Colloidal and Deposited Products of the Interaction of Tetrachloroauric Acid with Hydrogen Selenide and Hydrogen Sulfide in Aqueous Solutions


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
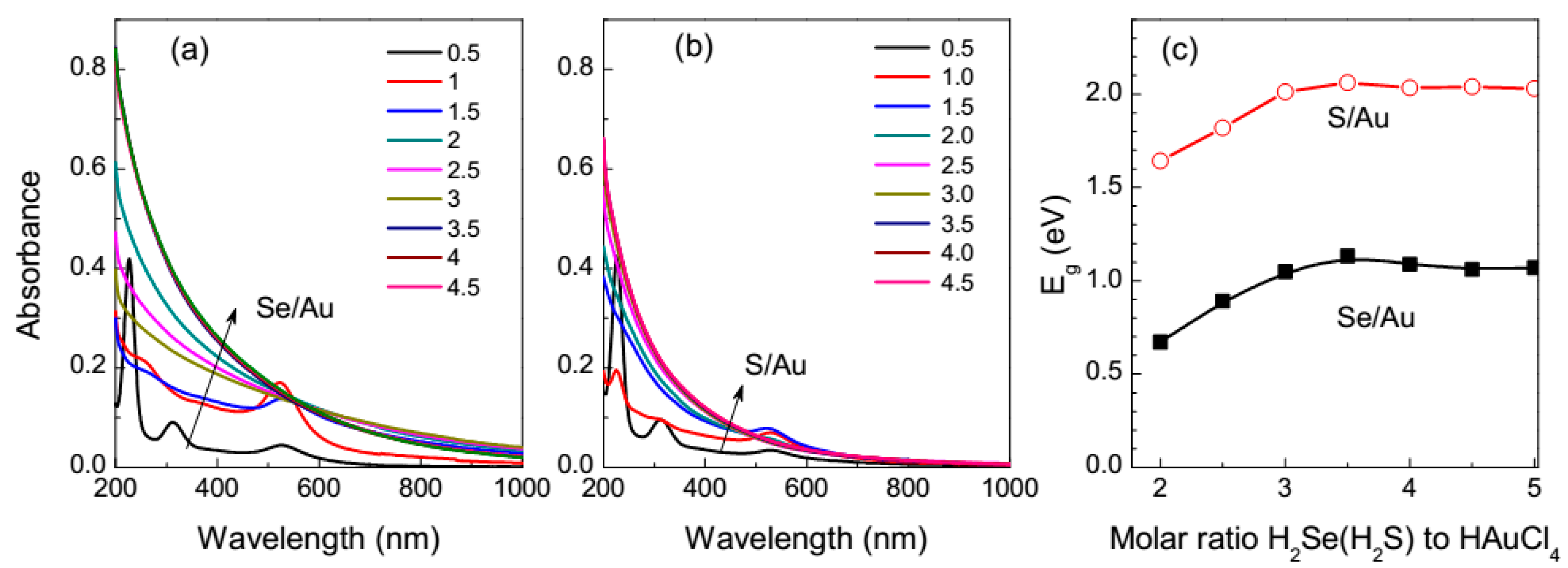
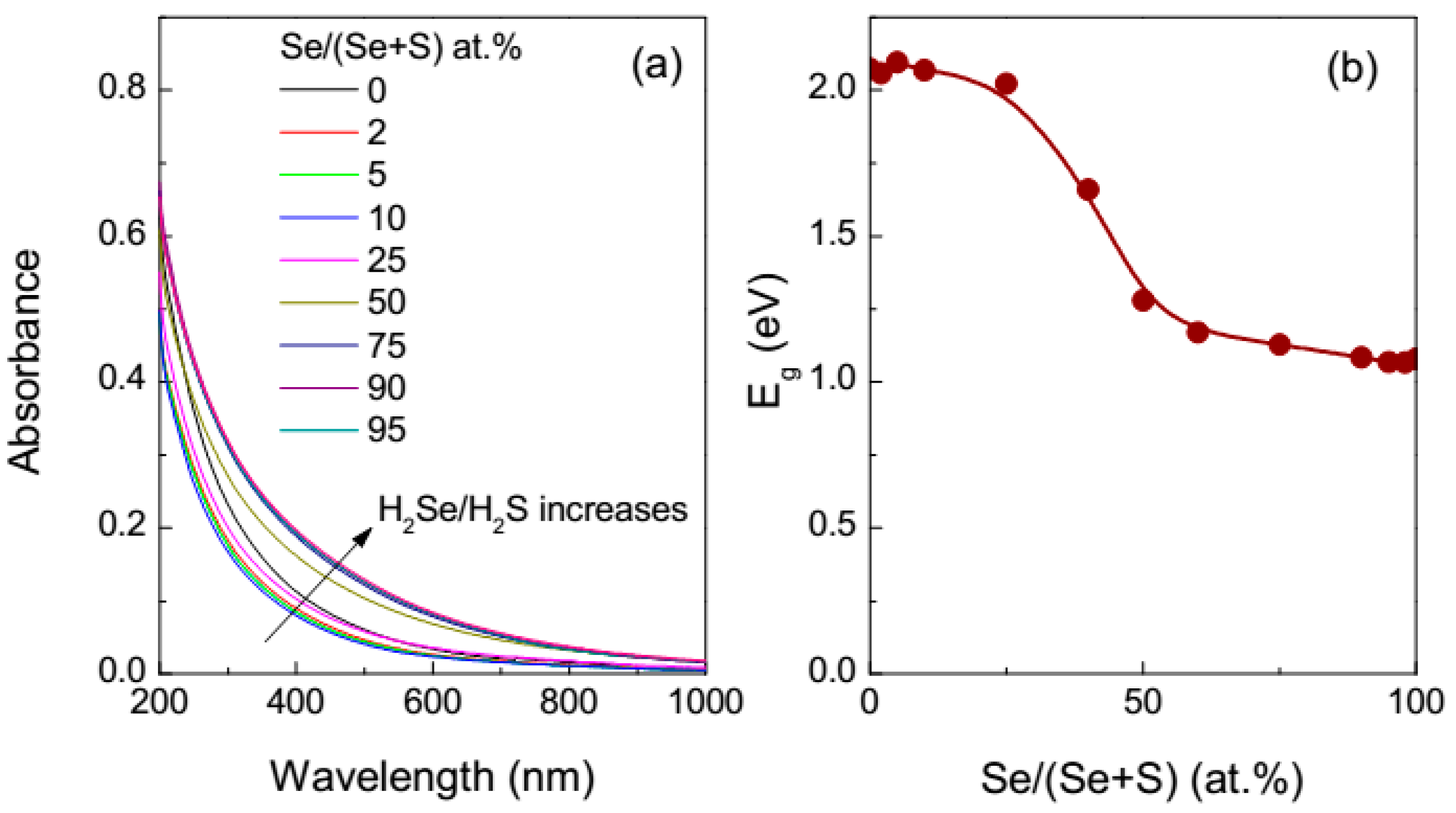
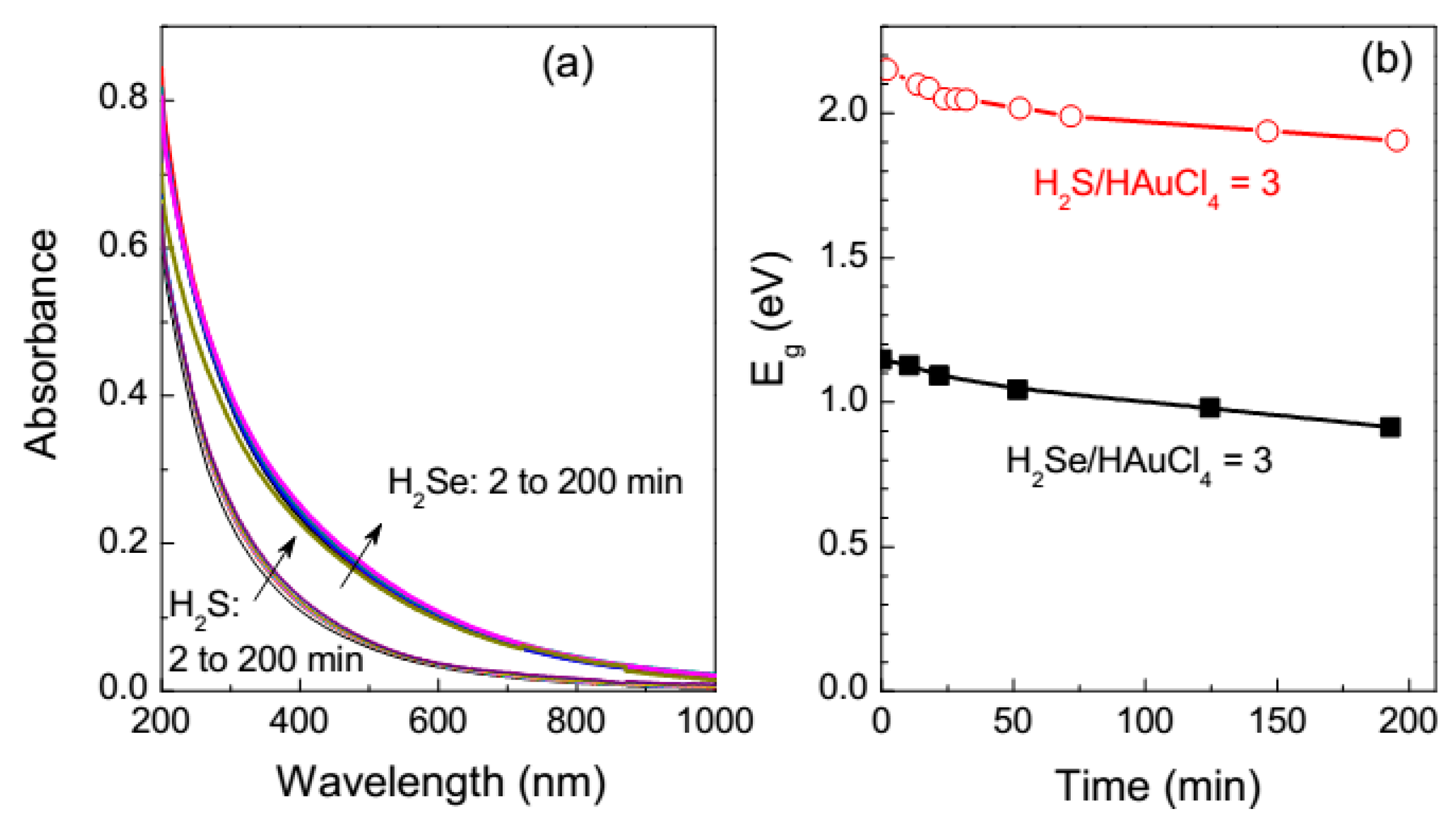
3.1. UV-Vis Absorption Spectroscopy
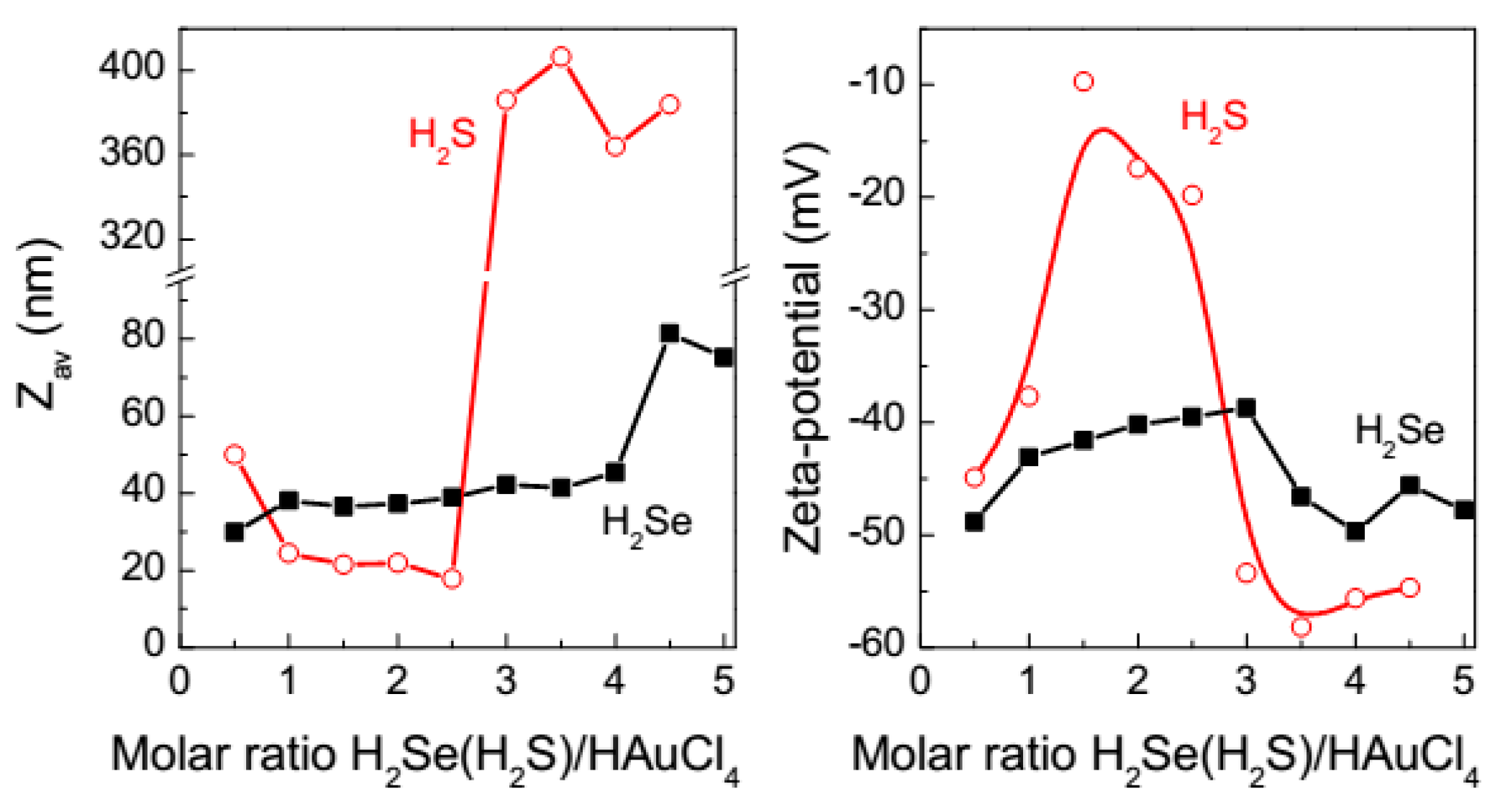
3.2. Dynamic Light Scattering and Zeta-Potential Measurement
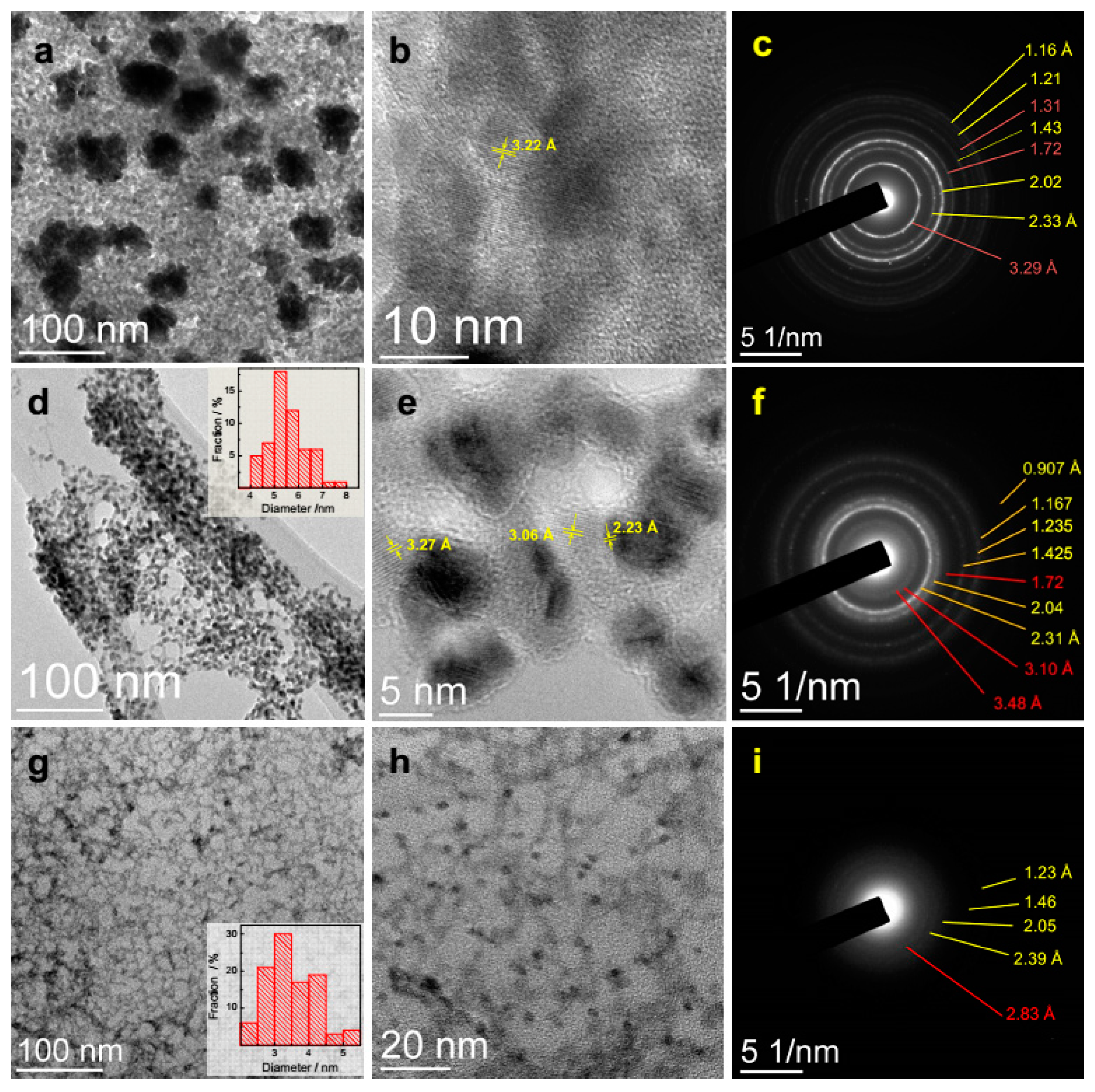
3.3. TEM
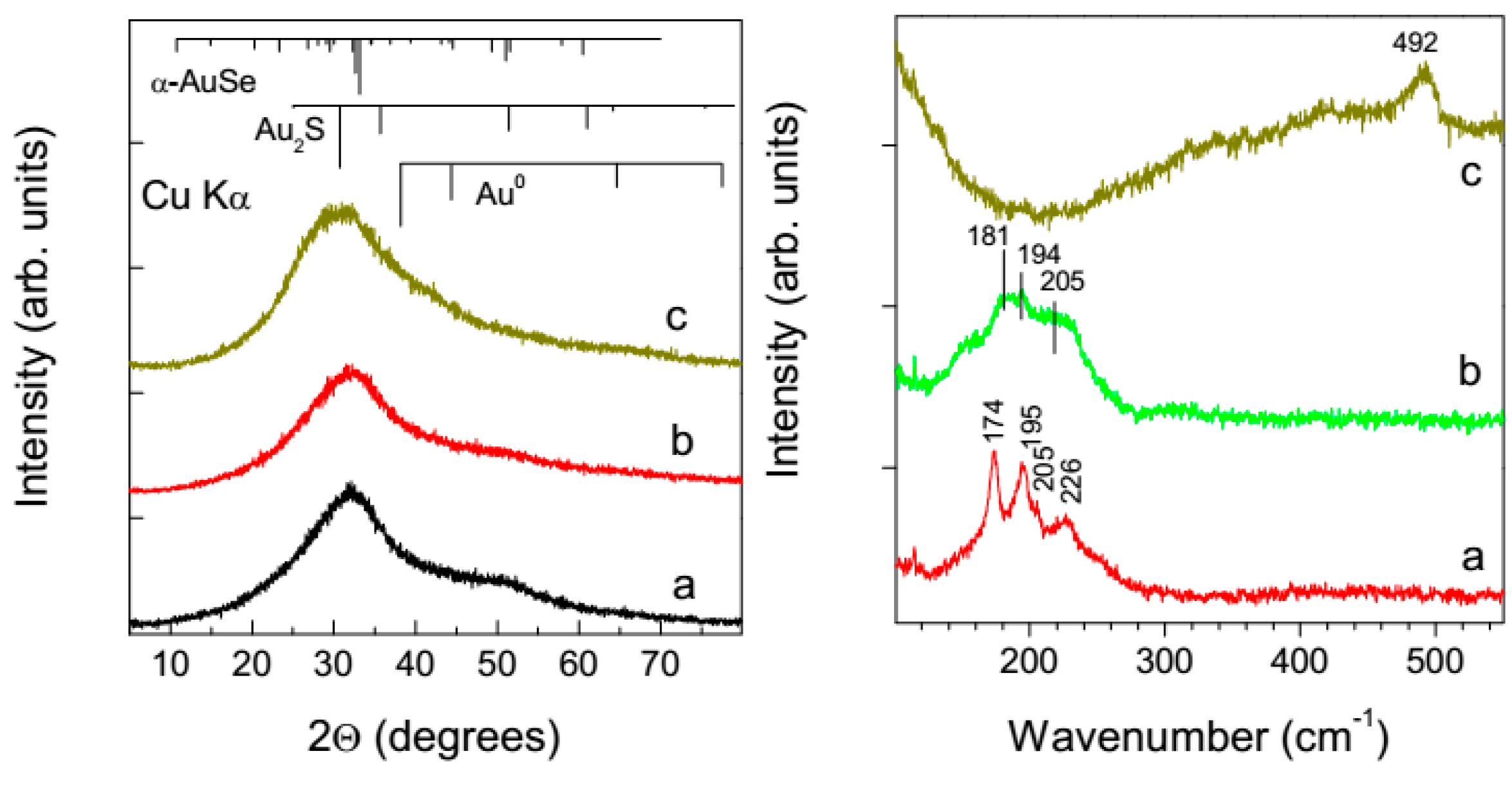
3.4. X-ray Diffraction Analysis and Raman Scattering
3.5. X-ray Photoelectron Spectroscopy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davidson, D.F. Selenium in Some Epithermal Deposits of Antimony, Mercury and Silver and Gold; Geol. Survey Bull: Reston, VA, USA, 1960; Volume 1112-A, pp. 1–16. [Google Scholar]
- Barton, M.D. The Ag–Au–S system. Econom. Geol. 1980, 75, 303–316. [Google Scholar] [CrossRef]
- Liu, J.; Liu, J.; Zheng, M.; Liu, X. Au–Se paragenesis in Cambrian stratabound gold deposits, Western Qinling Mountains, China. Int. Geol. Rev. 2000, 42, 1037–1045. [Google Scholar] [CrossRef]
- Bindi, L.; Cipriani, C. Structural and physical properties of fischesserite, a rare gold–silver selenide from the De Lamar mine, Owyhee County, Idaho, USA. Can. Mineral. 2004, 42, 1733–1737. [Google Scholar] [CrossRef]
- Pal’yanova, G.A.; Kokh, K.A.; Seryotkin, Y.V. Formation of gold and silver sulfides in the system Ag–Au–S. Russ. Geol. Geophys. 2011, 52, 443–449. [Google Scholar] [CrossRef]
- Cocker, H.A.; Mauk, J.L.; Rabone, S.D.C. The origin of Ag–Au–S–Se minerals in adularia-sericite epithermal deposits: Constraints from the Broken Hills deposit, Hauraki Goldfield, New Zealand. Miner. Depos. 2013, 48, 249–266. [Google Scholar] [CrossRef]
- Seryotkin, Y.V.; Pal’yanova, G.A.; Savva, N.E. Sulfur–selenium isomorphous substitution and morphotropic transition in the Ag3Au(Se,S)2 series. Russ. Geol. Geophys. 2013, 54, 646–651. [Google Scholar] [CrossRef]
- Palyanova, G.; Karmanov, N.; Savva, N. Sulfidation of native gold. Am. Mineral. 2014, 99, 1095–1103. [Google Scholar] [CrossRef]
- Pal’yanova, G.A.; Kravtsova, R.G.; Zhuravkova, T.V. Ag2(S,Se) solid solutions in the ores of the Rogovik gold–silver deposit (northeastern Russia). Russ. Geol. Geophys. 2015, 56, 1738–1748. [Google Scholar] [CrossRef]
- Bindi, L.; Stanley, C.J.; Seryotkin, Y.V.; Bakakin, V.V.; Pal’yanova, G.A.; Kokh, K.A. The crystal structure of uytenbogaardtite, Ag3AuS2, and its relationships with gold and silver sulfides–selenides. Mineral. Mag. 2016, 80, 1031–1040. [Google Scholar] [CrossRef]
- Palyanova, G.A.; Seryotkin, Y.V.; Kokh, K.A.; Bakakin, V.V. Isomorphism and solid solutions among Ag- and Au-selenides. J. Solid State Chem. 2016, 241, 157–163. [Google Scholar] [CrossRef]
- Palyanova, G.; Seryotkin, Y.; Kokh, K.; Bakakin, V.V. Sulfur–selenium isomorphous substitution in the AgAu(Se,S) series. J. Alloys Compd. 2016, 664, 385–391. [Google Scholar] [CrossRef]
- Palyanova, G.A.; Savva, N.E.; Zhuravkova, T.V.; Kolova, E.E. Gold and silver minerals in low-sulfidation ores of the Dzhulietta deposit (northeastern Russia). Russ. Geol. Geophys. 2016, 57, 1171–1190. [Google Scholar] [CrossRef]
- Tolstykh, N.; Vymazalová, A.; Tuhý, M.; Shapovalova, M. Conditions of formation of Au–Se–Te mineralization in the Gaching ore occurrence (Maletoivayam ore field), Kamchatka, Russia. Mineral. Mag. 2018, 82, 649–674. [Google Scholar] [CrossRef]
- Fang, C.M.; de Groot, R.A.; Wiegers, G.A. Ab initio band structure calculations of the low-temperature phases of Ag2Se, Ag2Te and Ag3AuSe2. J. Phys. Chem. Solids 2002, 63, 457–464. [Google Scholar] [CrossRef]
- Xiao, C.; Xu, J.; Li, K.; Feng, J.; Yang, J.; Xie, Y. Superionic phase transition in silver chalcogenide nanocrystals realizing optimized thermoelectric performance. J. Am. Chem. Soc. 2012, 134, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- Dalmases, M.; Ibáñez, M.; Torruella, P.; Fernàndez-Altable, V.; López-Conesa, L.; Cadavid, D.; Piveteau, I.; Nachtegaal, M.; Llorca, J.; Ruiz-González, M.L.; et al. Synthesis and thermoelectric properties of noble metal ternary chalcogenide systems of Ag–Au–Se in the forms of alloyed nanoparticles and colloidal nanoheterostructures. Chem. Mater. 2016, 28, 7017–7028. [Google Scholar] [CrossRef]
- Liu, M.; Zeng, H.C. General synthetic approach to heterostructured nanocrystals based on noble metals and I–VI, II–VI, and I–III–VI metal chalcogenides. Langmuir 2014, 30, 9838–9849. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, B.; Zheng, S.; Xu, Y.; Xue, H.; Pang, H. Syntheses and energy storage applications of MxSy (M = Cu, Ag, Au) and their composites: Rechargeable batteries and supercapacitors. Adv. Funct. Mater. 2017, 27, 1703949. [Google Scholar] [CrossRef]
- Benning, L.G.; Seward, T.M. Hydrosulphide complexing of Au(I) in hydrothermal solutions from 150–400 °C and 500–1500 bar. Geochim. Cosmochim. Acta 1996, 60, 1849–1871. [Google Scholar] [CrossRef]
- Tagirov, B.R.; Baranova, N.N.; Zotov, A.V.; Schott, J.; Bannykh, L.N. Experimental determination of the stabilities of Au2S(cr)at 25 °C and Au(HS)2− at 25–250 °C. Geochim. Cosmochim. Acta 2006, 70, 3689–3701. [Google Scholar] [CrossRef]
- Pokrovski, G.S.; Tagirov, B.R.; Schott, J.; Hazemanne, J.-L.; Proux, O. A new view on gold speciation in sulfur-bearing hydrothermal fluids from in situ X-ray absorption spectroscopy and quantum-chemical modeling. Geochim. Cosmochim. Acta 2009, 73, 5406–5427. [Google Scholar] [CrossRef]
- Zezin, D.Y.; Migdisov, A.A.; Williams-Jones, A.E. The solubility of gold in H2O–H2S vapour at elevated temperature and pressure. Geochim. Cosmochim. Acta 2011, 75, 5140–5153. [Google Scholar] [CrossRef]
- Liu, W.; Etschmann, B.; Testemale, D.; Hazemann, J.-L.; Rempel, K.; Müller, H.; Brugger, J. Gold transport in hydrothermal fluids: Competition among the Cl−, Br−, HS− and NH3(aq) ligands. Chem. Geol. 2014, 376, 11–19. [Google Scholar] [CrossRef]
- Trigub, A.L.; Tagirov, B.R.; Kvashnina, K.O.; Lafuerz, S.; Filimonova, O.N.; Nickolsky, M.S. Experimental determination of gold speciation in sulfide-rich hydrothermal fluids under a wide range of redox conditions. Chem. Geol. 2017, 471, 52–64. [Google Scholar] [CrossRef]
- Ishikawa, K.; Isonaga, T.; Wakita, S.; Suzuki, Y. Structure and electrical properties of Au2S. Solid State Ion. 1995, 79, 60–66. [Google Scholar] [CrossRef]
- Osadchii, E.G.; Rappo, O.A. Determination of standard thermodynamic properties of sulfides in the Ag–Au–S system by means of a solid-state galvanic cell. Am. Mineral. 2004, 89, 1405–1410. [Google Scholar] [CrossRef]
- Simon, G.; Essene, E.J. Phase relations among selenides, sulfides, tellurides, and oxides; I, Thermodynamic properties and calculated equilibria. Econ. Geol. 1996, 91, 1183–1208. [Google Scholar] [CrossRef]
- Echmaeva, E.A.; Osadchii, E.G. Determination of the thermodynamic properties of compounds in the Ag–Au–Se and Ag–Au–Te systems by the EMF method. Geol. Ore Depos. 2009, 51, 247–258. [Google Scholar] [CrossRef]
- Rabenau, A.; Rau, H.; Rosenstein, G. Phase relations in the gold-selenium system. J. Less Common Met. 1971, 24, 291–299. [Google Scholar] [CrossRef]
- Rabenau, A.; Schulz, H. The crystal structures of α-AuSe and β-AuSe. J. Less Common Met. 1976, 48, 89–101. [Google Scholar] [CrossRef]
- Cretier, J.E.; Wiegers, G.A. The crystal structure of the beta form of gold selenide, β-AuSe. Mat. Res. Bull. 1973, 8, 1427–1430. [Google Scholar] [CrossRef]
- Feng, D.; Taskinen, P. Thermodynamic stability of AuSe at temperature from (400 to 700) K by a solid state galvanic cell. J. Chem. Thermodyn. 2014, 71, 98–102. [Google Scholar] [CrossRef]
- Xu, X.L.; Chen, W.K.; Wang, X. Density functional study on adsorption of NO on AuSe (010) surface. Chin. J. Chem. 2008, 26, 107–112. [Google Scholar] [CrossRef]
- Wagner, F.E.; Palade, P.; Friedl, J.; Filoti, G.; Wang, N. 197Au Mössbauer study of gold selenide, AuSe. J. Phys. Conf. Ser. 2010, 217, 012039. [Google Scholar] [CrossRef]
- Ettema, A.R.H.F.; Stegink, T.A.; Haas, C. The valence of Au in AuTe2 and AuSe studied by X-ray absorption spectroscopy. Solid State Commun. 1994, 90, 211–213. [Google Scholar] [CrossRef]
- Lee, W.R.; Jung, D. Electronic structure study of gold selenides. Bull. Korean Chem. Soc. 1999, 20, 147–150. [Google Scholar]
- Perry, D.L. Handbook of Inorganic Compounds; CRC Press: Boca Raton, FL, USA, 2011; ISBN 9781439814611. [Google Scholar]
- Nath, S.; Ghosh, S.K.; Pal, T. Solution phase evolution of AuSe nanoalloys in Triton X-100 underUV-photoactivation. Chem. Commun. 2004, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Prokeš, L.; Kubáček, P.; Peña-Méndez, E.M.; Amato, F.; Conde, J.E.; Alberti, M.; Havel, J. Laser ablation synthesis of gold selenides by using a mass spectrometer as a synthesizer: Laser desorption ionization time-of-flight mass spectrometry. Chem.-Eur. J. 2016, 22, 11261–11268. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Cheng, R.; Liu, X.; Pan, X.; Kong, F.; Gao, W.; Xu, K.; Tang, B. A nanosensor for in vivo selenol imaging based on the formation of AuSe bonds. Biomaterials 2016, 92, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Machogo, L.F.E.; Tetyana, P.; Sithole, R.; Gqoba, S.S.; Phao, N.; Airo, M.; Shumbula, P.M.; Moloto, M.J.; Moloto, N. Unravelling the structural properties of mixed-valence α- and β-AuSe nanostructures using XRD, TEM and XPS. Appl. Surf. Sci. 2018, 456, 973–979. [Google Scholar] [CrossRef]
- Neumann, H.; Yakushev, M.V.; Tomlinson, R.D. Diffusion effects at the Au/p–CuInSe2 contact studied by XPS. Cryst. Res. Technol. 2003, 38, 676–683. [Google Scholar] [CrossRef]
- Haldar, K.K.; Sinha, G.; Lahtinen, J.; Patra, A. Hybrid colloidal Au–CdSe pentapod heterostructures synthesis and their photocatalytic properties. ACS Appl. Mater. Interfaces 2012, 4, 6266–6272. [Google Scholar] [CrossRef] [PubMed]
- de la Cueva, L.; Meyns, M.; Bastús, N.G.; Rodríguez-Fernández, J.; Otero, R.; Gallego, J.M.; Alonso, C.; Klinke, C.; Juárez, B.H. Shell or dots-precursor controlled morphology of Au−Se depositson CdSe nanoparticles. Chem. Mater. 2016, 28, 2704–2714. [Google Scholar] [CrossRef]
- Jia, J.; Bendounan, A.; Kotresh, H.M.N.; Chaouchi, K.; Sirotti, F.; Sampath, S.; Esaulov, V.A. Selenium adsorption on Au(111) and Ag(111) surfaces: Adsorbed seleniumand selenidefilms. J. Phys. Chem. C 2013, 117, 9835–9842. [Google Scholar] [CrossRef]
- Ruano, G.; Tosi, E.; Sanchez, E.; Abufager, P.; Martiarena, M.L.; Grizzi, O.; Zampieri, G. Stages of Se adsorption on Au (111): A combined XPS, LEED, TOF-DRS, and DFT study. Surf. Sci. 2017, 662, 113–122. [Google Scholar] [CrossRef]
- Yee, C.K.; Ulman, A.; Ruiz, J.D.; Parikh, A.; White, H.; Rafailovich, M. Alkyl selenide- and alkyl thiolate-functionalized gold nanoparticles: Chain packing and bond nature. Langmuir 2003, 19, 9450–9458. [Google Scholar] [CrossRef]
- Hohman, J.N.; Thomas, J.C.; Zhao, Y.; Auluck, H.; Kim, M.; Vijselaar, W.; Kommeren, S.; Terfort, A.; Weiss, P.S. Exchange reactions between alkanethiolates and alkaneselenols on Au{111}. J. Am. Chem. Soc. 2014, 136, 8110–8121. [Google Scholar] [CrossRef] [PubMed]
- Averitt, R.D.; Sarkar, D.; Halas, N.J. Plasmon resonance shifts of Au-coated Au2S nanoshells: Insight into multicomponent nanoparticle growth. Phys. Rev. Lett. 1997, 78, 4217–4220. [Google Scholar] [CrossRef]
- Morris, T.; Copeland, H.; Szulczewski, G. Synthesis and characterization of gold sulfide nanoparticles. Langmuir 2002, 18, 535–539. [Google Scholar] [CrossRef]
- Majimel, J.; Bacinello, D.; Durand, E.; Vallee, F.; Treguer-Delapierre, M. Synthesis of hybrid gold–gold sulfide colloidal particles. Langmuir 2008, 24, 4289–4294. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-L.; Huang, M.H. Hydrothermal synthesis of free-floating Au2S nanoparticle superstructures. J. Phys. Chem. C 2008, 112, 11661–11666. [Google Scholar] [CrossRef]
- Schwartzberg, A.M.; Grant, C.D.; van Buuren, T.; Zhang, J.Z. Reduction of HAuCl4 by Na2S revisited: The case for Au nanoparticle aggregates and against Au2S/Au core/shell particles. J. Phys. Chem. C 2007, 111, 8892–8901. [Google Scholar] [CrossRef]
- Mikhlin, Y.; Likhatski, M.; Karacharov, A.; Zaikovski, V.; Krylov, A. Formation of gold and gold sulfide nanoparticles and mesoscale intermediate structures in the reactions of aqueous HAuCl4 with sulfide and citrate ions. Phys. Chem. Chem. Phys. 2009, 11, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Mikhlin, Yu.; Likhatski, M.; Tomashevich, Ye.; Romanchenko, A.; Erenburg, S.; Trubina, S. XAS and XPS examination of the Au–S nanostructures produced via the reduction of aqueous gold (III) by sulfide ions. J. Electron Spectrosc. Relat. 2010, 177, 24–29. [Google Scholar] [CrossRef]
- Mikhlin, Y.; Karacharov, A.; Likhatski, M.; Podlipskaya, T.; Zizak, I. Direct observation of liquid pre-crystallization intermediates during the reduction of aqueous tetrachloroaurate by sulfide ions. Phys. Chem. Chem. Phys. 2014, 16, 4538–4543. [Google Scholar] [CrossRef] [PubMed]
- Likhatski, M.; Karacharov, A.; Kondrasenko, A.; Mikhlin, Y. On a role of liquid intermediates in nucleation of gold sulfide nanoparticles in aqueous media. Faraday Discuss. 2015, 179, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Dhayagude, A.C.; Maiti, N.; Debnath, A.K.; Joshi, S.S.; Kapoor, S. Metal nanoparticle catalyzed charge rearrangement in selenourea probed by surface-enhanced Raman scattering. RSC Adv. 2016, 6, 17405–17414. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Nasluzov, V.A.; Romanchenko, A.S.; Shor, A.M.; Pal’yanova, G.A. XPS and DFT studies of the electronic structures of AgAuS and Ag3AuS2. J. Alloys Compd. 2014, 617, 314–321. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Pal’yanova, G.A.; Tomashevich, Y.V.; Vishnyakova, E.A.; Vorobyev, S.A.; Kokh, K.A. XPS and Ag L3-edge XANES characterization of silver- and silver-gold sulfoselenides. J. Phys. Chem. Solids 2018, 116, 292–298. [Google Scholar] [CrossRef]
- Hough, R.M.; Noble, R.R.P.; Reich, M. Natural gold nanoparticles. Ore Geol. Rev. 2011, 42, 55–61. [Google Scholar] [CrossRef]
- Saunders, J.A.; Burke, M. Formation and aggregation of gold (electrum) nanoparticles in epithermal ores. Minerals 2017, 7, 163. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vorobyev, S.; Likhatski, M.; Romanchenko, A.; Maksimov, N.; Zharkov, S.; Krylov, A.; Mikhlin, Y. Colloidal and Deposited Products of the Interaction of Tetrachloroauric Acid with Hydrogen Selenide and Hydrogen Sulfide in Aqueous Solutions. Minerals 2018, 8, 492. https://doi.org/10.3390/min8110492
Vorobyev S, Likhatski M, Romanchenko A, Maksimov N, Zharkov S, Krylov A, Mikhlin Y. Colloidal and Deposited Products of the Interaction of Tetrachloroauric Acid with Hydrogen Selenide and Hydrogen Sulfide in Aqueous Solutions. Minerals. 2018; 8(11):492. https://doi.org/10.3390/min8110492
Chicago/Turabian StyleVorobyev, Sergey, Maxim Likhatski, Alexander Romanchenko, Nikolai Maksimov, Sergey Zharkov, Alexander Krylov, and Yuri Mikhlin. 2018. "Colloidal and Deposited Products of the Interaction of Tetrachloroauric Acid with Hydrogen Selenide and Hydrogen Sulfide in Aqueous Solutions" Minerals 8, no. 11: 492. https://doi.org/10.3390/min8110492
APA StyleVorobyev, S., Likhatski, M., Romanchenko, A., Maksimov, N., Zharkov, S., Krylov, A., & Mikhlin, Y. (2018). Colloidal and Deposited Products of the Interaction of Tetrachloroauric Acid with Hydrogen Selenide and Hydrogen Sulfide in Aqueous Solutions. Minerals, 8(11), 492. https://doi.org/10.3390/min8110492