
Apatite Biominerals

Abstract
:1. Introduction
2. Structure and Chemical Formulas
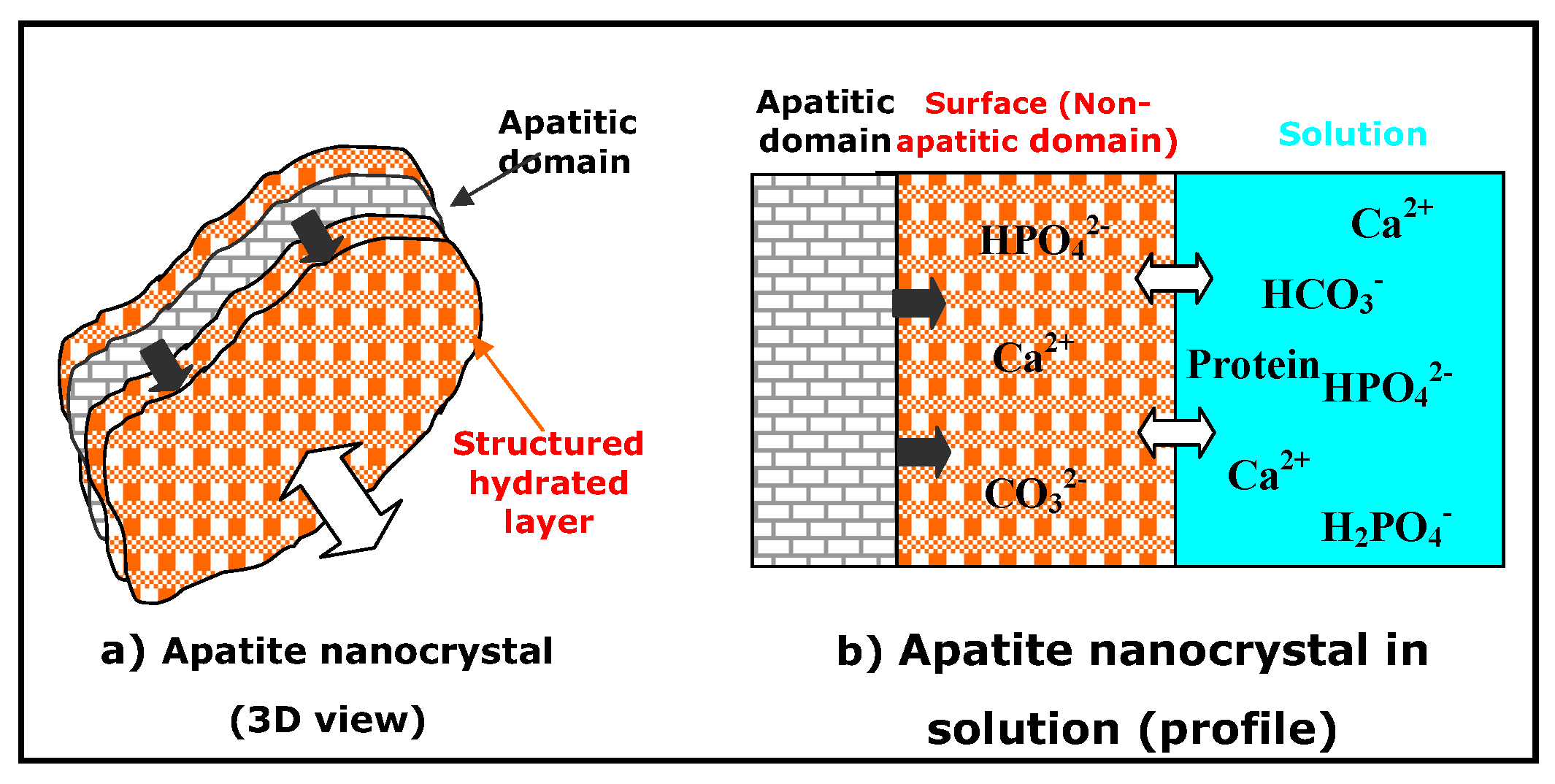
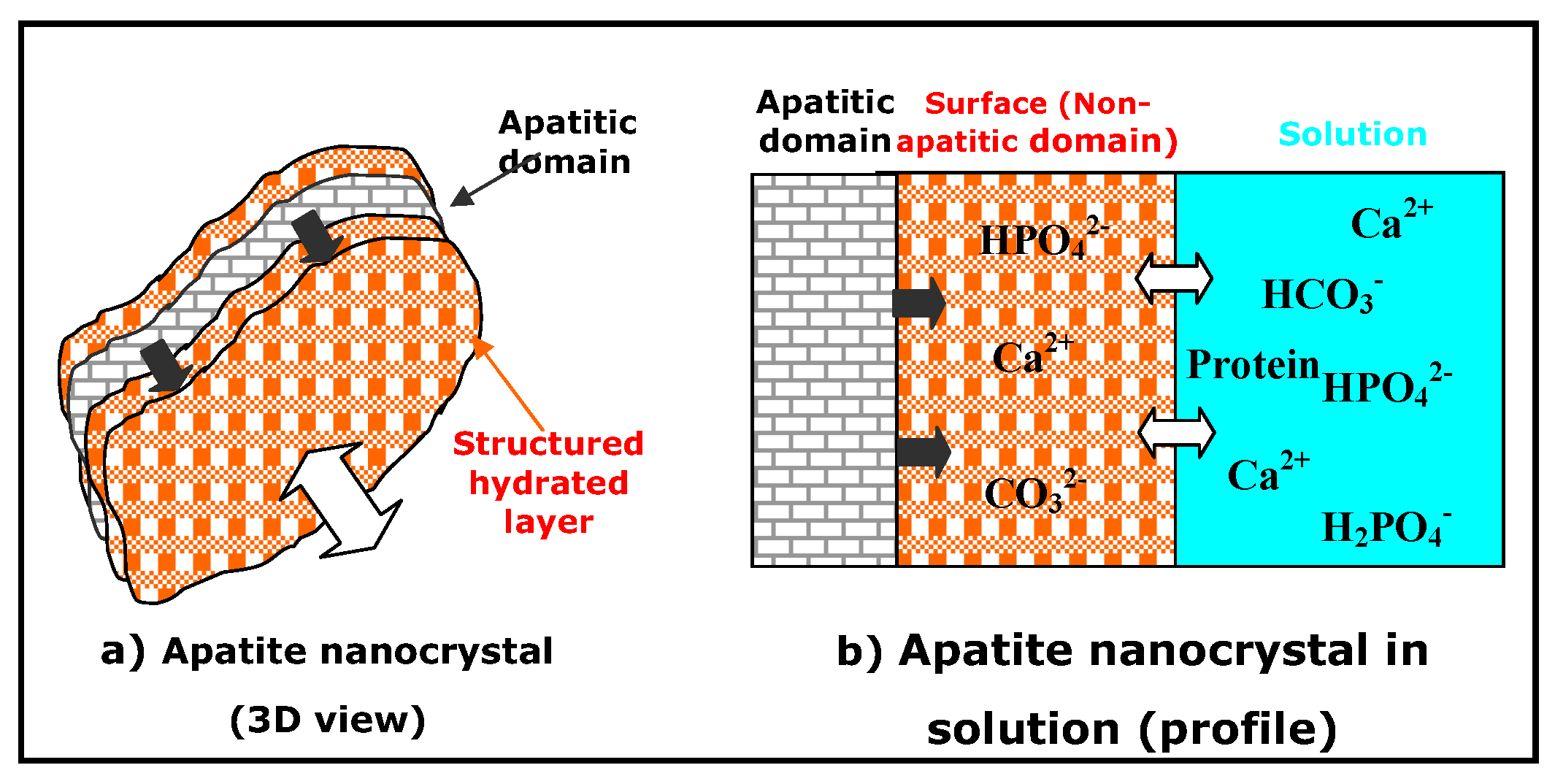
3. Non-Apatitic Environments and the Hydrated Layer
- (1)
- Apatite nanocrystals contain non-apatitic anionic and cationic chemical environments,
- (2)
- These environments strongly interact with hydrated domains,
- (3)
- Immature samples (freshly precipitated nanocrystalline apatites) show FTIR fine-band substructure changes upon drying without leading to long-range order modifications,
- (4)
4. Composition of the Main Mineralized Tissues
4.1. Main Elements
O2− + H2O → 2 OH−
4.2. Minor Elements
4.3. Trace Elements
5. Crystal Physical Characteristics
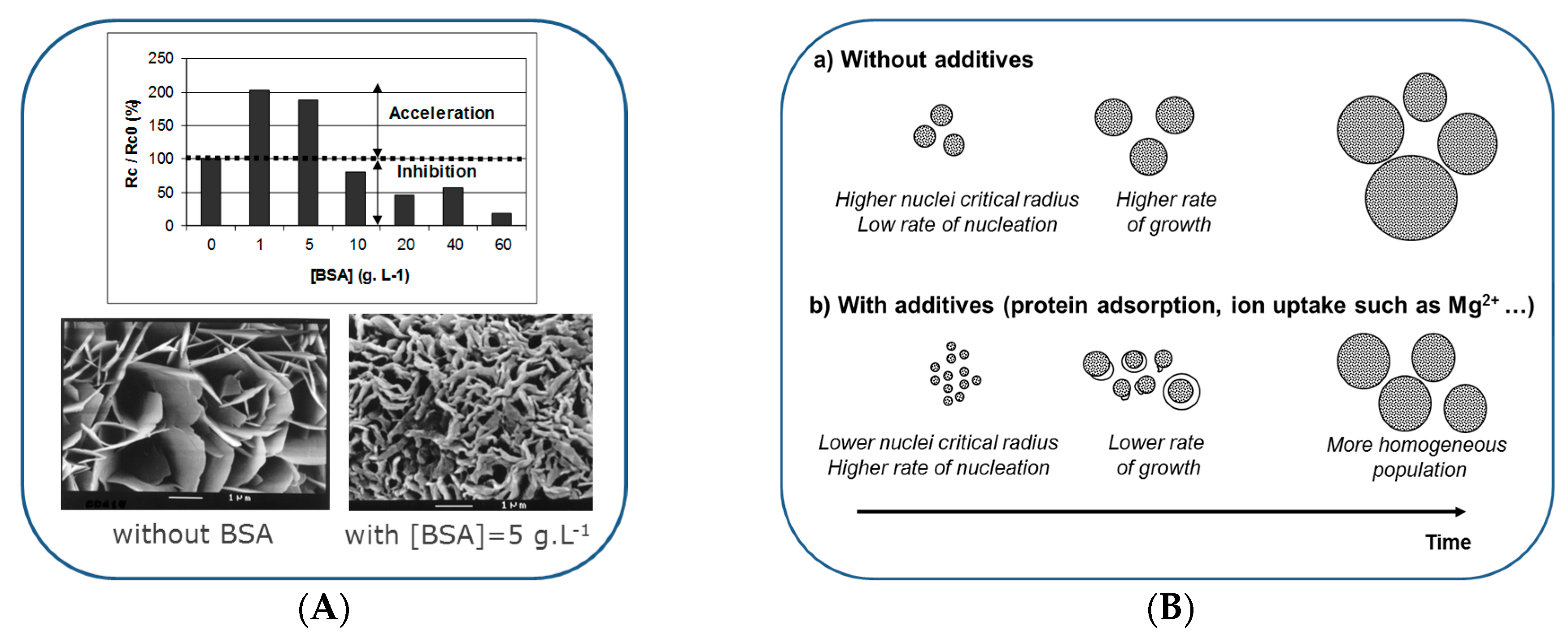
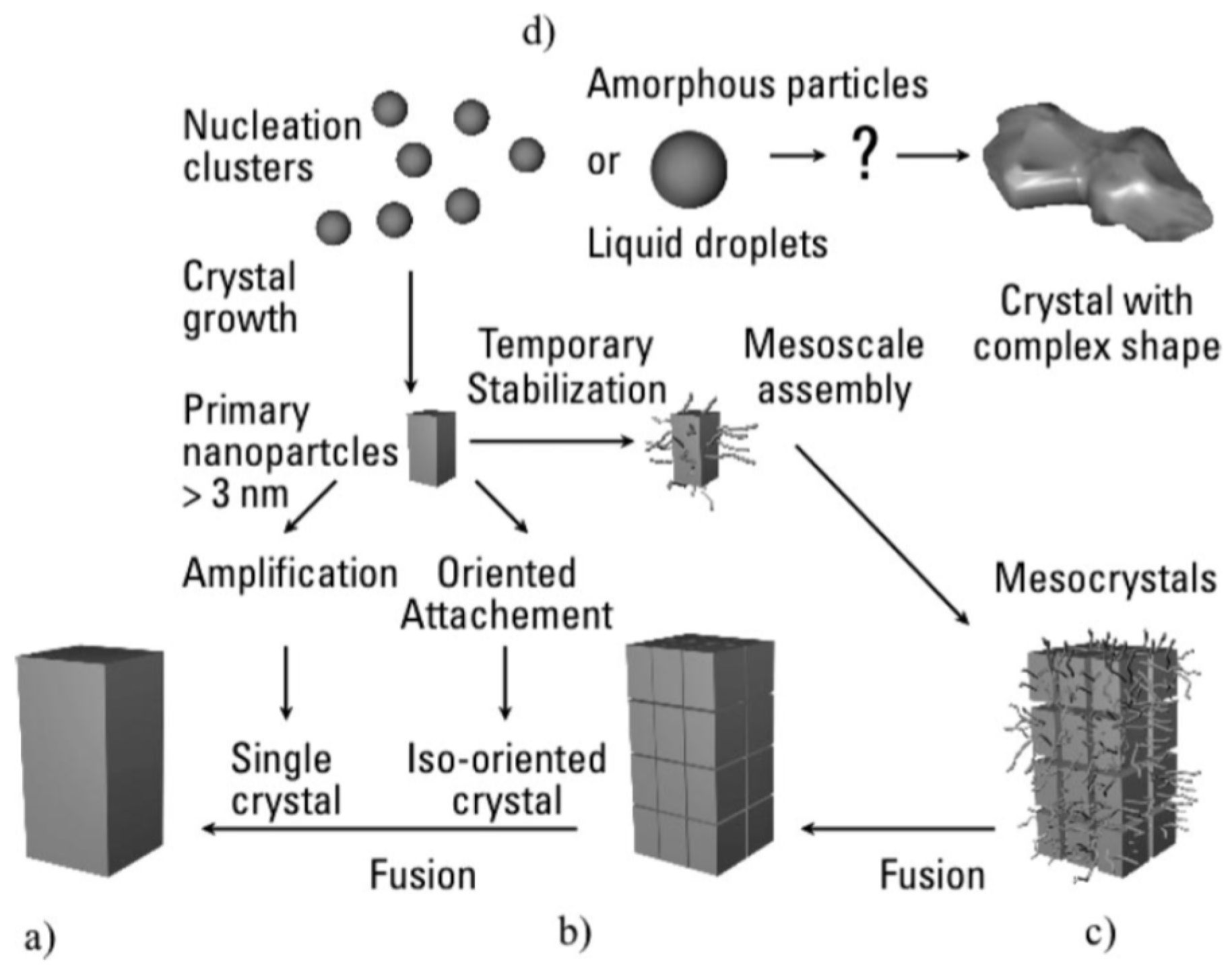
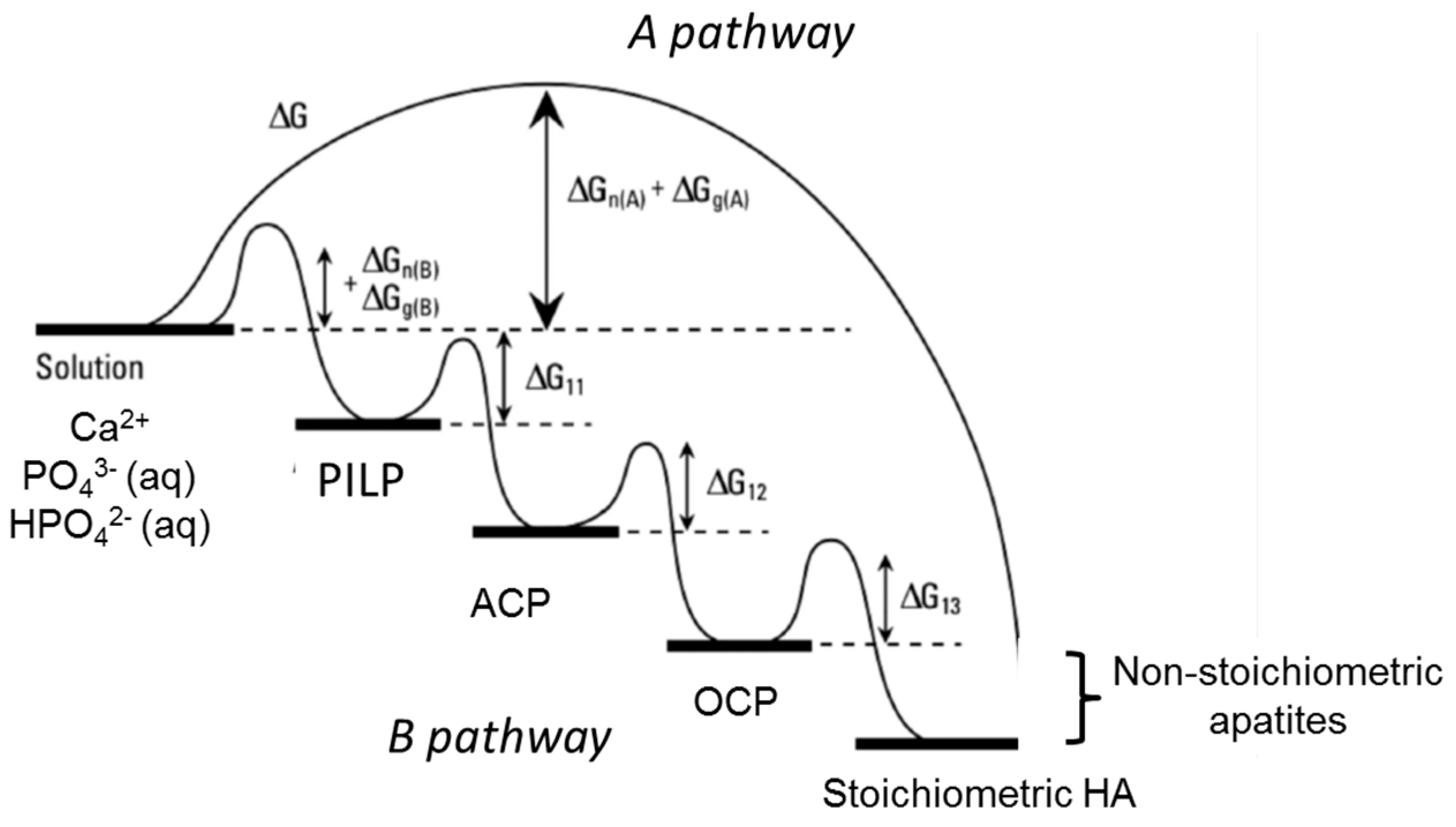
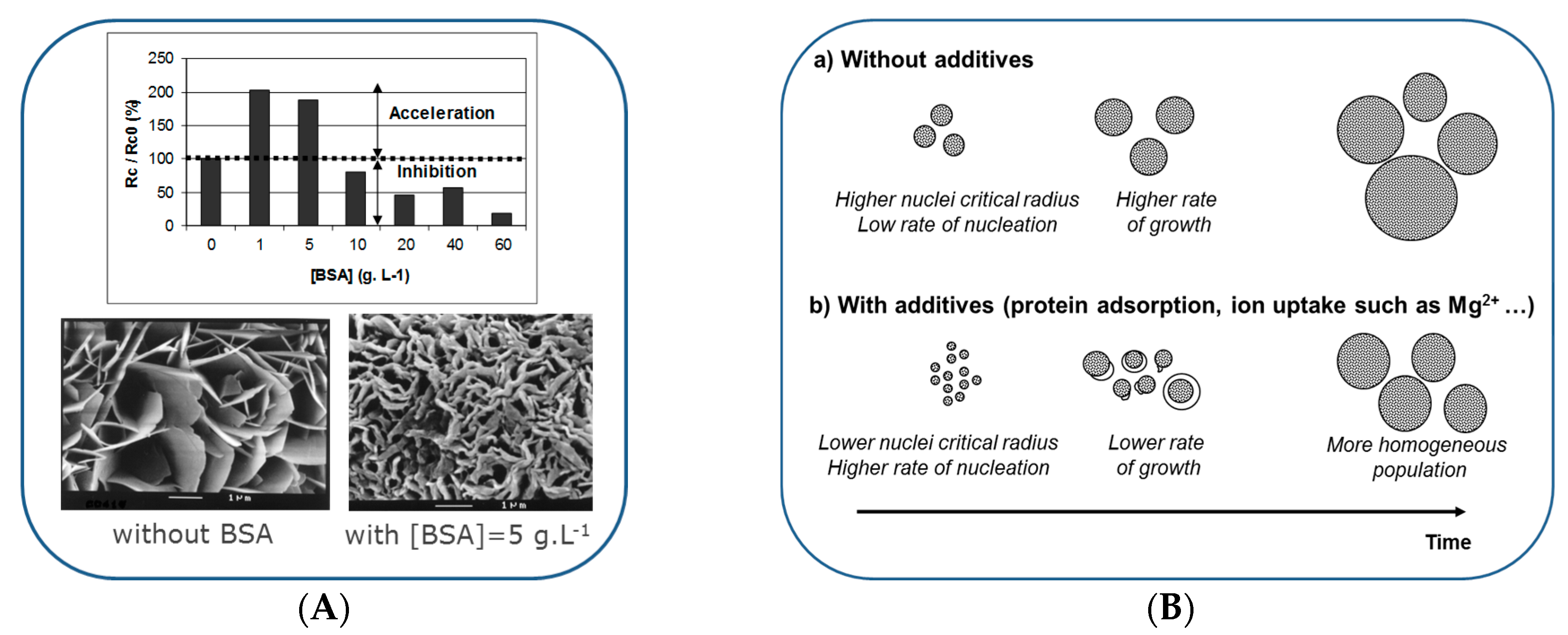
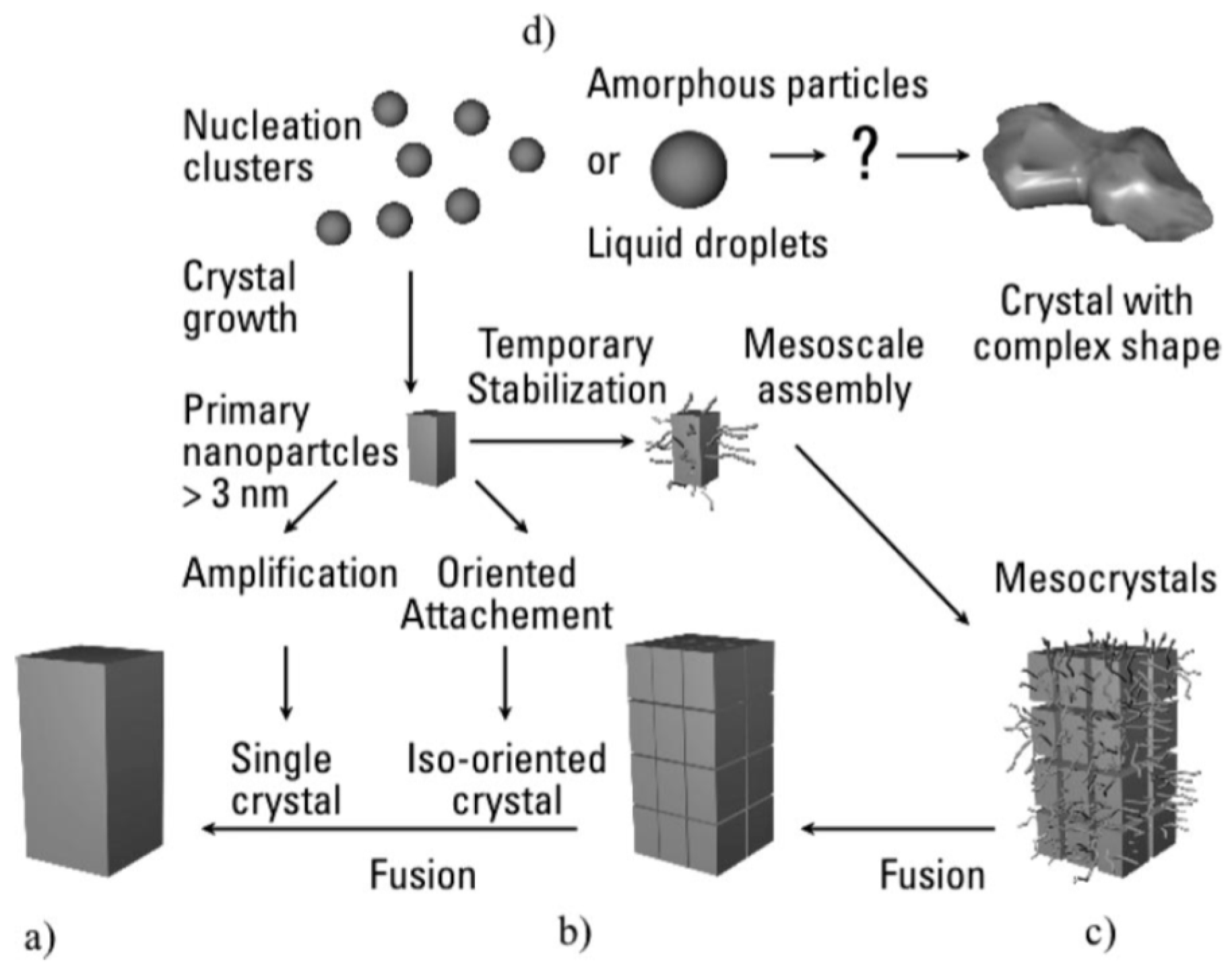
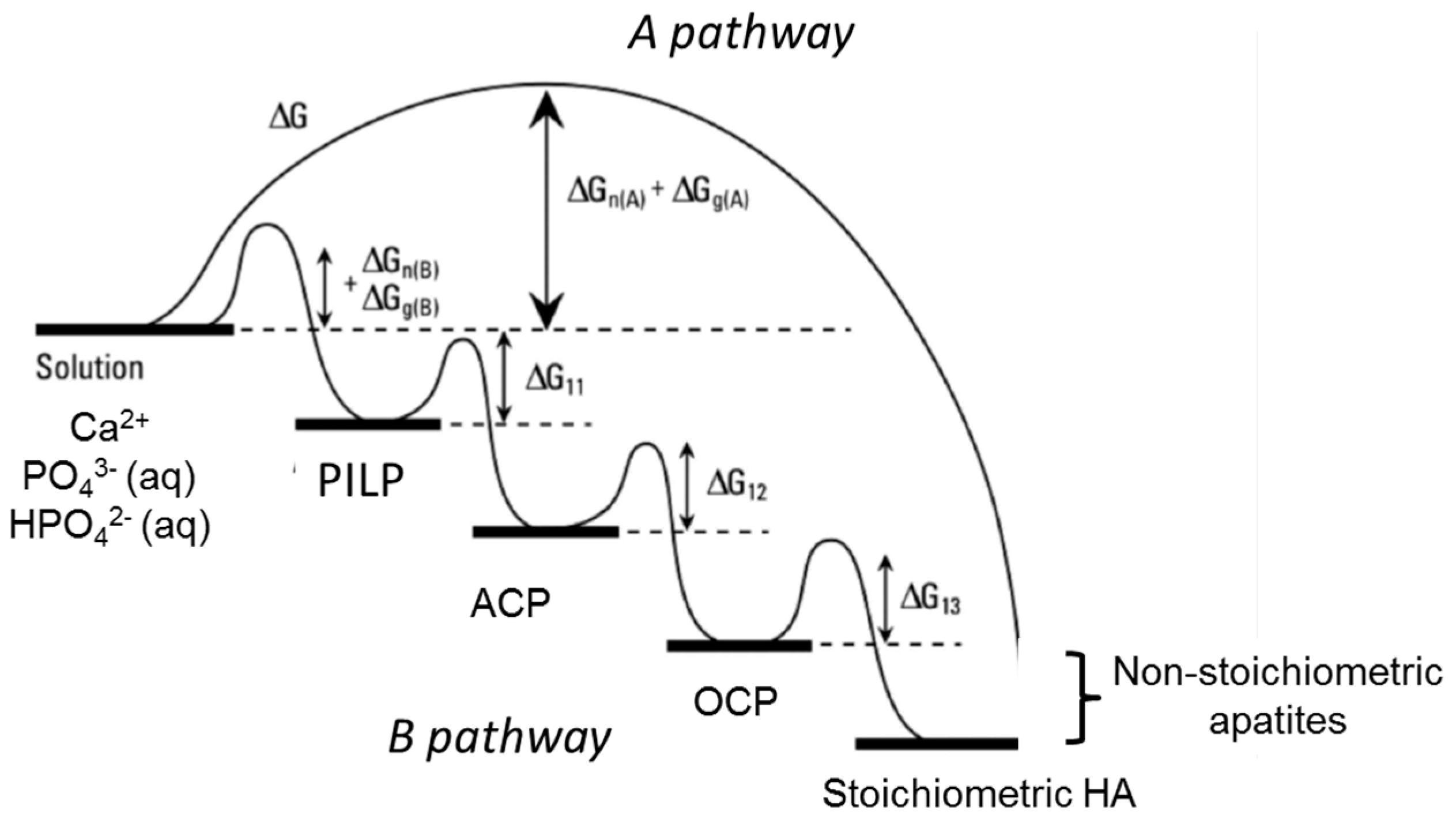
6. Formation
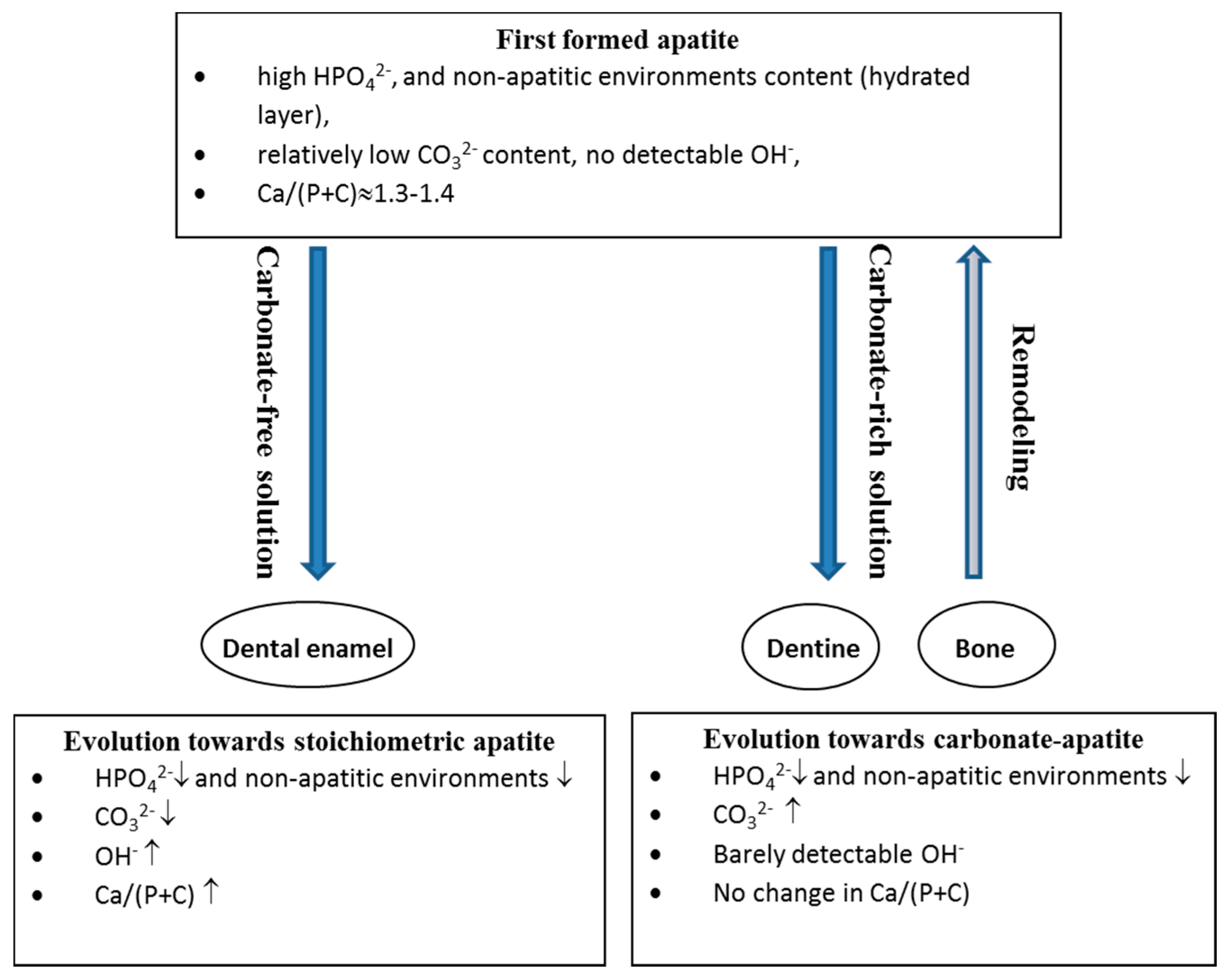
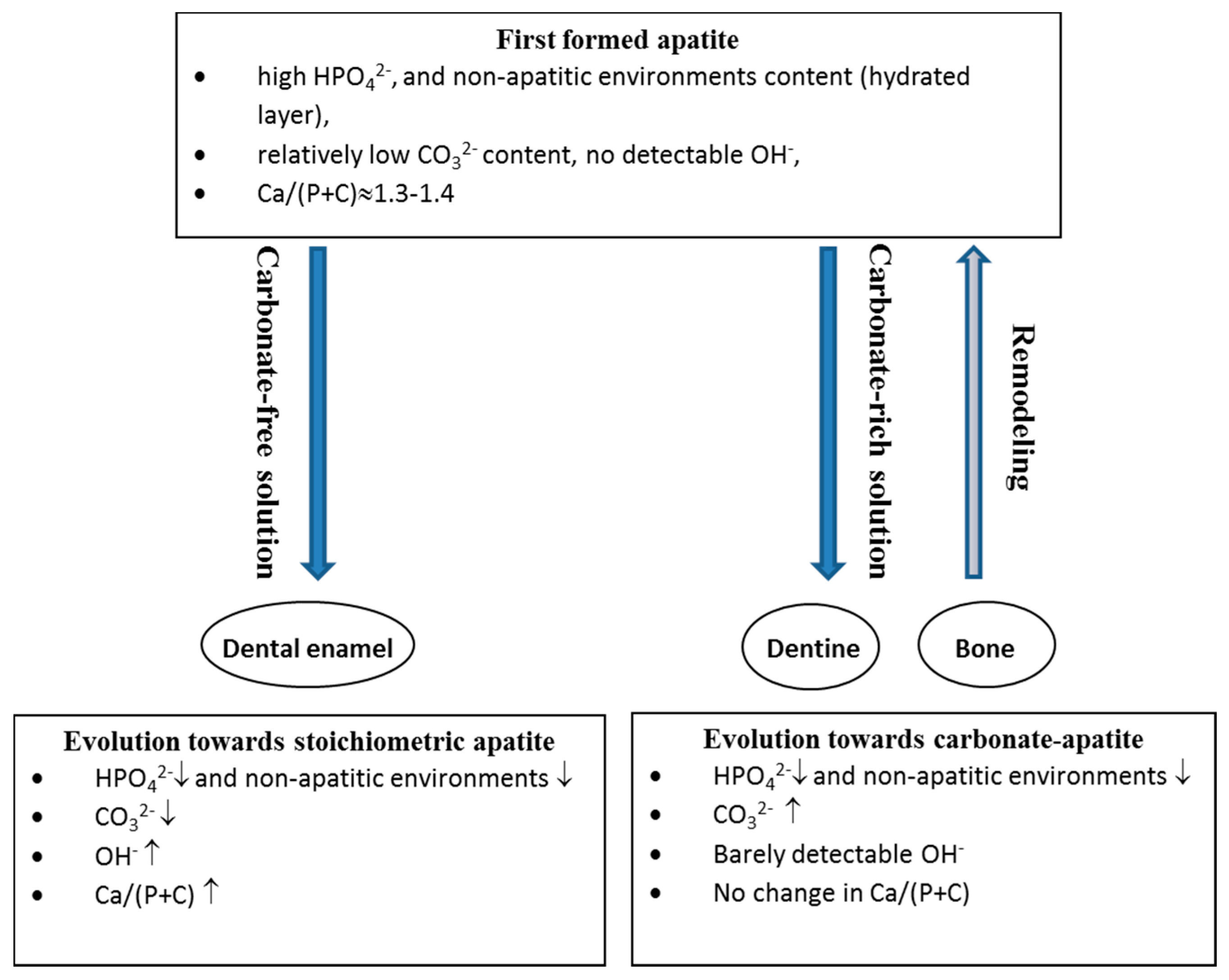
7. Maturation and Evolution
7.1. Bone
- a decrease in the labile environments;
- an increase in CO32− and a decrease in HPO42− with a constant Ca/(P + C) ratio; and
- the development of the apatite domains and an increase in their dimensions and crystallinity.
7.2. Enamel
8. Adaptability of Apatite Biominerals to Biological Function
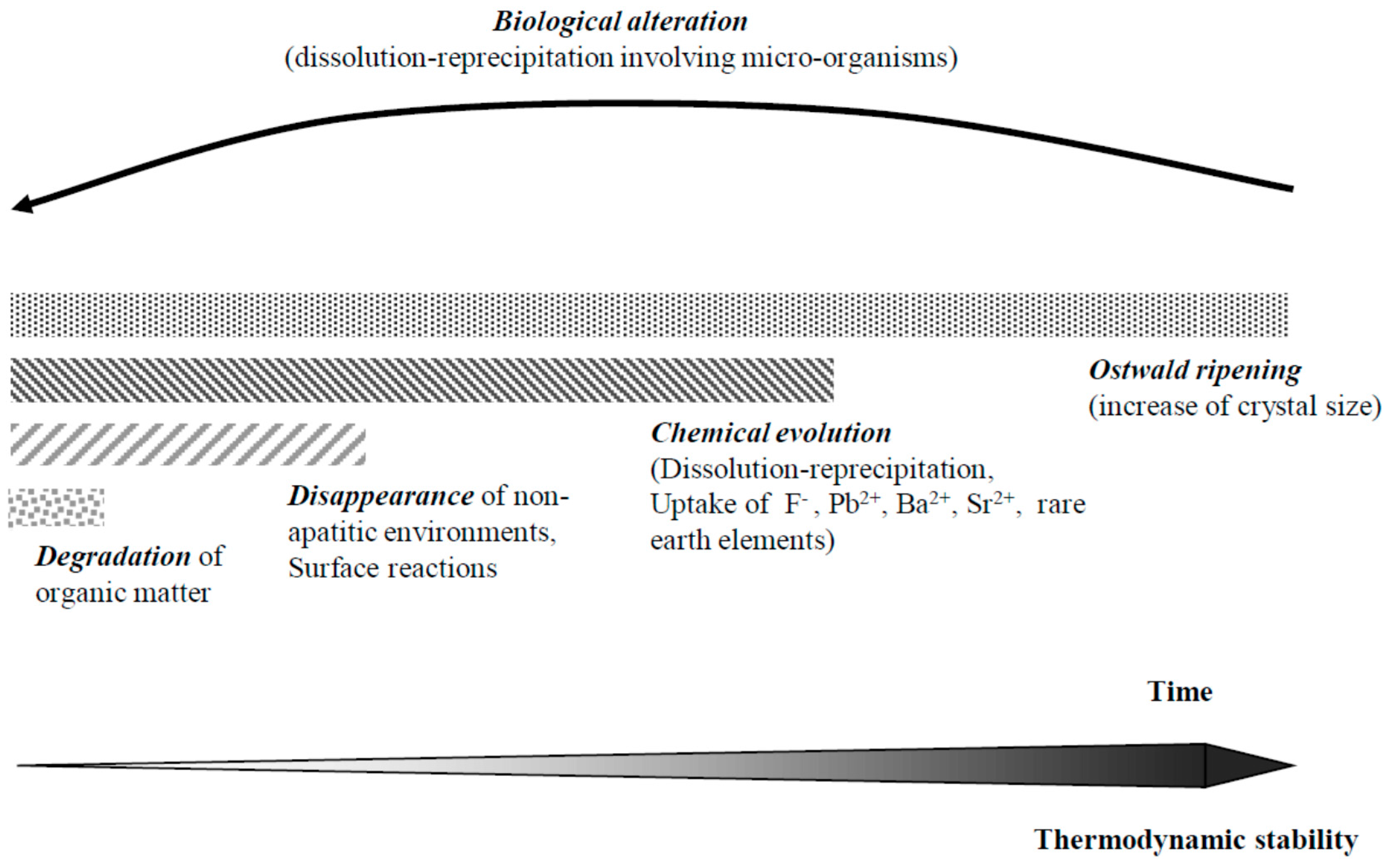
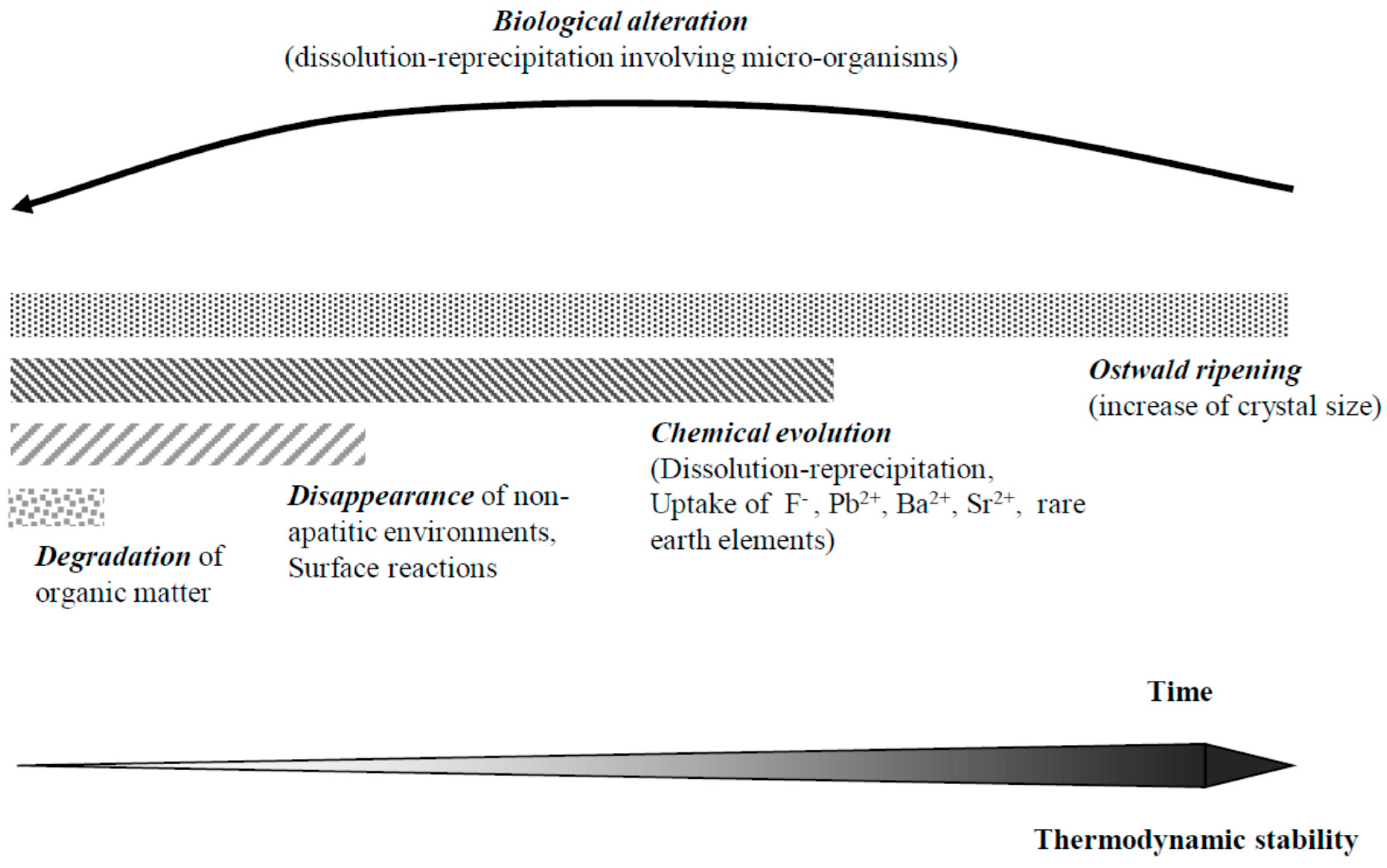
9. Diagenetic Evolution of Biological Apatite Fossils
10. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Lowenstam, H.A. Minerals formed by organisms. Science 1981, 211, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.C. Calcium phosphate biominerals. Rev. Mineral. Geochem. 2002, 48, 427–453. [Google Scholar] [CrossRef]
- Bazin, D.; Daudon, M.; Combes, C.; Rey, C. Characterization and some physicochemical aspects of pathological microcalcifications. Chem. Rev. 2012, 112, 5092–5120. [Google Scholar] [CrossRef] [PubMed]
- Carlstrom, D. A crystallographic study of vertebrates otoliths. Biol. Bull. 1963, 125, 441–463. [Google Scholar] [CrossRef]
- Li, Z.; Pasteris, J.D. Tracing the pathway of compositional changes in bone mineral with age: Preliminary study of bioapatite aging in hypermineralized dolphin’s bulla. Biochim. Biophys. Acta 2014, 1840, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Sakae, T.; Nakada, H.; LeGeros, J.P. Historical review of biological apatite crystallography. J. Hard Tissue Biol. 2015, 24, 111–122. [Google Scholar] [CrossRef]
- Uskokovic, V. The role of hydroxyl channel in defining selected physicochemical peculiarities exhibited by hydroxyapatite. RSC Adv. 2015, 5, 36614–36633. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Halter, T.J.; Borah, B.M.; Nancollas, G.H. Tracking amorphous precursor formation and transformation during induction stages of nucleation. Cryst. Growth Des. 2014, 14, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Omelon, S.; Ariganello, M.; Bonucci, E.; Grynpas, M.; Nanci, A. A review of phosphate mineral nucleation in biology and geobiology. Calcif. Tissue Int. 2013, 93, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L. Mineralization of bones and teeth. Elements 2007, 3, 385–391. [Google Scholar] [CrossRef]
- Gomez-Morales, J.; Iafisco, M.; Delgado-Lopez, J.M.; Sarda, S.; Drouet, C. Progress on the preparation of nanocrystalline apatites and surface characterization: Overview of fundamental and applied aspects. Prog. Cryst. Growth Charact. Mater. 2013, 59, 1–46. [Google Scholar] [CrossRef]
- Wopenka, B.; Pasteris, J.D. A mineralogical perspective on the apatite in bone. Mater. Sci. Eng. C 2005, 25, 131–143. [Google Scholar] [CrossRef]
- De Jong, W.F. La substance minérale dans les os. Recueil des Travaux Chimiques des Pays-Bas 1926, 45, 445–448. (In French) [Google Scholar] [CrossRef]
- White, T.J.; Zhili, D. Structural derivation and crystal chemistry of apatites. Acta Crystallogr. Sect. B Struct. Sci. 2003, B59, 1–16. [Google Scholar] [CrossRef]
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates; Elsevier: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Labarthe, J.C.; Bonel, G.; Montel, G. Structure and properties of B-type phosphocalcium carbonate apatites. Ann. Chim. 1973, 8, 289–301. [Google Scholar]
- LeGeros, R.Z. Calcium phosphates in oral biology and medecine. Monogr. Oral Sci. 1991, 15, 1–201. [Google Scholar] [PubMed]
- Aoba, T. Recent observations on enamel crystal formation during mammalian amelogenesis. Anat. Rec. 1996, 245, 208–218. [Google Scholar] [CrossRef]
- Fleet, M.E.; Liu, X. Coupled substitution of type A and B carbonate in sodium-bearing apatite. Biomaterials 2007, 28, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Combes, C.; Drouet, C.; Grossin, D. Bioactive ceramics: Physical chemistry. In Comprehensive Biomaterials; Ducheyne, P., Healy, K.E., Hutmacher, D.W., Grainger, D.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 1, pp. 187–221. [Google Scholar]
- Legros, R.; Balmain, N.; Bonel, G. Age-related changes in mineral of rat and bovine cortical bone. Calcif. Tissue Int. 1987, 41, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Glimcher, M.J.; Rey, C.; Ackerman, J.L. A unique protonated phosphate group in bone mineral not present in synthetic calcium phosphates. Identification by phosphorus-31 solid state NMR spectroscopy. J. Mol. Biol. 1994, 244, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Shimizu, M.; Collins, B.; Glimcher, M.J. Resolution-enhanced Fourier transform infrared spectroscopy study of the environment of phosphate ions in the early deposits of a solid phase of calcium phosphate in bone and enamel, and their evolution with age. I: Investigations in the ν4 PO4 domain. Calcif. Tissue Int. 1990, 46, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Winand, L. Etude physico-chimique du phosphate tricalcique hydraté et de l’hydroxyapatite. Ann. Chim. 1961, 6, 951–967. (In French) [Google Scholar]
- Montel, G.; Bonel, G.; Heughebaert, J.-C.; Trombe, J.-C.; Rey, C. New concepts in the composition, crystallization and growth of the mineral components of calcified tissues. J. Cryst. Growth 1981, 53, 74–79. [Google Scholar] [CrossRef]
- Rey, C.; Combes, C.; Drouet, C.; Sfihi, H.; Barroug, A. Physico-chemical properties of nanocrystalline apatites: Implications for biominerals and biomaterials. Mater. Sci. Eng. C 2007, 27, 198–205. [Google Scholar] [CrossRef]
- Rey, C.; Renugopalakrishnan, V.; Shimizu, M.; Collins, B.; Glimcher, M.J. A resolution-enhanced Fourier Transform Infrared spectroscopic study of the environment of the CO32− ion in the mineral phase of enamel during its formation and maturation. Calcif. Tissue Int. 1991, 49, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kourkoumelis, N.; Tzaphlidou, M. Spectroscopic assessment of normal cortical bone: Differences in relation to bone site and sex. Sci. World J. 2010, 10, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Jäger, C.; Welzel, T.; Meyer-Zaika, W.; Epple, M. A solid-state NMR investigation of the structure of nanocrystalline hydroxyapatite. Magn. Reson. Chem. 2006, 44, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Hina, A.; Tofighi, A.; Glimcher, M.J. Maturation of poorly crystalline apatites: Chemical and structural aspects in vivo and in vitro. Cells Mater. 1995, 5, 345–356. [Google Scholar]
- Nassif, N.; Martineau, F.; Syzgantseva, O.; Gobeaux, F.; Willinger, M.; Coradin, T.; Cassaignon, S.; Azaïs, T.; Giraud-Guille, M.-M. In vivo inspired conditions to synthesize biomimetic hydroxyapatite. Chem. Mater. 2010, 22, 3653–3663. [Google Scholar] [CrossRef]
- Cazalbou, S.; Combes, C.; Eichert, D.; Rey, C.; Glimcher, M.J. Poorly crystalline apatites: Evolution and maturation in vitro and in vivo. J. Bone Miner. Metab. 2004, 22, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Eichert, D.; Sfihi, H.; Combes, C.; Rey, C. Specific characteristics of wet nanocrystalline apatites. consequences on biomaterials and bone tissue. Key Eng. Mater. 2004, 254–256, 927–930. [Google Scholar] [CrossRef]
- Eichert, D.; Combes, C.; Drouet, C.; Rey, C. Formation and evolution of hydrated surface layers of apatites. Key Eng. Mater. 2005, 284–286, 3–6. [Google Scholar] [CrossRef]
- Cazalbou, S. Echanges Cationiques Impliquant des Apatites Nanocristallines Analogues au Minéral Osseux. Ph.D. Thesis, Institut National Polytechnique de Toulouse, Toulouse, France, 2000. [Google Scholar]
- Cazalbou, S.; Eichert, D.; Ranz, X.; Drouet, C.; Combes, C.; Harmand, M.F.; Rey, C. Ion exchanges in apatites for biomedical application. J. Mater. Sci. Mater. Med. 2005, 16, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Combes, C.; Drouet, C.; Lebugle, A.; Sfihi, H.; Barroug, A. Nanocrystalline apatites in biological systems: Characterisation, structure and properties. Materialwissenschaft und Werkstofftechnik 2007, 38, 996–1002. [Google Scholar] [CrossRef]
- Drouet, C.; Carayon, M.; Combes, C.; Rey, C. Surface enrichment of biomimetic apatites with biologically-active ions Mg2+ and Sr2+: A preamble to the activation of bone repair materials. Mater. Sci. Eng. C 2008, 28, 1544–1550. [Google Scholar] [CrossRef]
- Errassifi, F.; Menbaoui, A.; Autefage, H.; Benaziz, L.; Ouizat, S.; Santran, V.; Sarda, S.; Lebugle, A.; Combes, C.; Barroug, A.; et al. Adsorption onto nanocrystalline apatitic calcium phosphates. Applications to growth factors and drugs delivery. In Advances in Bioceramics and Biotechnologies; Narayan, R., McKittrick, J., Singh, M., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ouizat, S.; Barroug, A.; Legrouri, A.; Rey, C. Adsorption of bovine serum albumin on poorly crystalline apatite: Influence of maturation. Mater. Res. Bull. 2000, 34, 2279–2289. [Google Scholar] [CrossRef]
- Rey, C.; Combes, C. Physical chemistry of biological apatites. In Biomineralization and Biomaterials, Fundamentals and Applications; Aparicio, C., Pau Ginebra, M., Eds.; Woodhead publishing: Swanston, UK, 2015; pp. 95–128. [Google Scholar]
- Brown, W.E. Crystal structure of octacalcium phosphate. Nature 1962, 196, 1048–1050. [Google Scholar] [CrossRef]
- Eichert, D.; Drouet, C.; Sfihi, H.; Rey, C.; Combes, C. Nanocrystalline Apatite Based Biomaterials: Synthesis, Processing and Characterization; Eichert, D., Drouet, C., Sfihi, H., Rey, C., Combes, C., Eds.; Nova Science Publishers Inc.: New York, NY, USA, 2009. [Google Scholar]
- Rey, C.; Combes, C.; Drouet, C.; Cazalbou, S.; Grossin, D.; Brouillet, F.; Sarda, S. Surface properties of biomimetic nanocrystalline apatites; applications in biomaterials. Prog. Cryst. Growth Charact. Mater. 2014, 60, 63–73. [Google Scholar] [CrossRef]
- Driessens, F.C.M.; Verbeeck, R.M.H. Biominerals; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Iyengar, G.V.; Tandon, L. Minor and Trace Elements in Human Bones and Teeth; International Atomic Energy Agency: Vienna, Austria, 1999. [Google Scholar]
- Teruel, J.D.D.; Alcolea, A.; Hernández, A.; Ruiz, A.J.O. Comparison of chemical composition of enamel and dentine in human, bovine, porcine and ovine teeth. Arch. Oral Biol. 2015, 60, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.A.B.; Adachi, E.M.; Saiki, M. INAA of enamel and dentine samples of a group of children and adults: A comparative study. J. Radioanal. Nucl. Chem. 2007, 276, 49–52. [Google Scholar] [CrossRef]
- Zenóbio, M.A.F.; Tavares, M.S.N.; Zenóbio, E.G.; Silva, T.A. Elemental composition of dental biologic tissues: Study by means of different analytical techniques. J. Radioanal. Nucl. Chem. 2011, 289, 161–166. [Google Scholar] [CrossRef]
- Ishiguro, K.; Nakagaki, H.; Takeuchi, K.; Mukai, M.; Yoshika, I.; Miyauchi, K.; Robinson, C.; Weatherell, J.A. Distribution of fluoride in the dental tissues and their supporting mandibular bone from the same individual. Arch. Oral Biol. 1994, 39, 535–537. [Google Scholar] [CrossRef]
- Lakomaa, E.L.; Rytömaa, I. Mineral composition of enamel and dentin of primary and permanent teeth in Finland. Scand. J. Dent. Res. 1977, 85, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Saiki, M.; Adachi, L.K.; Adachi, E.M. Elemental comparison in sound and carious human teeth by instrumental neutron activation analysis. Eur. Radiol. 2014, 24, 29–32. [Google Scholar] [CrossRef]
- Hendricks, S.B.; Hill, W.L. The nature of bone and phosphate rocks. Proc. Natl. Acad. Sci. USA 1950, 36, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Glimcher, M.J. Three-dimensional spatial relationship between the collagen fibrils and the inorganic calcium phosphate crystals of pickerel (Americanus americanus) and herring (Clupea harengus) bone. J. Mol. Biol. 1991, 217, 487–501. [Google Scholar] [CrossRef]
- Bala, Y.; Farlay, D.; Boivin, G. Bone mineralization: From tissue to crystal in normal and pathological contexts. Osteoporos. Int. 2013, 24, 2153–2166. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Abrams, S.A.; David-Street, J.E.; Heer, M.; O’Brien, K.O.; Wastney, M.E.; Zwart, S.R. Fifty years of human space travel: Implications for bone and calcium research. Annu. Rev. Nutr. 2014, 34, 377–400. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, L.; Traub, W.; de Buffrenil, V.; Allizard, F.; Arad, T.; Weiner, S. Rostrum of a toothed whale: Ultrastructural study of a very dense bone. Bone 1998, 23, 241–247. [Google Scholar] [CrossRef]
- Rey, C.; Collins, B.; Goehl, T.; Dickson, I.R.; Glimcher, M.J. The carbonate environment in bone mineral: A resolution-enhanced fourier transform infrared spectroscopy study. Calcif. Tissue Int. 1989, 45, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.C.; Holcom, D.W.; Young, R.A. Infrared determination of the degree of substitution of hydroxyl by carbonate ion in human enamel. Calcif. Tissue Int. 1985, 37, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Penel, G.; Leroy, G.; Rey, C.; Bres, E. Micro-raman spectral study of the PO4 and CO3 vibrational modes in synthetic and biological apatites. Calcif. Tissue Int. 1998, 63, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Beshah, K.; Rey, C.; Glimcher, M.J.; Shimizu, M.; Griffin, R.G. Solid state carbon-13 and proton NMR studies of carbonate-containing calcium phosphates and enamel. J. Solid State Chem. 1990, 84, 71–81. [Google Scholar] [CrossRef]
- Combes, C.; Rey, C.; Mounic, S. Identification and evaluation of HPO4 ions in biomimetic poorly crystalline apatites and bone mineral. Key Eng. Mater. 2001, 192–195, 143–146. [Google Scholar] [CrossRef]
- Bohic, S.; Rey, C.; Legrand, A.; Sfihi, H.; Rohanizadeh, R.; Martel, C.; Barbier, A.; Daculsi, G. Characterization of the trabecular rat bone mineral: Effect of ovariectomy and bisphosphonate treatment. Bone 2000, 26, 341–348. [Google Scholar] [CrossRef]
- Wang, Y.; Von Euw, S.; Fernandes, F.M.; Cassaignon, S.; Selmane, M.; Laurent, G.; Pehau-Arnaudet, G.; Coelho, C.; Bonhomme-Coury, L.; Giraud-Guille, M.-M.; et al. Water-mediated structuring of bone apatite. Nat. Mater. 2013, 12, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.G.; Parker, S.F.; Mitchell, P.C.H. A study by high energy transfer inelastic neutron scattering spectroscopy of the mineral fraction of ox femur bone. J. Mol. Struct. 2003, 651–653, 123–126. [Google Scholar] [CrossRef]
- Cho, G.; Wu, Y.; Ackerman, J.L. Detection of hydroxyl ions in bone mineral by solid-state NMR spectroscopy. Science 2003, 300, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Pasteris, J.D.; Wopenka, B.; Freeman, J.J.; Rogers, K.; Valsami-Jones, E.; van der Houwen, J.; Silva, M.J. Lack of OH in nanocrystalline apatite as a function of degree of atomic order: Implications for bone and biomaterials. Biomaterials 2004, 25, 229–238. [Google Scholar] [CrossRef]
- Rey, C.; Miquel, J.L.; Facchini, L.; Legrand, A.P.; Glimcher, M.J. Hydroxyl groups in bone mineral. Bone 1995, 16, 583–586. [Google Scholar] [CrossRef]
- Grossin, D.; Rollin-Martinet, S.; Estournès, C.; Rossignol, F.; Champion, E.; Combes, C.; Rey, C.; Geoffroy, C.; Drouet, C. Biomimetic apatite sintered at very low temperature by spark plasma sintering: Physico-chemistry and microstructure aspects. Acta Biomater. 2010, 6, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Renugopalakrishnan, V.; Collins, B.; Glimcher, M.J. Fourier transform infrared spectroscopic study of the carbonate ions in bone mineral during aging. Calcif. Tissue Int. 1991, 49, 251–258. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.; Trautz, O.R.; LeGeros, J.P.; Klein, E. Carbonate substitution in the apatite structure. Bull. Soc. Chim. Fr. 1968, 1712–1718. [Google Scholar]
- Laurencin, D.; Wong, A.; Chrzanowski, W.; Knowles, J.C.; Qiu, D.; Pickup, D.M.; Newport, R.J.; Gan, Z.; Duer, M.J.; Smith, M.E. Probing the calcium and sodium local environment in bones and teeth using multinuclear solid state NMR and X-ray absorption spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Dykes, E.; Elliott, J.C. The occurrence of chloride ions in the apatite lattice of Holly Springs hydroxyapatite and dental enamel. Calcif. Tissue Res. 1971, 7, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Burnell, J.M.; Teubner, E.J.; Miller, A.G. Normal maturational changes in bone matrix, mineral, and crystal size in the rat. Calcif. Tissue Int. 1980, 31, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Landis, W.J.; Lee, D.D.; Brenna, J.T.; Chandra, S.; Morrison, G.H. Detection and localization of silicon and associated elements in vertebrate bone tissue by imaging ion microscopy. Calcif. Tissue Int. 1986, 38, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Code, R.F.; Gelman, R.L.; Armstrong, R.S.; Hallsworth, C.; Lemaire, C.; Cheng, P.T. Field dependence of 19F NMR in rat bone powders. Phys. Med. Biol. 1990, 35, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Grynpas, M.; Rey, C. The effect of fluoride treatment on bone mineral crystals in the rat. Bone 1992, 13, 423–429. [Google Scholar] [CrossRef]
- Boivin, G.; Meunier, P.J. Fluoride and bone: Toxic effects and therapeutic role. In Therapeutic Uses of Trace Elements; Nève, J., Chappuis, P., Lamand, M., Eds.; Springer: Berlin, Germany; Heidelberg, Germany, 1996; pp. 283–295. [Google Scholar]
- Nakagaki, H.; Koyama, Y.; Sakakibara, Y.; Weatherell, J.A.; Robinson, C. Distribution of fluoride across human dental enamel, dentine and cementum. Arch. Oral Biol. 1987, 32, 651–654. [Google Scholar] [CrossRef]
- Cazalbou, S.; Hina, A.; Rey, C. Interactions between trace elements and bone mineral matrix. In New Aspects of Trace Element Research; Abdulla, M., Bost, M., Gamon, S., Arnaud, P., Chazot, G., Eds.; Smith-Gordon: London, UK, 1999; pp. 58–62. [Google Scholar]
- Glimcher, M.J. Bone: Nature of the calcium phosphate crystals and cellular, structural, and physical chemical mechanisms in their formation. Rev. Mineral. Geochem. 2006, 64, 223–282. [Google Scholar] [CrossRef]
- Landis, W.J.; Song, M.J.; Leith, A.; McEwen, L.; McEwen, B. Mineral and organic matrix interaction in normally calcifying tendon visualized in 3 dimensions by high-voltage electron-microscopic tomography and graphic image-reconstruction. J. Struct. Biol. 1993, 110, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Nikel, O.; Laurencin, D.; Bonhomme, C.; Sroga, G.E.; Besdo, S.; Lorenz, A.; Vashishth, D. Solid state NMR investigation of intact human bone quality: Balancing issues and insight into the structure at the organic-mineral interface. J. Phys. Chem. C Nanomater. Interfaces 2012, 116, 6320–6331. [Google Scholar] [CrossRef] [PubMed]
- Skwarek, E.; Janusz, W.; Sternik, D. Adsorption of citrate ions on hydroxyapatite synthetized by various methods. J. Radioanal. Nucl. Chem. 2013, 299, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.; Müller, K.H.; Wong, W.C.; Pickard, C.J.; Reid, D.G.; Skepper, J.N.; Duer, M.J. Citrate bridges between mineral platelets in bone. Proc. Natl. Acad. Sci. USA 2014, 111, E1354–E1363. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Combes, C. What bridges mineral platelets of bone? BoneKEy Rep. 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.C.; Jackson, D.C. Lactate uptake by skeletal bone in anoxic turtles, trachemys scripta. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 146, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Landis, W.J. The strength of a calcified tissue depends in part on the molecular structure and organization of its constituent mineral crystals in their organic matrix. Bone 1995, 16, 533–544. [Google Scholar] [CrossRef]
- Morris, D.C.; Vaananen, H.K.; Anderson, H.C. Matrix vesicle calcification in rat epiphyseal growth palte cartilage prepared anhydrously for electron microscopy. Metab. Bone Dis. Relat. Res. 1983, 5, 131–137. [Google Scholar] [CrossRef]
- Kim, H.-M.; Rey, C.; Glimcher, M.J. X-ray diffraction, electron microscopy, and fourier transform infrared spectroscopy of apatite crystals isolated from chicken and bovine cartilage. Calcif. Tissue Int. 1996, 59, 58–63. [Google Scholar]
- McNally, E.A.; Schwarcz, H.P.; Botton, G.A.; Arsenault, L.A. A model for the ultrastructure of bone based on electron microscopy of ion-milled sections. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Nylen, M.U.; Eanes, E.D.; Omnell, K.A. Crystal growth in rat enamel. J. Cell Biol. 1963, 18, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Frazier, P.D. Adult human enamel—An SEM study of crystallite size and morphology. J. Ultrastruct. Res. 1968, 22, 1–11. [Google Scholar] [CrossRef]
- Santo, A.R.E.; Line, S.R.P. The enamel organic matrix: Structure and function. Braz. J. Oral Sci. 2005, 4, 716–724. [Google Scholar]
- King, J.C.; Shames, D.M.; Woodhouse, L.R. Zinc and health: Current status and future directions zinc homeostasis in humans 1. J. Nutr. 2000, 130, 1360S–1366S. [Google Scholar] [PubMed]
- Burger, C.; Zhou, H.W.; Wang, H.; Sics, I.; Hsiao, B.S.; Chu, B.; Graham, L.; Glimcher, M.J. Lateral packing of mineral crystals in bone collagen fibrils. Biophys. J. 2008, 95, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Kim, H.M.; Gerstenfeld, L.; Glimcher, M.J. Characterization of the apatite crystals of bone and their maturation in osteoclast cell culture: Comparison with native bone crystals. Connect. Tissue Res. 1996, 35, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Hodge, A.J. Molecular models illustrating the possible distribution of holes in simple systematically staggered arrays of type I collagen molecules in native-type fibrils. Connect. Tissue Res. 1989, 21, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, J.; Landis, W.J. A contribution with review to the description of mineralization of bone and other calcified tissues in vivo. Anat. Rec. 1991, 230, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Eidelman, N.; Chow, L.C.; Brown, W.E. Calcium phosphate saturation levels in ultrafiltered serum. Calcif. Tissue Int. 1987, 40, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Combes, C.; Rey, C. Adsorption of proteins and calcium phosphate materials bioactivity. Biomaterials 2002, 23, 2817–2823. [Google Scholar] [CrossRef]
- Combes, C.; Rey, C.; Frèche, M. In vitro crystallization of octacalcium phosphate on type I collagen: Influence of serum albumin. J Mater. Sci. Mater. Med. 1999, 10, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y.-Y.; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone structure and formation: A new perspective. Mater. Sci. Eng. R Rep. 2007, 58, 77–116. [Google Scholar] [CrossRef]
- Veis, A.; Dorvee, J.R. Biomineralization mechanisms: A new paradigm for crystal nucleation in organic matrices. Calcif. Tissue Int. 2013, 93, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, F.C.; Cölfen, H. Controlling mineral morphologies and structures in biological and synthetic systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef] [PubMed]
- Gower, L.B. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar] [CrossRef] [PubMed]
- Colfen, H.; Antonietti, M. Mesocrystals and Non Classical Crystallization; John Wiley & Sons Ltd.: Chichester, UK, 2008. [Google Scholar]
- Drouet, C. A comprehensive guide to experimental and predicted thermodynamic properties of phosphate apatite minerals in view of applicative purposes. J. Chem. Thermodyn. 2015, 81, 143–159. [Google Scholar] [CrossRef]
- Rollin-Martinet, S.; Navrotsky, A.; Champion, E.; Grossin, D.; Drouet, C. Thermodynamic basis for evolution of apatite in calcified tissues. Am. Mineral. 2013, 98, 2037–2045. [Google Scholar] [CrossRef]
- Weiner, S. Transient precursor strategy in mineral formation of bone. Bone 2006, 39, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, F.; Pieterse, K.; George, A.; Bomans, P.H.H.; Friedrich, H.; Brylka, L.J.; Hilbers, P.A.J.; de With, G.; Sommerdijk, N.A.J.M. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat. Mater. 2010, 9, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Bodier-Houllé, P.; Steuer, P.; Meyer, J.M.; Bigeard, L.; Cuisinier, F.J. High-resolution electron-microscopic study of the relationship between human enamel and dentin crystals at the dentinoenamel junction. Cell Tissue Res. 2000, 301, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Moradian-oldak, J. Control of apatite crystal growth in a fluoride containing amelogenin-rich matrix. Biomaterials 2005, 26, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Hanaizumi, Y.; Ohshima, H. Occurrence of amorphous and crystalline mineral deposits at the epithelial-mesenchymal interface of incisors in the calcium-loaded rat: Implication of novel calcium binding domains. Anat. Rec. 1996, 245, 174–185. [Google Scholar] [CrossRef]
- Beniash, E.; Metzler, R.A.; Lam, R.S.K.; Gilbert, P.U.P.A. Transient amorphous calcium phosphate in forming enamel. J. Struct. Biol. 2009, 166, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Spevak, L.; Flach, C.R.; Hunter, T.; Mendelsohn, R.; Boskey, A. Fourier transform infrared spectroscopic imaging parameters describing acid phosphate substitution in biologic hydroxyapatite. Calcif. Tissue Int. 2013, 92, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Bonar, L.C.; Roufosse, A.H.; Sabine, W.K.; Grynpas, M.D.; Glimcher, M.J. X-ray diffraction studies of the crystallinity of bone mineral in newly synthesized and density fractionated bone. Calcif. Tissue Int. 1983, 35, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Heaney, R.P. How does bone support calcium homeostasis? Bone 2003, 33, 264–268. [Google Scholar] [CrossRef]
- Baig, A.A.; Fox, J.L.; Wang, Z.; Higuchi, W.I.; Miller, S.C.; Barry, A.M.; Otsuka, M. Metastable equilibrium solubility behavior of bone mineral. Calcif. Tissue Int. 1999, 64, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Eichert, D. Etude de la Réactivité de Surface D’apatites de Synthèse Nanocristallines. Ph.D’s Thesis, Institut National Polytechnique de Toulouse, Toulouse, France, 2001. [Google Scholar]
- Bala, Y.; Farlay, D.; Delmas, P.D.; Meunier, P.J.; Boivin, G. Time sequence of secondary mineralization and microhardness in cortical and cancellous bone from ewes. Bone 2010, 46, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Busa, B.; Miller, L.M.; Rubin, C.T.; Qin, Y.-X.; Judex, S. Rapid establishment of chemical and mechanical properties during lamellar bone formation. Calcif. Tissue Int. 2005, 77, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Meunier, P.J. The degree of mineralization of bone tissue measured by computerized quantitative contact microradiography. Calcif. Tissue Int. 2002, 70, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Currey, J.D. The design of mineralised hard tissues for their mechanical functions. J. Exp. Biol. 1999, 202, 3285–3294. [Google Scholar] [PubMed]
- Moreno, E.; Kresak, M.; Zahradnik, R.T. Fluoridated hydroxyapatite solubility and caries formation. Nature 1974, 247, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Hina, A. Etude de la Réactivité en Milieu Aqueux, D’apatites Phosphocalciques D’intérêt Biologique. Ph.D. Thesis, Institut National Poytechnique de Toulouse, Toulouse, France, 1996. [Google Scholar]
- Talmage, R.V.; Talmage, D.W. Calcium homeostasis: Solving the solubility problem. J. Musculoskelet. Neuronal Interact. 2006, 6, 402–407. [Google Scholar] [PubMed]
- Shashi, A.; Singla, S. Parathyroid function in osteofluorosis. World J. Med. Sci. 2013, 8, 67–73. [Google Scholar]
- Zhu, Y.; Zhu, Z.; Zhao, X.; Liang, Y.; Huang, Y. Characterization, dissolution and solubility of lead hydroxypyromorphite [Pb5(PO4)3OH] at 25–45 °C. J. Chem. 2015, 2015. [Google Scholar] [CrossRef]
- Christoffersen, J.; Christoffersen, M.R.; Kolthoff, N.; Bärenholdt, O. Effects of strontium ions on growth and dissolution of hydroxyapatite and on bone mineral detection. Bone 1997, 20, 47–54. [Google Scholar] [CrossRef]
- Farlay, D.; Boivin, G.; Panczer, G.; Lalande, A.; Meunier, P.J. Long-term strontium ranelate administration in monkeys preserves characteristics of bone mineral crystals and degree of mineralization of bone. J. Bone Miner. Res. 2005, 20, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Leggett, R.W. An age-specific kinetic model of lead metabolism in humans. Eviron. Heal. Perspect. 1993, 101, 598–616. [Google Scholar] [CrossRef]
- Flaherty, E.J.O. A physiologically based kinetic model for lead in children and adults. Environ. Health Perspect. 1998, 106, 1495–1503. [Google Scholar] [CrossRef]
- Simmer, J.P.; Hu, J.C.-C. Dental enamel formation and its impact on clinical dentistry. J. Dent. Educ. 2001, 65, 896–905. [Google Scholar] [PubMed]
- Smith, C.E. Cellular and chemical events during enamel maturation. Crit. Rev. Oral Biol. Med. 1998, 9, 128–161. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.; Kirkham, J.; Brookes, S.J.; Bonass, W.A.; Shore, R.C. The chemistry of enamel development. Int. J. Dev. Biol. 1995, 39, 145–152. [Google Scholar] [PubMed]
- Lynch, R.J.M. The primary and mixed dentition, post-eruptive enamel maturation and dental caries: A review. Int. Dent. J. 2013, 63, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Arends, J. Mechanism of enamel dissolution and its prevention. J. Biol. Buccale 1977, 5, 219–237. [Google Scholar] [PubMed]
- Kuhn, L.T.; Grynpas, M.D.; Rey, C.C.; Wu, Y.; Ackerman, J.L.; Glimcher, M.J. A comparison of the physical and chemical differences between cancellous and cortical bovine bone mineral at two ages. Calcif. Tissue Int. 2008, 83, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Jalvo, Y.; Sanchez-Chillon, B.; Andrews, P.; Fernandez-Lopez, S.; Alcala Martinez, I. Morphological taphonomic transformations of fossil bones in continental environments, and repercussions on their chemical composition. Archaeometry 2002, 44, 353–361. [Google Scholar] [CrossRef]
- Kohn, M.J.; Schoeninger, M.; Barker, W.W. Altered states: Effects of diagenesis on fossil tooth chemistry. Geochim. Cosmochim. Acta 1999, 63, 2737–2747. [Google Scholar] [CrossRef]
- Clementz, M.T. New insight from old bones: Stable isotope analysis of fossil mammals. J. Mammal. 2012, 93, 368–380. [Google Scholar] [CrossRef]
- Kohn, M.J.; Law, J.M. Stable isotope chemistry of fossil bone as a new paleoclimate indicator. Geochim. Cosmochim. Acta 2006, 70, 931–946. [Google Scholar] [CrossRef]
- Cazalbou, S.; Eichert, D.; Drouet, C.; Combes, C.; Rey, C. Minéralisations biologiques à base de phosphate de calcium. Comptes Rendus Palevol 2004, 3, 563–572. (In French) [Google Scholar] [CrossRef]
- Grunenwald, A.; Keyser, C.; Sautereau, A.M.; Crubézy, E.; Ludes, B.; Drouet, C. Novel contribution on the diagenetic physicochemical features of bone and teeth minerals, as substrates for ancient DNA typing. Anal. Bioanal. Chem. 2014, 406, 4691–4704. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, L.; Trueman, C.N.; Palmer, M.R. Protracted diagenetic alteration of REE contents in fossil bioapatites: Direct evidence from Lu-Hf isotope systematics. Geochim. Cosmochim. Acta 2010, 74, 6077–6092. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements or Ions | Bone | Dentine | Dental Enamel | References |
|---|---|---|---|---|
| Major elements (wt %) | ||||
| C (total) | 16.7 | 11.8 | 1.4 | bone [41]; dentine and enamel [49] |
| CO32− | 5.6 | 4.6 | 3.2 | bone [53]; dentine and enamel [45] |
| N | 4.9 | 4.0 | 0.32 | dentine and enamel [49] |
| Ca | 25.4 | 26.9 | 36.6 | dentine and enamel [45] |
| P | 11.6 | 13.2 | 17.7 | dentine and enamel [45] |
| Minor elements (wt %) | ||||
| Cl | 0.13 | 0.065 | 0.37 | dentine and enamel [46,47,48,49,51,52] |
| K | 0.0047 | 0.024 | 0.070 | dentine and enamel [46,47,51] |
| Mg | 0.27 | 0.74 | 0.29 | dentine and enamel [46,47,48,51,52] |
| Na | 0.53 | 0.76 | 0.77 | dentine and enamel [47,48,49,51,52] |
| S | 0.08 | 0.070 | 0.021 | dentine and enamel [47] |
| Main trace elements (ppm) | ||||
| Al | 29 | 210 | 55 | dentine and enamel [46,49,51] |
| B | 22 | - | 11 | enamel [45] |
| F | 400 | 215 | 50 | all values from Ishiguro et al. [50] |
| Fe | 76 | 44 | 34 | dentine and enamel [46,47,51,52] |
| Pb | 4.4 | 15 | 17 | all values computed from [46] |
| Sr | 70 | 145 | 173 | dentine and enamel [46,47,48,49,52] |
| Zn | 205 | 148 | 170 | dentine and enamel [46,47,48,49,51,52] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Combes, C.; Cazalbou, S.; Rey, C. Apatite Biominerals. Minerals 2016, 6, 34. https://doi.org/10.3390/min6020034
Combes C, Cazalbou S, Rey C. Apatite Biominerals. Minerals. 2016; 6(2):34. https://doi.org/10.3390/min6020034
Chicago/Turabian StyleCombes, Christèle, Sophie Cazalbou, and Christian Rey. 2016. "Apatite Biominerals" Minerals 6, no. 2: 34. https://doi.org/10.3390/min6020034
APA StyleCombes, C., Cazalbou, S., & Rey, C. (2016). Apatite Biominerals. Minerals, 6(2), 34. https://doi.org/10.3390/min6020034