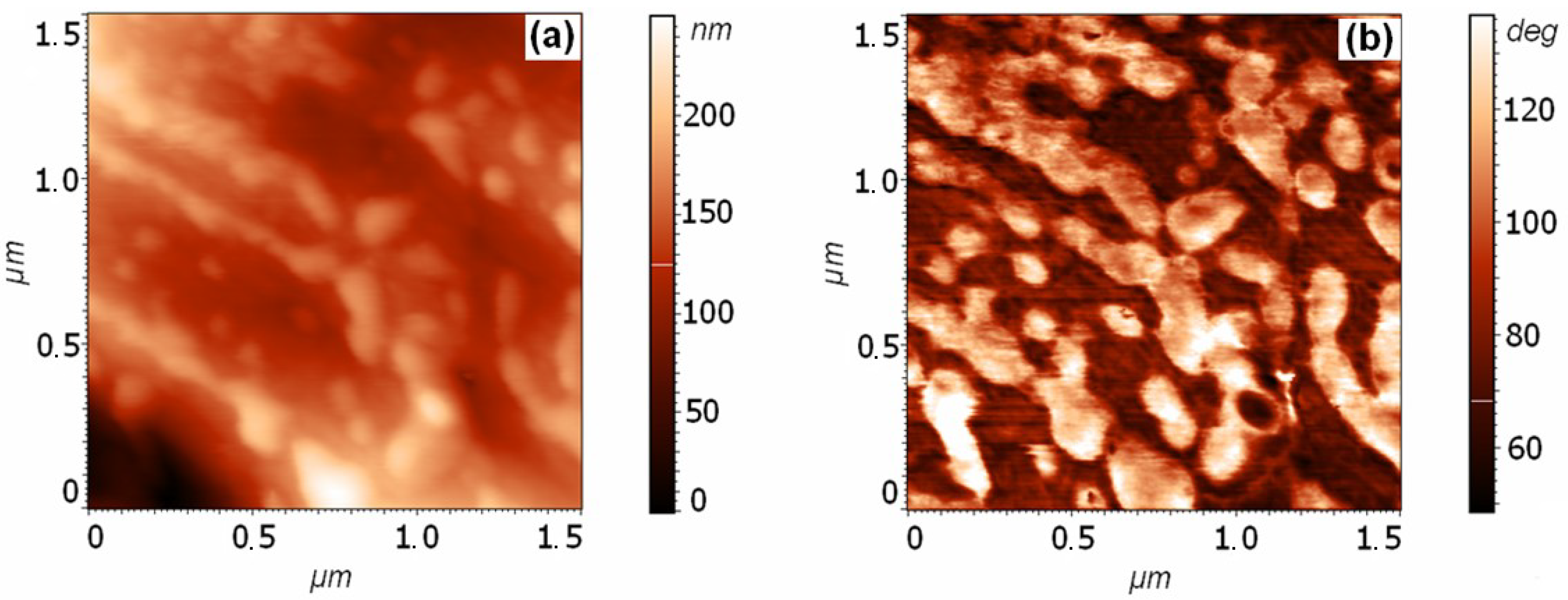
3.1. Analysis of the Surface of Pyrite and Pyrrhotite Using X-ray Photoelectron Spectroscopy and Atomic Force Microscopy
3.2. Deposition of Platinum Revealed by XPS
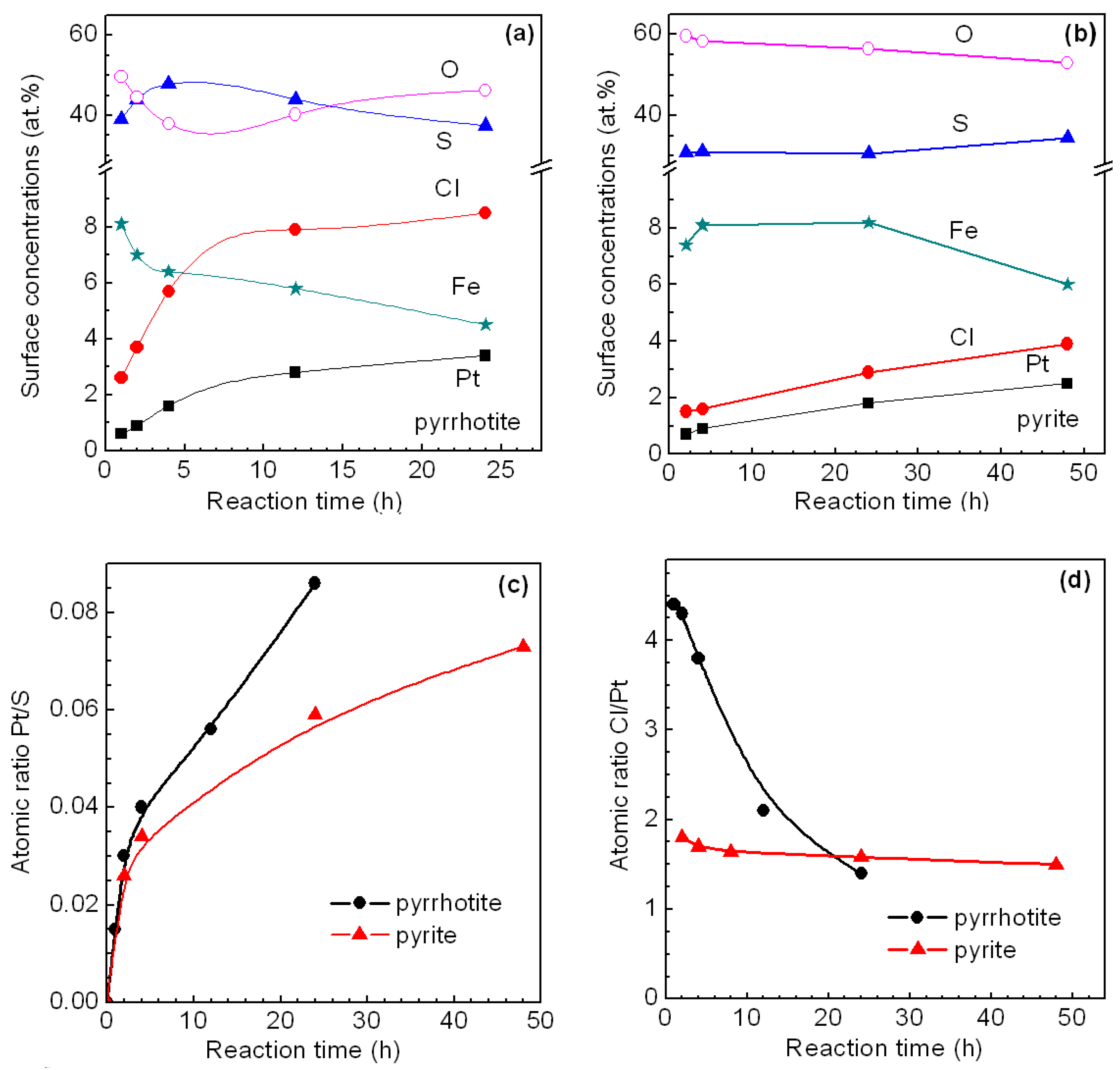
3.2.1. Pyrrhotite and Pyrite
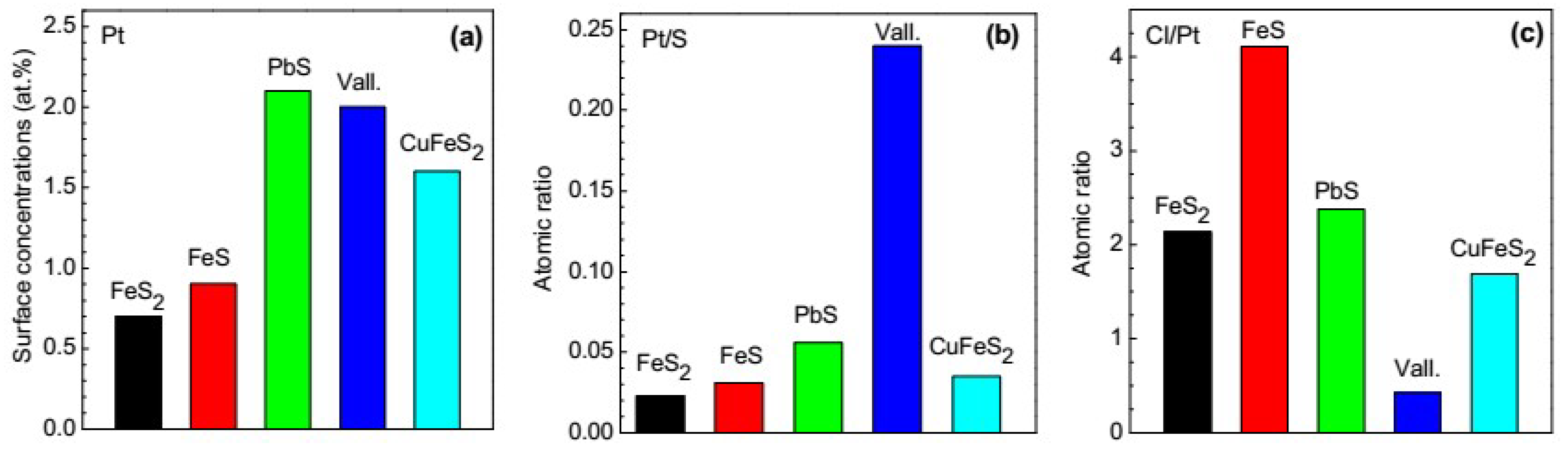
3.2.2. Galena, Chalcopyrite and Valleriite
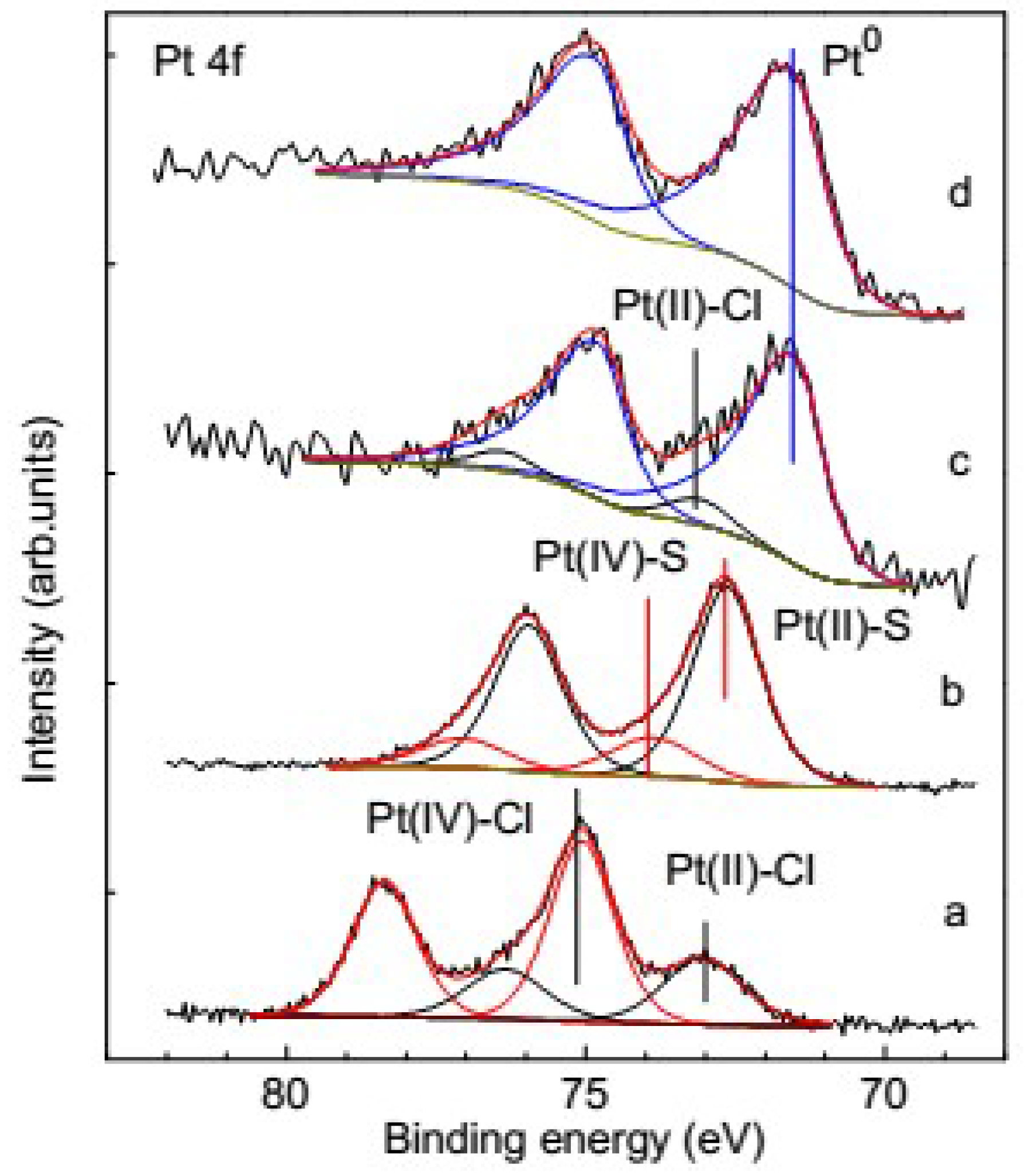
3.3. Species of the Deposited Platinum
3.3.1. XPS of the Reference Materials
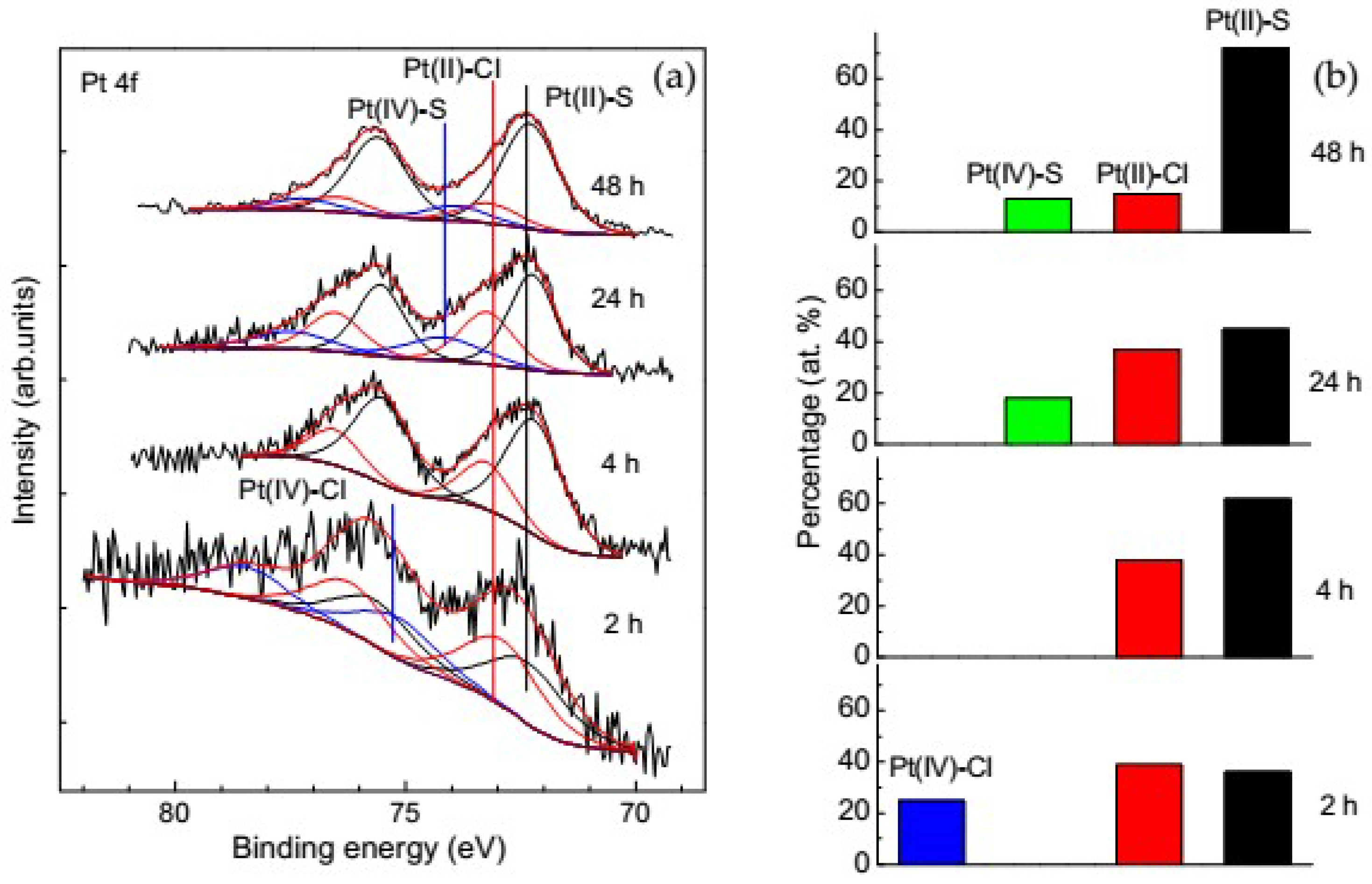
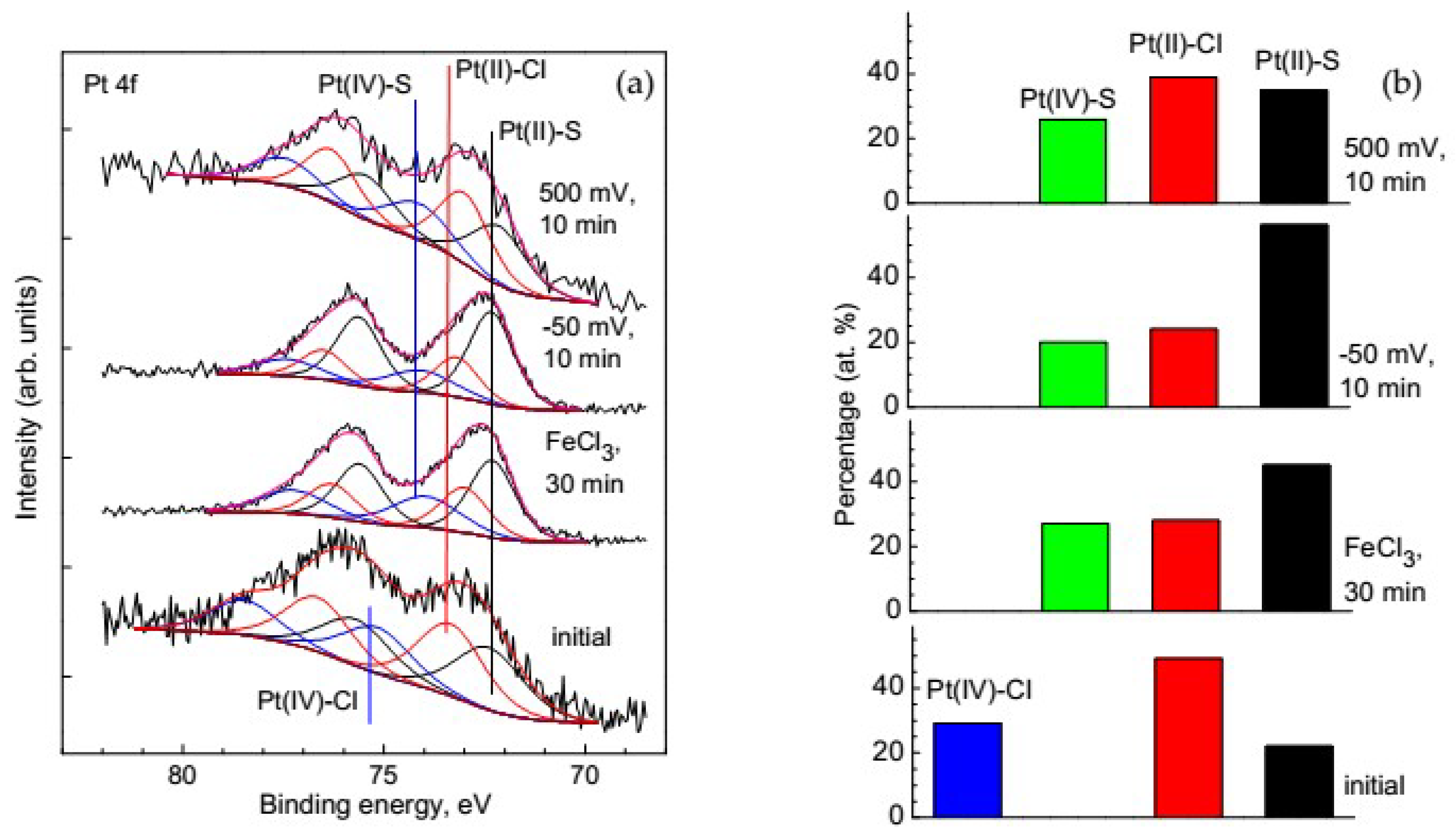
3.3.2. Change of Platinum Species in the Deposition Process on Pyrite
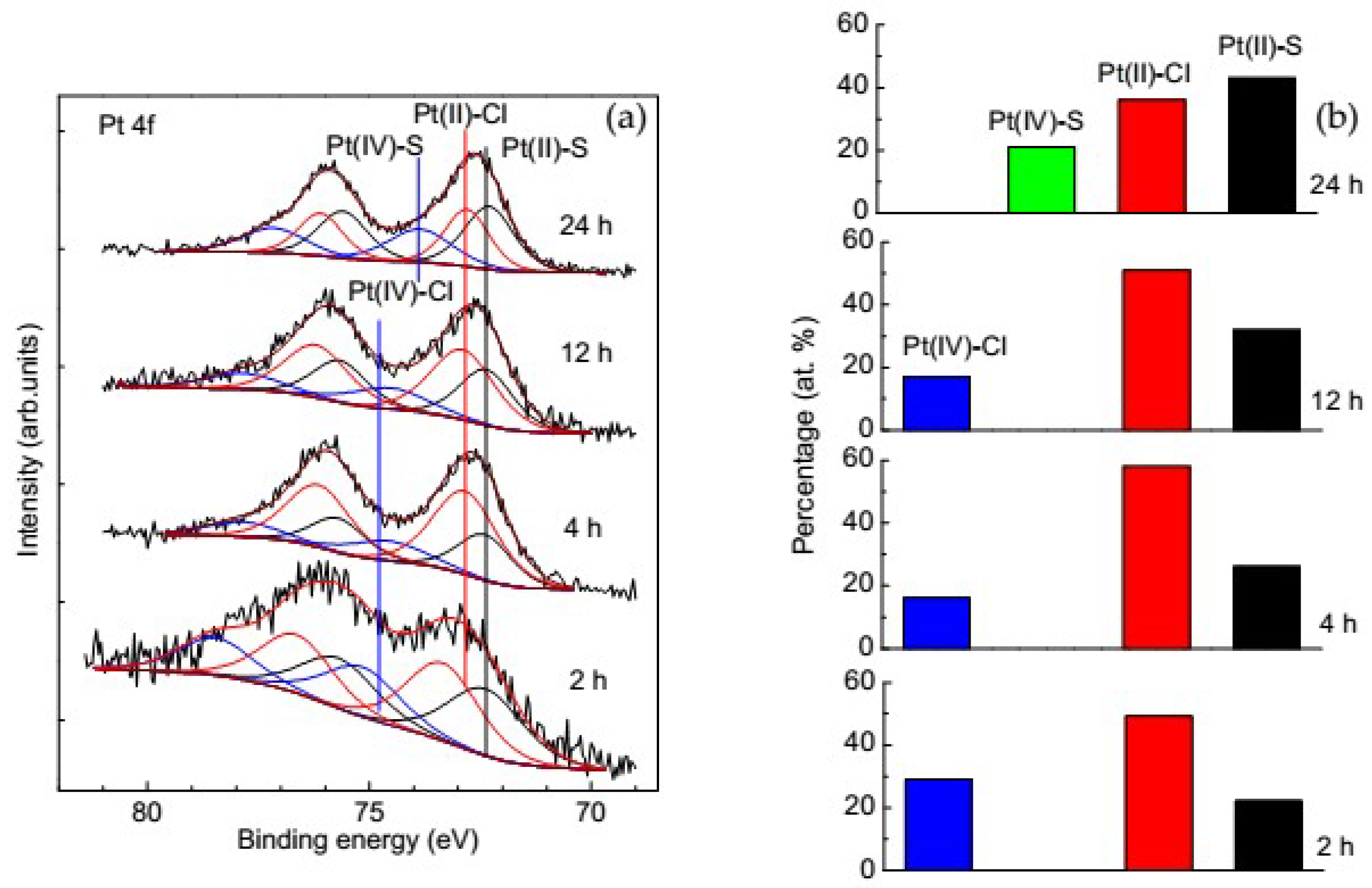
3.3.3. Change of Platinum Species in the Deposition Process on Pyrrhotite
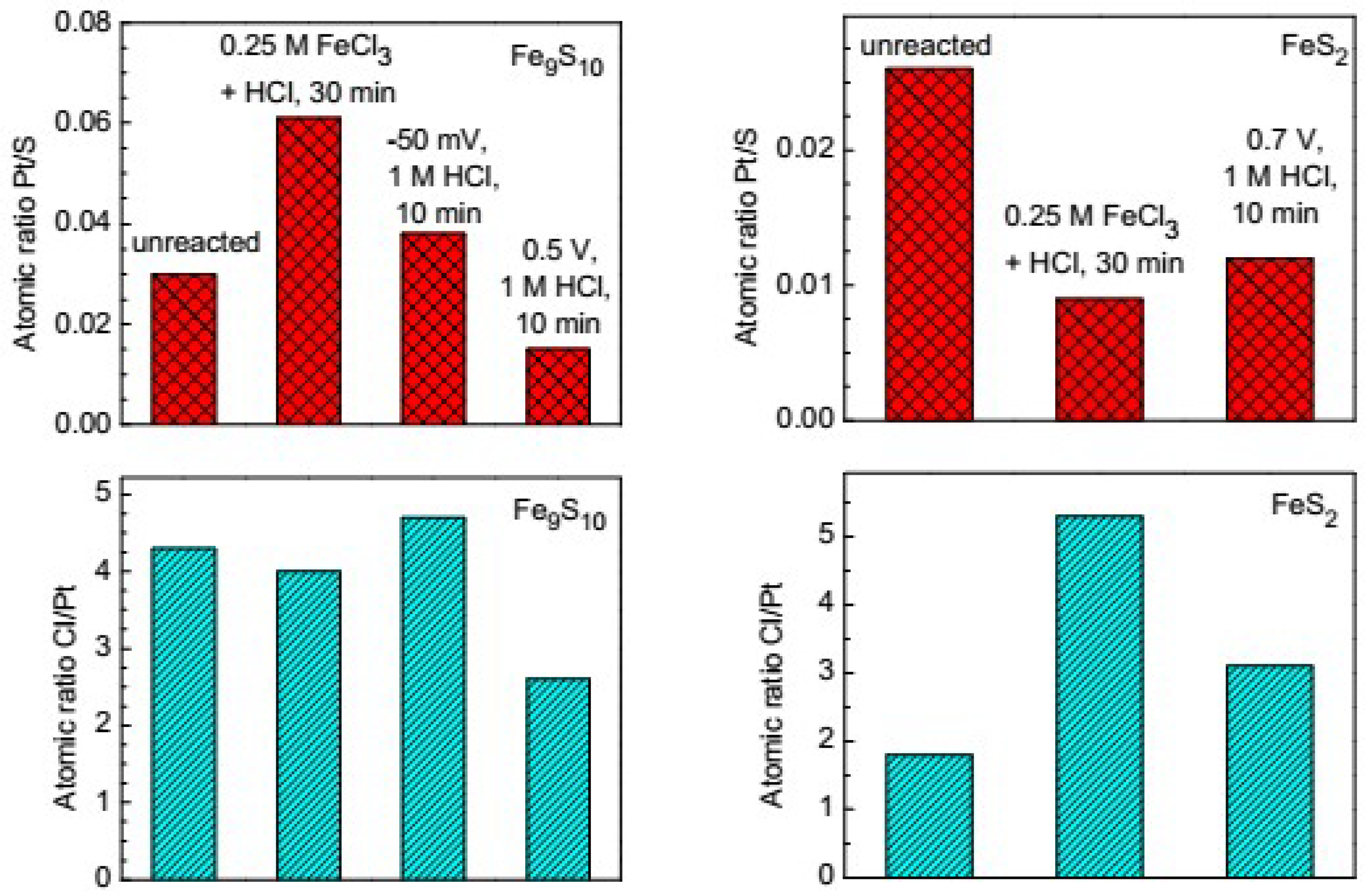
3.4. Influence of the Preliminary Modification of the Mineral Surface on the Platinum Deposition
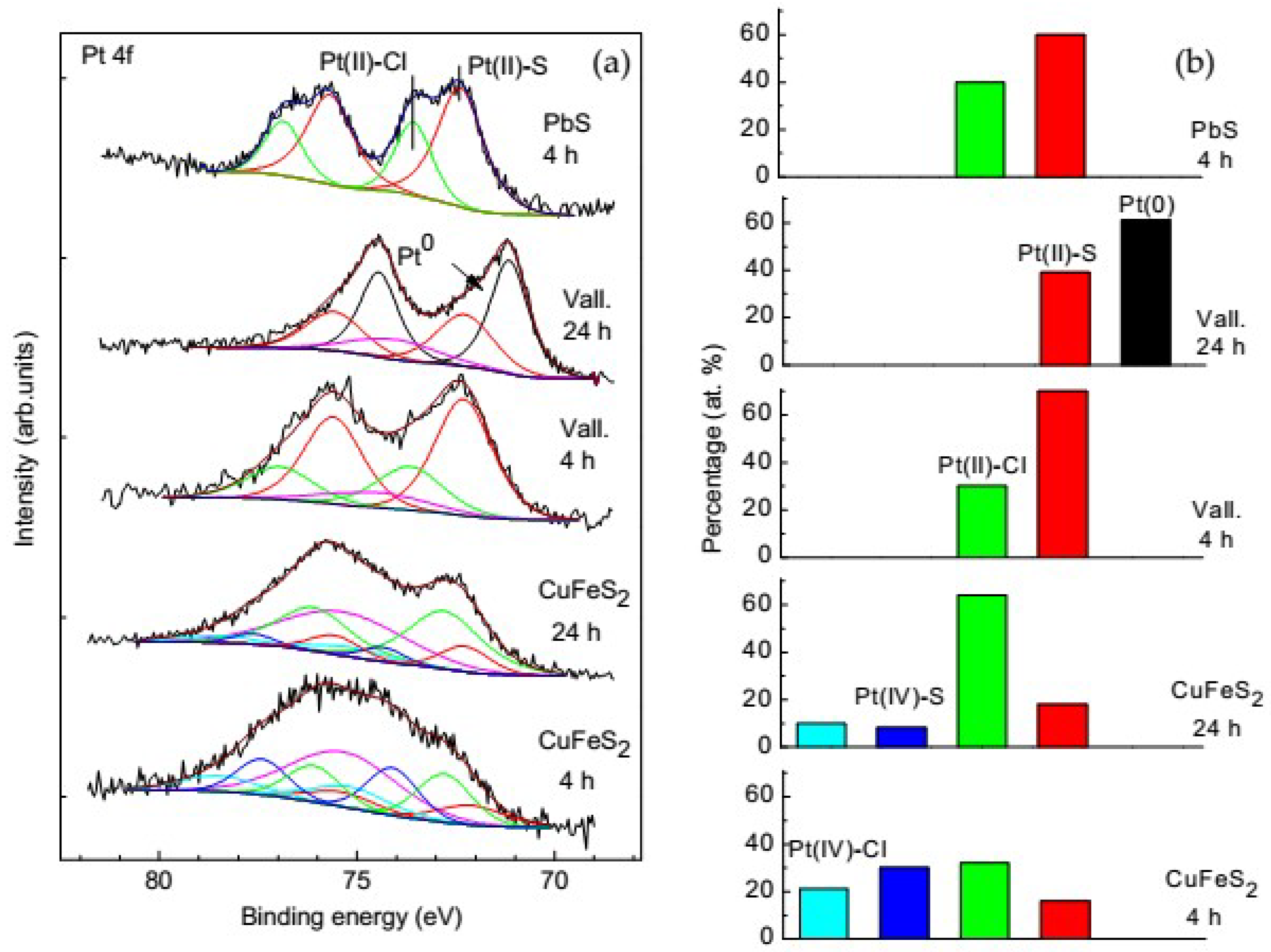
3.5. Platinum Species on Different Sulfide Minerals
4. Discussion
5. Conclusions
- The interaction of the aqueous solutions of H2PtCl6 with the surface of sulfide minerals results in the deposition of Pt on their surface. The amount of the deposited Pt increases with time.
- The highest rate of Pt uptake is observed on galena and valleriite.
- The preliminary moderate oxidation and non-oxidative leaching of pyrrhotite creating metal-deficient surface layers usually promotes Pt deposition. In the case of pyrite, the preliminary oxidation decreases the amount of the deposited Pt.
- The main Pt phases immobilized on sulfide minerals are sulfides and chloride complexes of Pt(II), and, to a smaller extent, chloride complexes of tetravalent Pt; no Pt(IV) was observed on galena and valleriite. As the reaction progresses, the amount of Pt(IV) usually decreases while the amount of the sulfide forms increases.
- Metallic Pt was found to form only on valleriite, probably owing to the negative charge localized at sulfidic nanolayers.
- The di- and polysulfide surface species arising on sulfide minerals upon their oxidation are capable of oxidizing immobilized Pt(II)–S to Pt(IV)–S species.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dey, S.; Jain Vimal, K. Platinum Group Metal Chalcogenides: Their syntheses and applications in catalysis and materials science. Platin. Metals Rev. 2004, 48, 16–29. [Google Scholar]
- Chia, X.; Adriano, A.; Lazar, P.; Sofer, Z.; Luxa, J.; Pumera, M. Layered platinum dichalcogenides (PtS2, PtSe2, and PtTe2) electrocatalysis: Monotonic dependence on the chalcogen size. Adv. Funct. Mater. 2016, 26, 4306–4318. [Google Scholar] [CrossRef]
- Bhatt, R.; Bhattacharya, S.; Basu, R. Growth of Pd4S, PdS and PdS2 films by controlled sulfurization of sputtered Pd on native oxide of Si. Thin Solid Films 2013, 539, 41–46. [Google Scholar] [CrossRef]
- Jha, M.K.; Lee, J.; Kim, M.; Jeong, J.; Kim, B.; Kumar, V. Hydrometallurgical recovery/recycling of platinum by the leaching of spent catalysts: A review. Hydrometallurgy 2013, 133, 23–32. [Google Scholar] [CrossRef]
- Junge, M.; Oberthür, T.; Kraemer, D.; Melcher, F.; Pina, R.; Derrey, I.T.; Manyeruke, T.; Strauss, H. Distribution of platinum-group elements in pristine and near-surface oxidized Platreef ore and the variation along strike, northern Bushveld Complex, South Africa. Miner. Depos. 2018. [Google Scholar] [CrossRef]
- Kraemer, D.; Junge, M.; Oberthür, T.; Bau, M. Improving recoveries of platinum and palladium from oxidized Platinum-Group Element ores of the Great Dyke, Zimbabwe, using the biogenic siderophore Desferrioxamine B. Hydrometallurgy 2015, 152, 169–177. [Google Scholar] [CrossRef]
- Kraemer, D.; Junge, M.; Oberthür, T.; Bau, M. Oxidized ores as future resource for platinum group metals: Current state of research. Chem. Ing. Tech. 2017, 89, 53–63. [Google Scholar] [CrossRef]
- Gammons, C.H.; Bloom, M.S.; Yu, Y. Experimental investigation of the hydrothermal geochemistry of platinum and palladium: I. Solubility of platinum and palladium sulfide minerals in NaCl/H2SO4 solutions at 300 °C. Geochim. Cosmochim. Acta 1992, 56, 3881–3894. [Google Scholar] [CrossRef]
- McDonald, I.; Ohnenstetter, D.; Rowe, J.P.; Tredoux, M.; Pattrick, R.A.D.; Vaughan, D.J. Platinum precipitation in the Waterberg deposit, Naboomspruit, South Africa. S. Afr. J. Geol. 1999, 102, 184–191. [Google Scholar]
- Oberthür, T.; Melcher, F.; Fusswinkel, T.; van den Kerkhof, A.M.; Sosa, G.M. The hydrothermal Waterberg platinum deposit, Mookgophong (Naboomspruit), South Africa. Part 1: Geochemistry and ore mineralogy. Mineral. Mag. 2018, 82, 725–749. [Google Scholar] [CrossRef]
- Gammons, C.H.; Bloom, M.S. Experimental investigation of the hydrothermal geochemistry of platinum and palladium: II. The solubility of PtS and PdS in aqueous sulfide solutions to 300 °C. Geochim. Cosmochim. Acta 1992, 57, 2451–2467. [Google Scholar] [CrossRef]
- Mountain, B.W.; Wood, S.A. Chemical controls on the solubility, transport, and deposition of platinum and palladium in hydrothermal solutions: A thermodynamic approach. Econ. Geol. 1988, 83, 492–510. [Google Scholar] [CrossRef]
- Gammons, C.H. Experimental investigations of the hydrothermal geochemistry of platinum and palladium: V. Equilibria between platinum metal, Pt(II), and Pt (IV) chloride complexes at 25 to 300 °C. Geochim. Cosmochim. Acta 1992, 59, 1655–1667. [Google Scholar] [CrossRef]
- Gammons, C.H. Experimental investigation of the hydrothermal geochemistry of platinum and palladium: IV. The stoichiometry of Pt (IV) and Pd (II) chloride complexes at 100 to 300 °C. Geochim. Cosmochim. Acta 1994, 59, 3881–3894. [Google Scholar] [CrossRef]
- Godel, B.; Barnes, S.-J. Platinum-group elements in sulfide minerals and the whole rocks of the JM Reef (Stillwater Complex): Implication for the formation of the reef. Chem. Geol. 2008, 248, 272–294. [Google Scholar] [CrossRef]
- Hyland, M.M.; Bancroft, G.M. Palladium sorption and reduction on sulphide mineral surfaces: An XPS and AES study. Geochim. Cosmochim. Acta 1989, 54, 117–130. [Google Scholar] [CrossRef]
- Plyusnina, L.P.; Likhoidov, G.G.; Shcheka, Z.A. Experimental modeling of platinum behavior under hydrothermal conditions (300–500 °C and 1 kbar). Geochem. Int. 2007, 45, 1124–1130. [Google Scholar] [CrossRef]
- Wood, S.A.; Mountain, B.W.; Pan, P. The aqueous geochemistry of platinum, palladium and gold: Recent experimental constraints and a re-evaluation of theoretical prediction. Can. Mineral. 1992, 30, 955–982. [Google Scholar]
- Jaireth, S. The calculated solubility of platinum and gold in oxygen-saturated fluids and the genesis of platinum-palladium and gold mineralization in the unconformity-related uranium deposits. Miner. Depos. 1992, 27, 42–54. [Google Scholar] [CrossRef]
- Watkinson, D.H.; Melling, D.R. Hydrothermal origin of platinum-group mineralization in low-temperature copper sulfide-rich assemblages, Salt Chuck Intrusion, Alaska. Econ. Geol. 1992, 87, 175–184. [Google Scholar] [CrossRef]
- Reith, F.; Zammit, C.M.; Shar, S.S.; Etschmann, B.; Bottrill, R.; Southam, G.; Ta, C.; Kilburn, M.; Oberthür, T.; Ball, A.S.; et al. Biological role in the transformation of platinum-group mineral grains. Nat. Geosci. 2016, 9, 1–6. [Google Scholar] [CrossRef]
- Reith, O.F.; Campbell, S.G.; Ball, A.S.; Pring, A.; Southam, G. Platinum in Earth surface environments. Earth-Sci. Rev. 2014, 131, 1–21. [Google Scholar] [CrossRef]
- Pawlak, J.; Łodyga-Chruścińska, E.; Chrustowicz, J. Fate of platinum metals in the environment. J. Trace Elem. Med. Biol. 2014, 28, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Reith, F.; Shuster, J. Geomicrobiology and Biogeochemistry of Precious Metals; MDPI: Basel, Switzerland, 2018; pp. 114–131. ISBN 978-3-03897-347-8. [Google Scholar]
- Piña, R.; Gervilla, F.; Barnes, S.-J.; Oberthür, T.; Lunar, R. Platinum-group element concentrations in pyrite from the Main Sulfide Zone of the Great Dyke of Zimbabwe. Miner. Depos. 2016, 51, 853–872. [Google Scholar] [CrossRef]
- Dare, S.A.S.; Barnes, S.-J.; Prichard, H.M.; Fisher, P.C. Chalcophile and platinum-group element (PGE) concentrations in the sulfide minerals from the McCreedy East deposit, Sudbury, Canada, and the origin of PGE in pyrite. Miner. Depos. 2011, 46, 381–407. [Google Scholar] [CrossRef]
- Junge, M.; Wirth, R.; Oberthür, T.; Melcher, F.; Schreiber, A. Mineralogical siting of platinum-group elements in pentlandite from the Bushveld Complex, South Africa. Miner. Depos. 2015, 50, 41–54. [Google Scholar] [CrossRef]
- Kubrakova, I.V.; Tyutyunnik, O.A.; Koshcheeva, I.Y.; Sadagov, A.Y.; Nabiullina, S.N. Migration behavior of platinum group elements in natural and technogeneous systems. Geochem. Int. 2017, 55, 108–124. [Google Scholar] [CrossRef]
- Torres, R.; Lapidus, G.T. Platinum, palladium and gold leaching from magnetite ore, with concentrated chloride solutions and ozone. Hydrometallurgy 2016, 166, 185–194. [Google Scholar] [CrossRef]
- Fuchs, W.A.; Rose, A.W. The geochemical behavior of platinum and palladium in the weathering cycle in the Stillwater complex, Montana. Econ. Geol. 1974, 69, 332–346. [Google Scholar] [CrossRef]
- Adams, M.; Liddell, K.; Holohan, T. Hydrometallurgical processing of Platreef flotation concentrate. Miner. Eng. 2011, 24, 545–550. [Google Scholar] [CrossRef]
- Liddell, K.S.; Adams, M.D. Kell hydrometallurgical process for extraction of platinum group metals and base metals from flotation concentrates. J. S. Afr. Inst. Min. Metall. 2012, 112, 31–36. [Google Scholar]
- Liddell, K.; Newton, T.; Adams, M.; Muller, B. Energy consumption for Kell hydrometallurgical refining versus conventional pyrometallurgical smelting and refining of PGM concentrates. J. S. Afr. Inst. Min. Metall. 2011, 111, 127–132. [Google Scholar]
- Mpinga, C.N.; Eksteen, J.J.; Aldrich, C.; Dyer, L. Direct leach approaches to Platinum Group Metal (PGM) ores and concentrates: A review. Miner. Eng. 2015, 78, 93–113. [Google Scholar] [CrossRef]
- Mwase, J.M.; Petersen, J.; Eksteen, J.J. A conceptual flowsheet for heap leaching of platinum group metals (PGMs) from a low-grade ore concentrate. Hydrometallurgy 2012, 111, 129–135. [Google Scholar] [CrossRef]
- Mwase, J.M.; Petersen, J.; Eksteen, J.J. Assessing a two-stage heap leaching process for Platreef flotation concentrate. Hydrometallurgy 2012, 129, 74–81. [Google Scholar] [CrossRef]
- Mwase, J.M.; Petersen, J.; Eksteen, J.J. A novel sequential heap leach process for treating crushed Platreef ore. Hydrometallurgy 2014, 141, 97–104. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Romanchenko, A.S. Gold deposition on pyrite and the common sulfide minerals: An STM/STS and SR-XPS study of surface reactions and Au nanoparticles. Geochim. Cosmochim. Acta 2007, 71, 5985–6001. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Romanchenko, A.S.; Likhatski, M.N.; Karacharov, A.; Erenberg, S.B.; Trubina, S.V. Understanding the initial stages of precious metals precipitation: Nanoscale metallic and sulfidic species of gold and silver on pyrite surfaces. Ore Geol. Rev. 2011, 42, 47–54. [Google Scholar] [CrossRef]
- Mycroft, J.R.; Bancroft, G.M.; McIntyre, N.S.; Lorimer, J.W. Spontaneous deposition of gold on pyrite from solutions containing Au (III) and Au (I) chlorides. Part I: A surface study. J. Electroanal. Chem. 1990, 292, 139–152. [Google Scholar] [CrossRef]
- Romanchenko, A.S.; Mikhlin, Y.L. An XPS study of products formed on pyrite and pyrrhotine by reacting with palladium(II) chloride solutions. J. Struct. Chem. 2015, 56, 531–537. [Google Scholar] [CrossRef]
- Gulyaev, R.V.; Stadnichenko, A.I.; Slavinskaya, E.M.; Koscheev, S.V.; Ivanova, A.S.; Boronin, A.I. In situ preparation and investigation of Pd/CeO2 catalysts for the low-temperature oxidation of CO. Appl. Catal. A Gen. 2012, 439, 41–50. [Google Scholar] [CrossRef]
- Kibis, L.S.; Stadnichenko, A.I.; Koscheev, S.V.; Zaikovskii, V.I.; Boronin, A.I. Highly oxidized palladium nanoparticles comprising Pd4+ species: Spectroscopic and structural aspects, thermal stability, and reactivity. J. Phys. Chem. C 2012, 116, 19342–19348. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Tomashevich, Y.V.; Pashkov, G.L.; Okotrub, A.V.; Asanov, I.P.; Mazalov, L.N. Electronic structure of the non-equilibrium iron-deficient layer of hexagonal pyrrhotite. Appl. Surf. Sci. 1998, 125, 73–84. [Google Scholar] [CrossRef]
- Mikhlin, Y. Reactivity of pyrrhotite surfaces: An electrochemical study. Phys. Chem. Chem. Phys. 2000, 2, 5672–5677. [Google Scholar] [CrossRef]
- Mikhlin, Y.; Romanchenko, A.; Shagaev, A. Scanning probe microscopy studies of PbS surfaces oxidized in air and etched in aqueous acid solutions. Appl. Surf. Sci. 2006, 252, 5245–5258. [Google Scholar] [CrossRef]
- Mikhlin, Y.; Kuklinskiy, A.; Mikhlina, E.; Kargin, V.; Asanov, I. Electrochemical behaviour of galena (PbS) in aqueous nitric acid and perchloric acid solutions. J. Appl. Electrochem. 2004, 34, 37–46. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Tomashevich, Y.V.; Asanov, I.P.; Okotrub, A.V.; Varnek, V.A.; Vyalikh, D.V. Spectroscopic and electrochemical characterization of the surface layers of chalcopyrite (CuFeS2) reacted in acidic solutions. Appl. Surf. Sci. 2004, 225, 395–409. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Romanchenko, A.S.; Tomashevich, E.V.; Volochaev, M.N.; Laptev, Y.V. XPS and XANES study of layered mineral valleriite. J. Struct Chem. 2017, 58, 1137–1143. [Google Scholar] [CrossRef]
- Laptev, Y.V.; Shevchenko, V.S.; Urakaev, F.K. Sulphidation of valleriite in SO2 solutions. Hydrometallurgy 2009, 98, 201–205. [Google Scholar] [CrossRef]
- Muhler, M.; Paal, Z. Sulfided platinum black by XPS. Surf. Sci. Spectra 1996, 4, 125–129. [Google Scholar] [CrossRef]
- Dembowski, J.; Marosi, L.; Essig, M. Platinum disulfide by XPS. Surf. Sci. Spectra 1994, 2, 133–137. [Google Scholar] [CrossRef]
- Karhu, H.; Kalantar, A.; Väyrynen, I.J.; Salmi, T.; Murzin, D.Y. XPS analysis of chlorine residues in supported Pt and Pd catalysts with low metal loading. Appl. Catal. A 2003, 247, 283–294. [Google Scholar] [CrossRef]
- Mikhlin, Y.; Tomashevich, Y.; Vorobyev, S.; Saikova, S.; Romanchenko, A.; Félix, R. Hard X-ray photoelectron and X-ray absorption spectroscopy characterization of oxidized surfaces of iron sulfides. Appl. Surf. Sci. 2016, 387, 796–804. [Google Scholar] [CrossRef]
- Mikhlin, Y.; Nasluzov, V.; Romanchenko, A.; Tomashevich, Y.; Shor, A.; Félix, R. Layered structure of the near-surface region of oxidized chalcopyrite (CuFeS2): Hard X-ray photoelectron spectroscopy, X-ray absorption spectroscopy and DFT+U studies. Phys. Chem. Chem. Phys. 2017, 19, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Mikhlin, Y.; Romanchenko, A.; Tomashevich, Y.; Shurupov, V. Near-surface regions of electrochemically polarized chalcopyrite (CuFeS2) as studied using XPS and XANES. Phys. Procedia 2016, 84, 390–396. [Google Scholar] [CrossRef]
- Bonnington, K.J.; Jennings, M.C.; Puddephatt, R.J. Oxidative addition of S-S bonds to dimethylplatinum(II) complexes: Evidence for a binuclear mechanism. Organometallics 2008, 27, 6521–6530. [Google Scholar] [CrossRef]
- McCready, M.S.; Puddephatt, R.J. Oxidative addition of functional disulfides to platinum(II): Formation of chelating and bridging thiolate–carboxylate complexes of platinum(IV). Inorg. Chem. Commun. 2011, 14, 210–212. [Google Scholar] [CrossRef]
- Yakubchuk, A.; Nikishin, A. Noril’sk-Talnakh Cu-Ni-PGE deposits: A revised tectonic model. Miner. Depos. 2004, 39, 125–142. [Google Scholar] [CrossRef]
- Harris, D.C.; Cabry, L.J.; Stewart, J.M. A “valleriite-type” mineral from Noril’sk, Western Siberia. Am. Mineral. 1970, 55, 2110–2114. [Google Scholar]
- Evans, H.T.; Allman, R. The crystal structure and crystal chemistry of valleriite. Zeitschrift fur Kristallographie 1968, 127, 73–93. [Google Scholar] [CrossRef]
- Hughes, A.E.; Kakos, G.A.; Turney, T.W.; Williams, T.B. Synthesis and structure of valleriite, a layered Metal Hydroxide/Sulfide Composite. J. Solid State Chem. 1993, 104, 422–436. [Google Scholar] [CrossRef]
- Li, R.; Cui, L. Investigations on valleriite from Western China: Crystal chemistry and separation properties. Int. J. Miner. Process. 1994, 41, 271–283. [Google Scholar] [CrossRef]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanchenko, A.; Likhatski, M.; Mikhlin, Y. X-ray Photoelectron Spectroscopy (XPS) Study of the Products Formed on Sulfide Minerals Upon the Interaction with Aqueous Platinum (IV) Chloride Complexes. Minerals 2018, 8, 578. https://doi.org/10.3390/min8120578
Romanchenko A, Likhatski M, Mikhlin Y. X-ray Photoelectron Spectroscopy (XPS) Study of the Products Formed on Sulfide Minerals Upon the Interaction with Aqueous Platinum (IV) Chloride Complexes. Minerals. 2018; 8(12):578. https://doi.org/10.3390/min8120578
Chicago/Turabian StyleRomanchenko, Alexander, Maxim Likhatski, and Yuri Mikhlin. 2018. "X-ray Photoelectron Spectroscopy (XPS) Study of the Products Formed on Sulfide Minerals Upon the Interaction with Aqueous Platinum (IV) Chloride Complexes" Minerals 8, no. 12: 578. https://doi.org/10.3390/min8120578
APA StyleRomanchenko, A., Likhatski, M., & Mikhlin, Y. (2018). X-ray Photoelectron Spectroscopy (XPS) Study of the Products Formed on Sulfide Minerals Upon the Interaction with Aqueous Platinum (IV) Chloride Complexes. Minerals, 8(12), 578. https://doi.org/10.3390/min8120578