1. Introduction
Selenium is a trace element, but it is widely spread on the Earth’s surface. Although its distribution is very heterogeneous, it has been estimated that, on average, the lithosphere contains about 0.05 ppm of Se. Some extremely polluted soils in USA, Ireland, and India reach 100 mg of selenium per kg, but concentrations over 0.1 mg per kg are uncommon [
1,
2,
3]. In general, high concentration of selenium is associated with mining, ore treatment, or industrial activities [
4]. Selenium is used as a pigment in the glass industry and is also of interest in electronics because of its photo-electronic and semiconducting properties. Moreover, as Se is an essential element for human beings, it is commercialized as a nutritional supplement and is also used as a soil fertilizer and in the cosmetics industry [
5].
In nature, Se is frequently associated with sulfide minerals such as pyrite, chalcopyrite or sphalerite [
5], high sulfur-content carbon [
6], black shales [
7], volcanic ash [
8], and some areas where geothermal activity is high [
9].
Oxidation states +1, +2, +3, and +5 of Se are not observed in nature [
10]. The selenide anion (Se
2−) is stable in very reducing ambient, where metal selenides and sulfides occur [
5]. Se(IV) and Se(VI) ions form soluble oxyanions that are frequently found in surface waters under reducing and oxidizing ambient [
11]. Selenate (SeO
42−) is found in more oxidizing ambient, and its capacity for adsorption and precipitation on mineral surfaces is low compared with selenite (SeO
32−), which is also very soluble under less oxidizing conditions [
12], and whose mobility is influenced by pH-dependent sorption/desorption phenomena [
13].
Selenium is an essential bio-element that is involved in different biological processes. [
14]. However, a thin line separates beneficial from toxic levels of Se, which makes it a pollutant [
15]. In nature, sorption processes involving soil mineral components play a critical role in selenium bioavailability. Different studies have focused on the sorption of selenium onto iron and aluminum oxides and hydroxides [
16,
17]. Calcium carbonate minerals have fewer sorption sites compared with iron and aluminum oxide minerals, but they are still important sorbents in calcareous systems [
18]. Along with specific adsorption, Se co-precipitation with carbonate minerals can occur [
19]. In fact, under supersaturated conditions, Se co-precipitation with calcite is the dominant sequestration mechanism. Experimental studies have shown that co-precipitation with calcite occurs through a series of adsorption and entrapment events [
20].
Taking this into account, the mobility of dissolved selenium on the Earth’s surface is of great environmental concern. The study of this mobility requires knowledge of the different species of selenium at different pH and redox conditions and therefore their interaction with the different mineral species present on the Earth’s crust. In carbonate media, it is important to know the influence of carbonate species CO32−, HCO3−, H2CO30, and cations such as Ca2+ and Mg2+, as possible factors affecting selenium mobility.
Calcite is a ubiquitous mineral in the Earth’s crust. Several studies have shown its potential for the immobilization of contaminants such as, Sr, Mn, and Cd present in aqueous solutions [
21,
22,
23,
24]. Several experimental studies have investigated the interaction of aqueous solutions containing selenium, especially Se(IV) and Se(VI), with calcium carbonate. Many of them show that selenium and sulfur play a similar role in their interaction with carbonates and that they are even competitors in terms of their incorporation into the crystal structure of carbonates. For example, the possibility of incorporating small amounts of selenate into the calcite structure in substitution of carbonate in a similar way to sulphate has been verified [
25,
26], as has the unequal incorporation of sulphate and selenate on the dominant face of calcite [
27]. In previous research [
26,
28], we have verified the importance of the presence of SO
42− and SeO
42− in the precipitation water on CaCO
3 polymorphism since both favor vaterite stabilization.
Although more scarce, there are also studies on the interaction and co-precipitation of CaCO
3 and CaSeO
3. In an experimental study, Aurelio et al. [
12] precipitated calcium carbonate under ambient conditions in the presence of small amounts of selenite (in concentrations between 0.1 and 12 mM). They found that it is possible to incorporate Se(IV) into the calcite structure up to 27.5 mmol per kg and also found that there is some preferential incorporation into the solid with respect to the aqueous phase. In addition, in experiments with higher Se(IV) concentrations, the precipitation of vaterite instead of calcite was observed. Other studies from the same research group [
29] have proved that Se(IV) in the calcite structure substitutes the CO
32− groups and that such substitution produces an increase in the glass crystallographic c-axis. Moreover, under higher pressure and temperature conditions (20 bar and 30 °C), they have also incorporated significant amounts of selenite in calcite and confirmed its structural position in substitution of carbonate [
30]. The dimensions and shape of the SeO
32− anion are considerably different from those of CO
32−; however, substitution is possible, even under ambient conditions, despite the considerable c-elongation of the crystal structure.
However, except for the indication of vaterite precipitation at 12 mM Se(IV) concentrations in the aqueous solution [
12], these studies have not addressed the influence of SeO
42− on the polymorphism of calcium carbonate or on the evolution of calcium carbonates in contact with selenium-containing carbonate waters. This work aims to advance knowledge in this regard, following a protocol similar to that used in previous works [
27], which basically consists of monitoring in the laboratory the aging of precipitated calcium carbonate in the presence of foreign ions in the aqueous solution left over from precipitation.
2. Materials and Methods
2.1. Aging Experiments
Four different sets of experiments of CaCO
3 precipitation from aqueous solutions in the presence of different amounts of Se (IV) were performed. In all of them, 50 mL of a 0.05 M aqueous solution of CaCl
2 was mixed with 50 mL of a second (Na
2CO
3 + Na
2SeO
3) solution in a glass beaker. Immediately after mixing, precipitation took place, the beaker was sealed to avoid evaporation, and the precipitate was maintained under continuous stirring at 400 rpm in the remaining solution for a predefined aging period under ambient conditions. Initial concentration of the parent solutions for each experiment are shown in
Table 1. All the solutions were prepared with ultrapure Milli-Q
® water and analytical grade reactants.
For each initial mixture, a number of nine identical experiments were addressed with the following aging periods: 10 min, 1 h, 6 h, 15 h, 1 day, 4 days, 7 days, 14 days, 30 days.
After aging, the solid was filtered with Millpore® 0.45 μm filters and rinsed twice with ultrapure water to eliminate solution remains. Furthermore, immediately after filtration, the aqueous solution pH was measured.
2.2. Characterization of Precipitates and Modelling of the Aqueous Solutions
The identification and characterization of the phases present in the precipitated solids, after drying at room conditions, were carried out by using different instrumental techniques.
Crystalline phases were identified by powder X-ray diffraction. Moreover, this technique was used in order to determine their structural properties, such as crystallinity and cell parameters. Samples of all the precipitates were pulverized in an agate mortar and analyzed in a Philips X’PertPro diffractometer equipped with programmable divergence slit, direct beam dimmer, secondary monochromator, and X-ray tube with Cu-anode (Kα λ = 1.5406 Å), of the X-ray Diffraction Laboratory of the Scientific and Technical Services of the University of Oviedo (Spain). Silicon was used as external standard to ensure the correction of eventual experimental systematic errors. The powder diagrams, including phase identification and crystallographic determinations, were handled with software programs X’Pert Plus 1.0 by Philips (Amsterdam, The Netherlands) and X’Pert HighScore Plus 2.2.4 by PANalytical (Almelo, The Netherlands).
The morphology of precipitates was observed under a JEOL-6610LV scanning electron microscope (SEM) of the Electron Microscopy Laboratory of the Scientific and Technical Services of the University of Oviedo (Spain). Moreover, the instrument was equipped with a backscattered electron detector and an INCA-Energy 350-Xmax EDX microanalyzer (Oxford Instruments, Oxfordshire, England, UK) that allowed investigation of the chemical composition of the solids and the detection of compositional heterogeneities. After discarding compositional heterogeneities in the region on which the analyses were performed, at least five analyses were performed on each of the morphologies identified in the sample. Samples of all the precipitates were attached to sample holders with a double-faced adhesive carbon tape and sputter-coated with gold in a Balzers SCD 004 instrument of the Electron Microscopy Laboratory of the Scientific and Technical Services of the Uni-versity of Oviedo (Spain). Oxford Instruments INCA 4.15 software (Oxfordshire, England, UK) was used to process data obtained from the characterization by SEM, especially the spectra from which the chemical composition of the different solid phases was determined.
Geochemical modeling of dissolutions was carried out with the Phreeqc 3.0 program [
31], which is available on the USGS website. The sit.dat database, version 9b0, which includes data compiled by Lawrence Livermore National Laboratory (Livermore, CA, USA) and covers the main selenium aqueous species, was used for modelling.
In order to improve knowledge about the behavior of the system over time, with regard to carbonates precipitation, the evolution of the pH of the solutions was monitored. Measurements were taken immediately after filtration by using a BASIC 20 pH meter, which was calibrated at each session. CRISON buffers of pH 4.0, 7.0, and 9.1 were used as standards.
3. Results
3.1. Phases Identification
The analysis by X-ray diffraction allowed the identification of the crystalline phases in the precipitated solids for the different aging times.
Table 2 summarizes the sequence of phases found in the four sets of experiments.
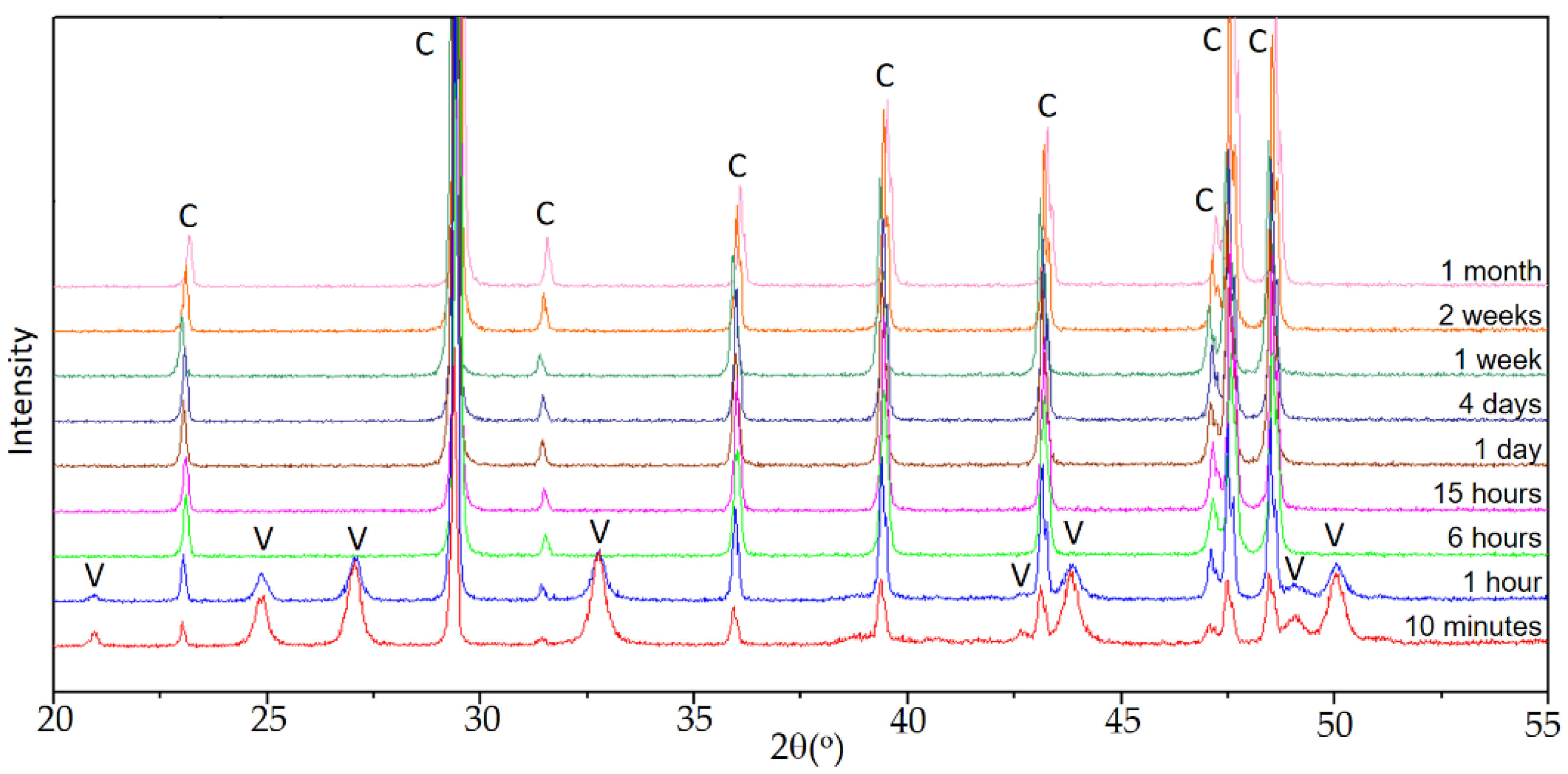
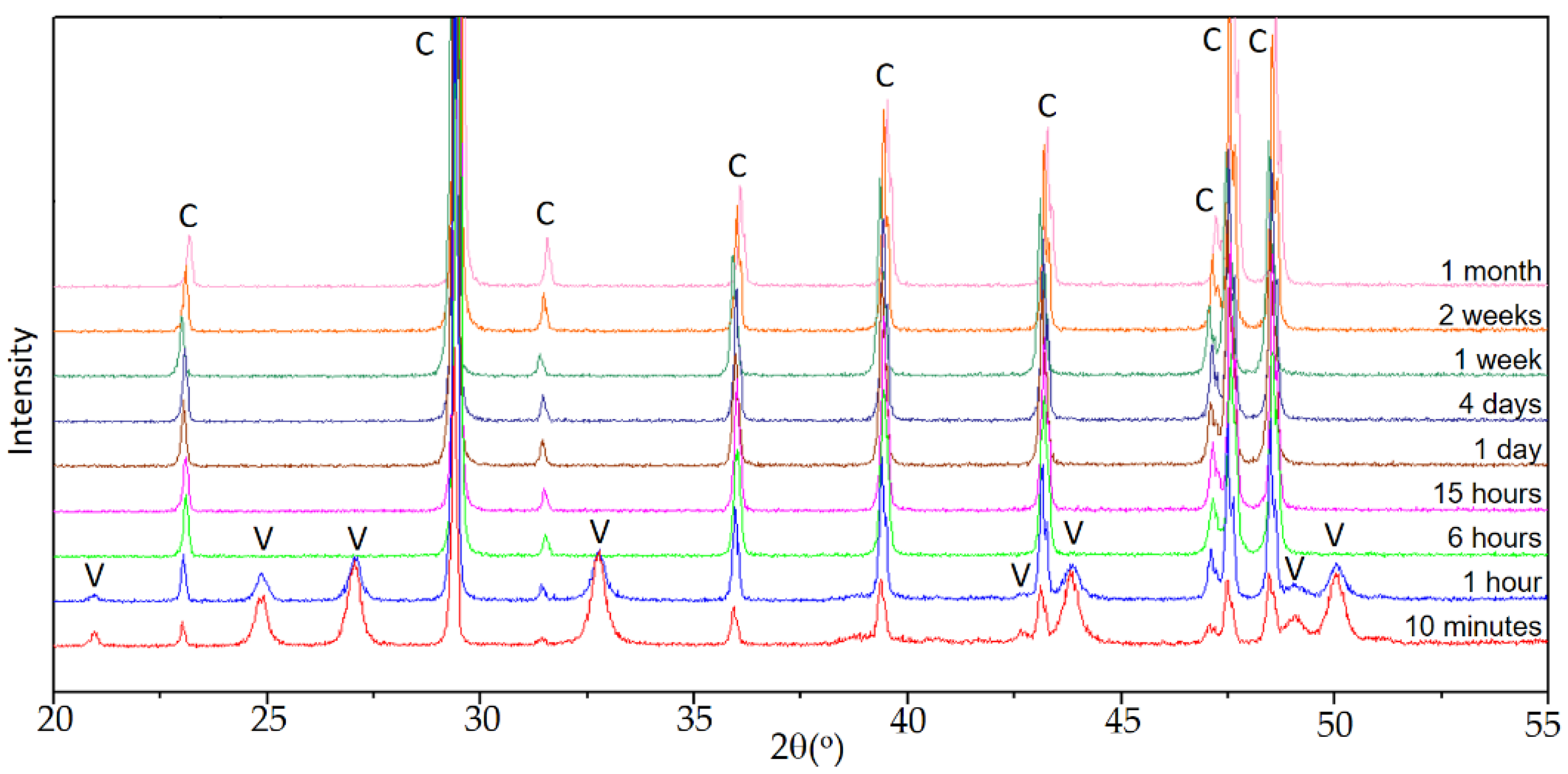
Figure 1 shows the sequence of diffractograms in experiment E0, used as a control.
When calcium carbonate precipitation occurs in the absence of Se(IV), the sequence is the one observed in previous works [
27], carried out under similar conditions. In the early stages of aging, vaterite and calcite were identified, the latter as the majority phase. The intensity of the reflections corresponding to calcite increased with time until, after 6 h, when calcite was the only phase in precipitated solids. As discussed in the previously referenced works, vaterite is the precursor phase of calcite, which is the stable polymorph of calcium carbonate under ambient conditions.
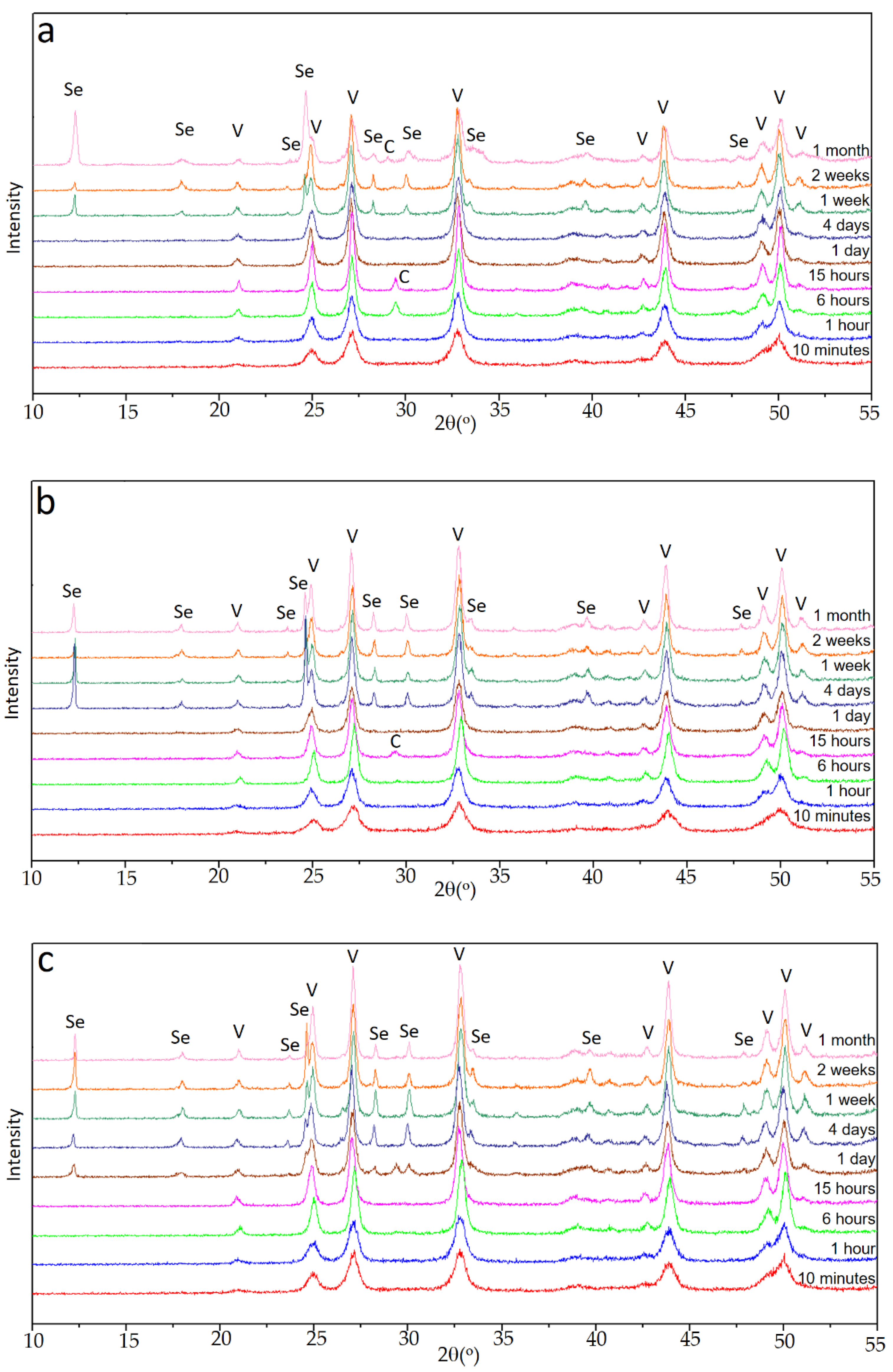
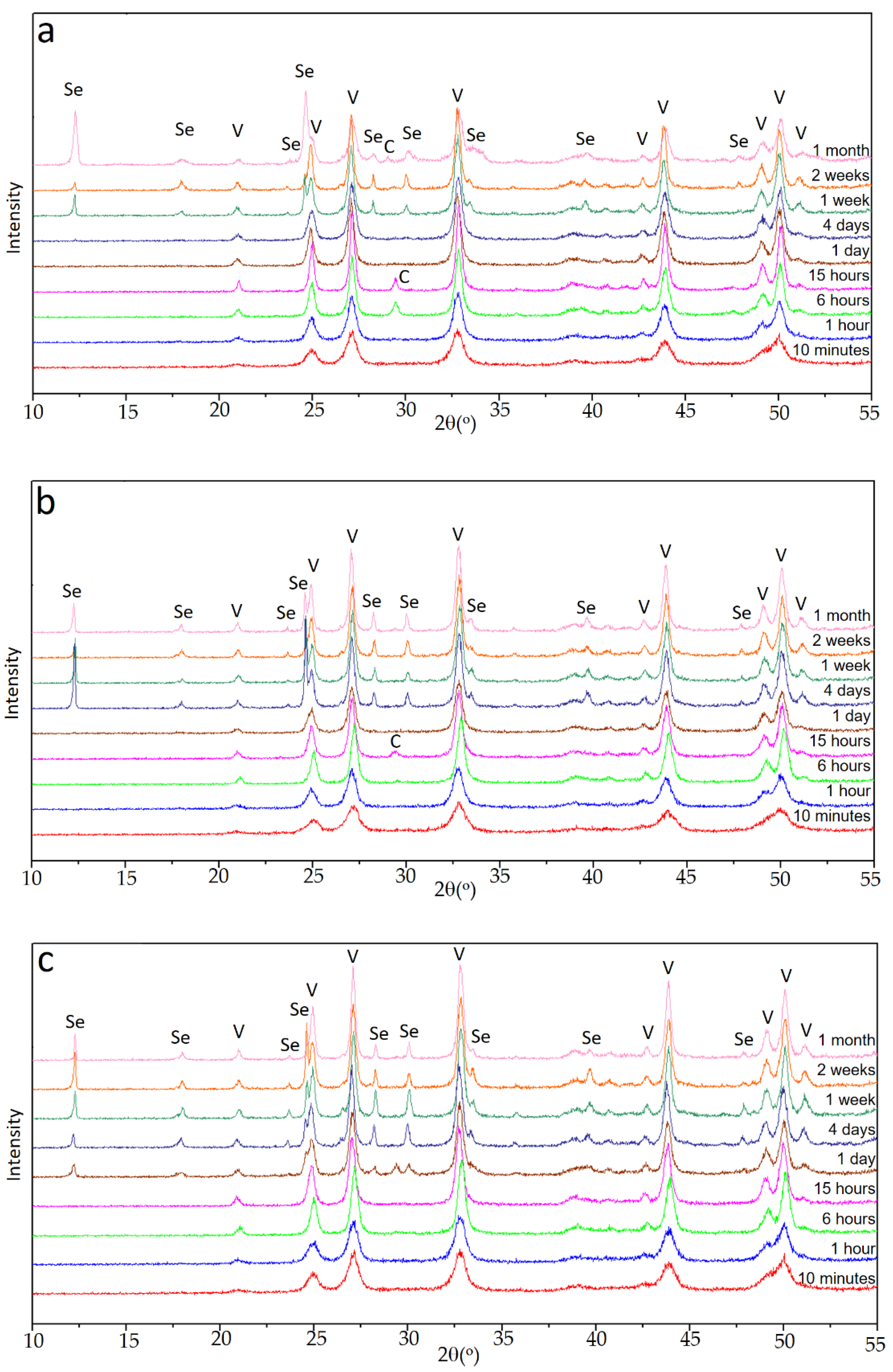
In the case of experiments carried out in the presence of Se(IV), the evolution observed in the X-ray diagrams was more complex. Depending on the initial amount of Se (IV), different sequences of appearance and disappearance of phases in the precipitated solids were observed. The quality of the diagrams allowed a good qualitative tracing of the sequences described below.
Figure 2a shows the sequence in set E1, with an initial Se(IV) content of 0.010 M. As can be seen, for aging up to 1 h, the only crystalline phase present in the solid was vaterite. Between 6 and 15 h, reflections corresponding to calcite were observed together with those of vaterite, but calcite was clearly a minority phase. In the diagrams corresponding to 1 and 4 days, the only crystalline phase was again vaterite. Between 1 and 2 weeks of aging time, the majority phase was still vaterite, but a hydrated selenite CaSeO
3·H
2O was also identified in the solid. Finally, after one month of aging time, vaterite was still the dominant phase, CaSeO
3·H
2O was also present, and peaks corresponding to calcite of low relative intensity were also observed.
As shown in
Figure 2b, the sequence of phases was significantly different in the set of experiment E2, with an initial concentration of Se(IV) 0.015M. As in the previous case, the first phase observed was vaterite, which was identified for aging time between 10 min and 1 h. Between 6 and 15 h, the presence of calcite together with vaterite in the sample was clear, but by far, vaterite was the dominant phase. After that moment, the only calcium polymorph present in the dominant phase was vaterite, although after 4 days it coexisted with CaSeO
3·H
2O.
Finally,
Figure 2c shows the sequence of diffractograms collected for the E3 set of experiments. For aging times below 1 day, the only phase detected was vaterite. From this time on, although vaterite was clearly the majority phase, CaSeO
3·H
2O and a relatively very small amount of calcite precipitated. The latter phase was no longer detected after 4 days of aging. The proportion of CaSeO
3·H
2O in the sample increased slightly up to two weeks, but in the final stages, it decreased again with respect to vaterite.
These sequences of appearance and disappearance of different phases were complex, but clearly pointed to an increase in the stability of vaterite with respect to experiment E0, in the absence of Se(IV). In all the three experiments E1, E2, and E3, and for all aging times, vaterite was the predominant phase; and calcite, when it appeared at intermediate times, was always very scarce and even tended to disappear or to lose relative importance with respect to vaterite or even to CaSeO3·H2O. This latter phase always appeared after a considerable aging time, which was smaller the higher the amount of Se(IV) in the initial solution. Although the relative amount of it with respect to vaterite could become important, it tended to be lower for the longer aging times considered in this work.
3.2. Phases Crystallinity
A detailed study of the X-ray diagrams provided information on the crystallinity of the identified phases. Typically, increasing aging times would result in an improvement in crystallinity that is a consequence of dissolution–recrystallization processes and precipitation of the solid in conditions progressively closer to equilibrium. This increase in crystallinity results in X-ray diagrams showing sharper reflections, i.e., narrower and more intense peaks. However, other factors can affect the full width at half maximum (FWHM) and intensity of the peaks. For example, heterogeneities in the chemical composition of the sample can increase the width of a peak, and in samples with more than one phase, the intensity of the reflections depends on their relative abundance in the sample. Taking this into account, and according to the results discussed below on the chemical composition of the solids, it can be seen that the crystallinity of the calcium carbonates—especially vaterite—obtained in the different experiments varied over the aging time.
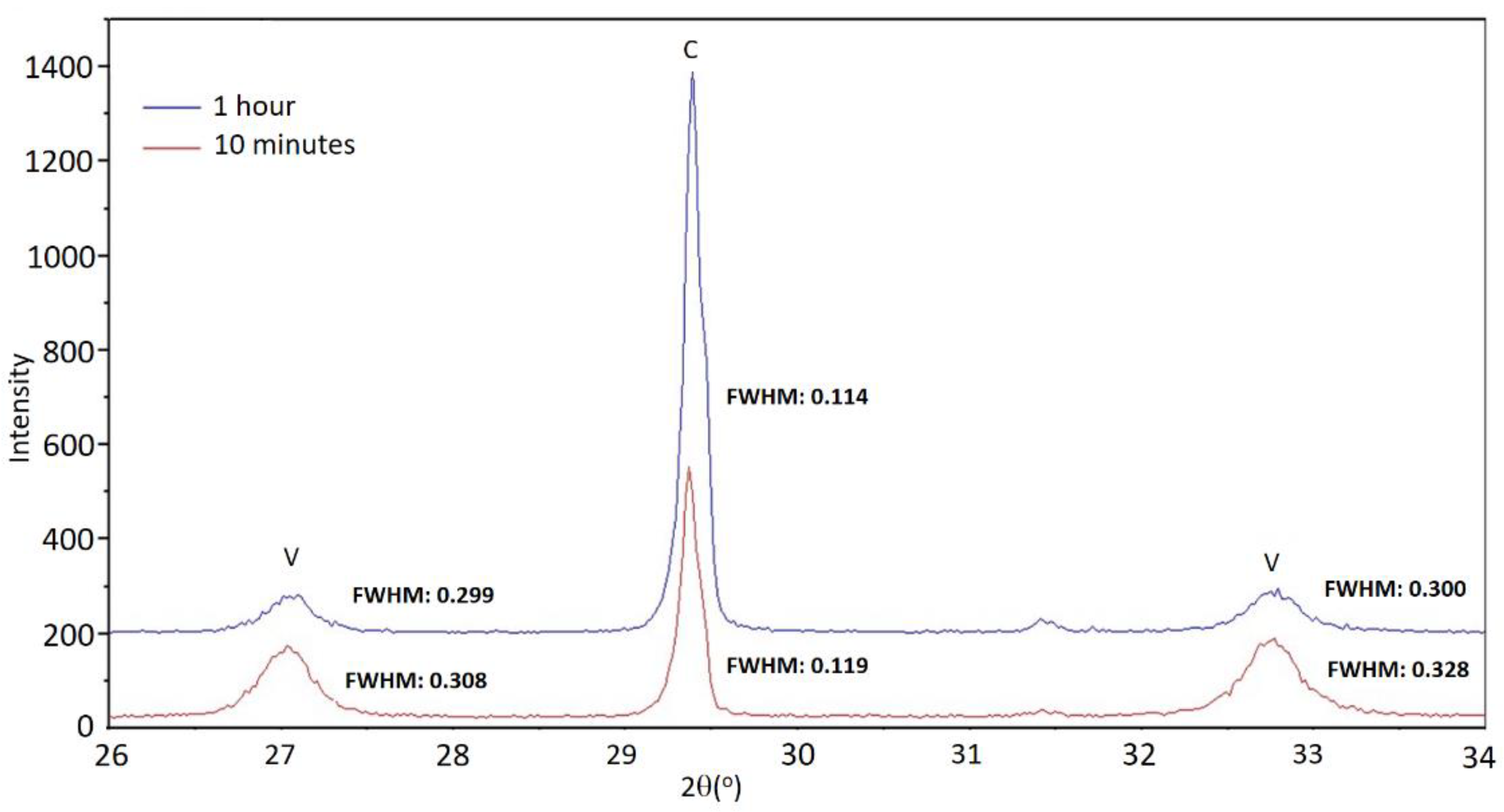
In the diffractograms of the control set E0, shown in
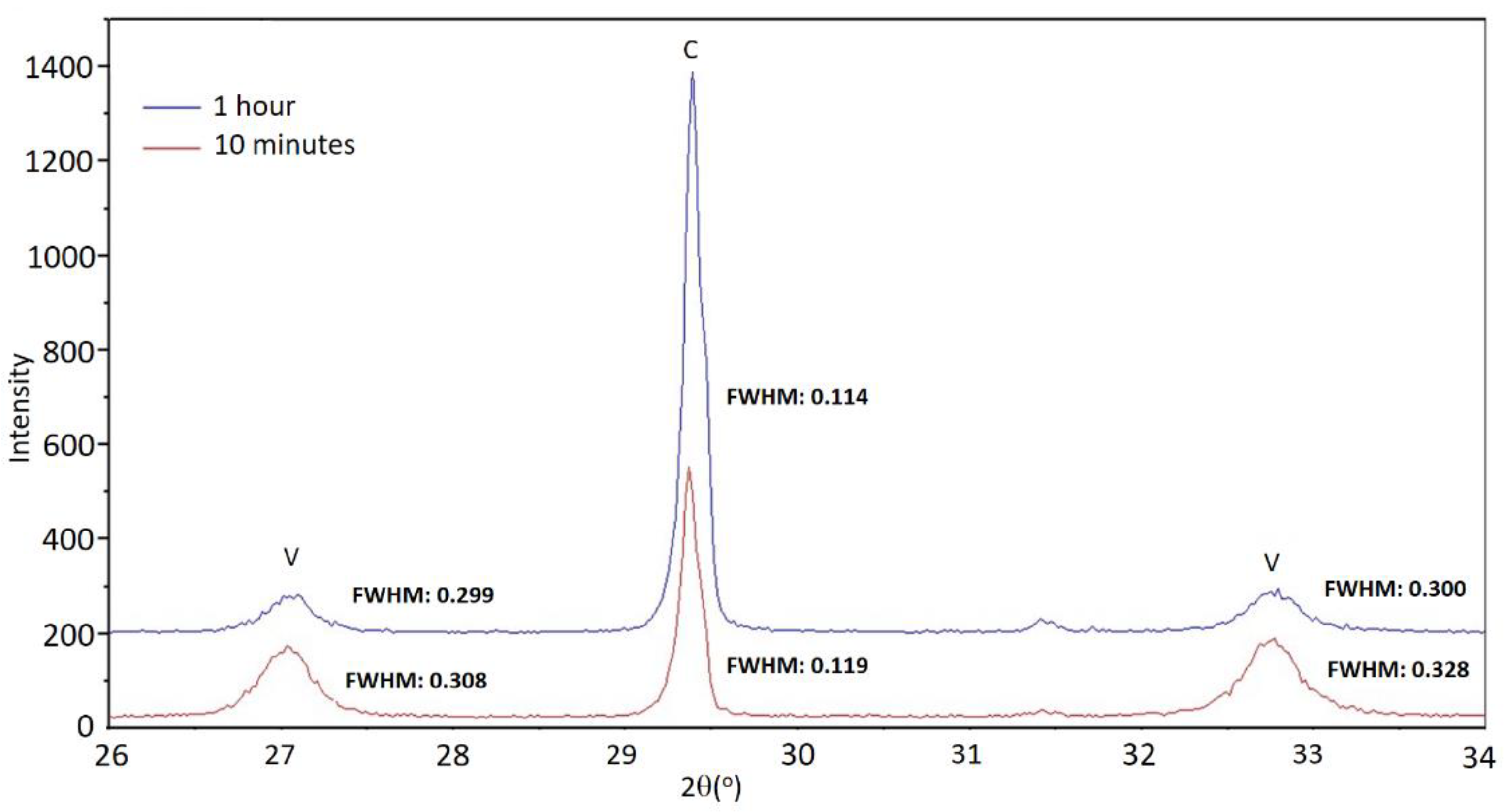
Figure 1, reflections corresponding to calcite are very well defined, indicating that it had a high degree of crystallinity. In general, it can be observed that the reflections of calcite are somewhat narrower and of greater intensity at longer aging times. The reflections of vaterite are noticeably wider, so the crystallinity of vaterite was poorer. As an example,
Figure 3 shows fragments of the diffractograms corresponding to sample E0 for the times of 10 min and 1 h, in which the width at half height of the reflections is indicated. The scarce persistence of this phase prevents drawing conclusions about the evolution of vaterite crystallinity in this set of experiments.
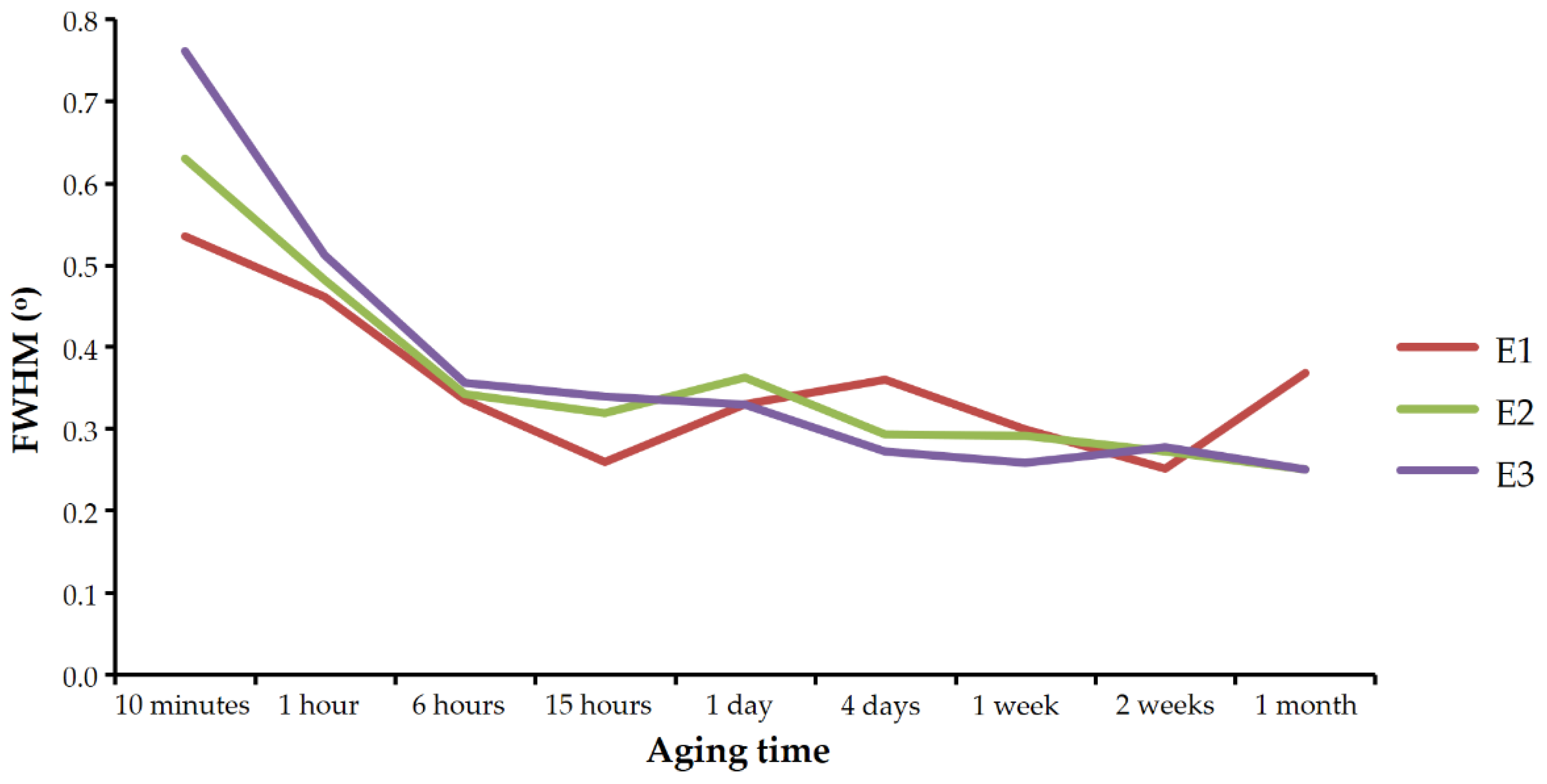
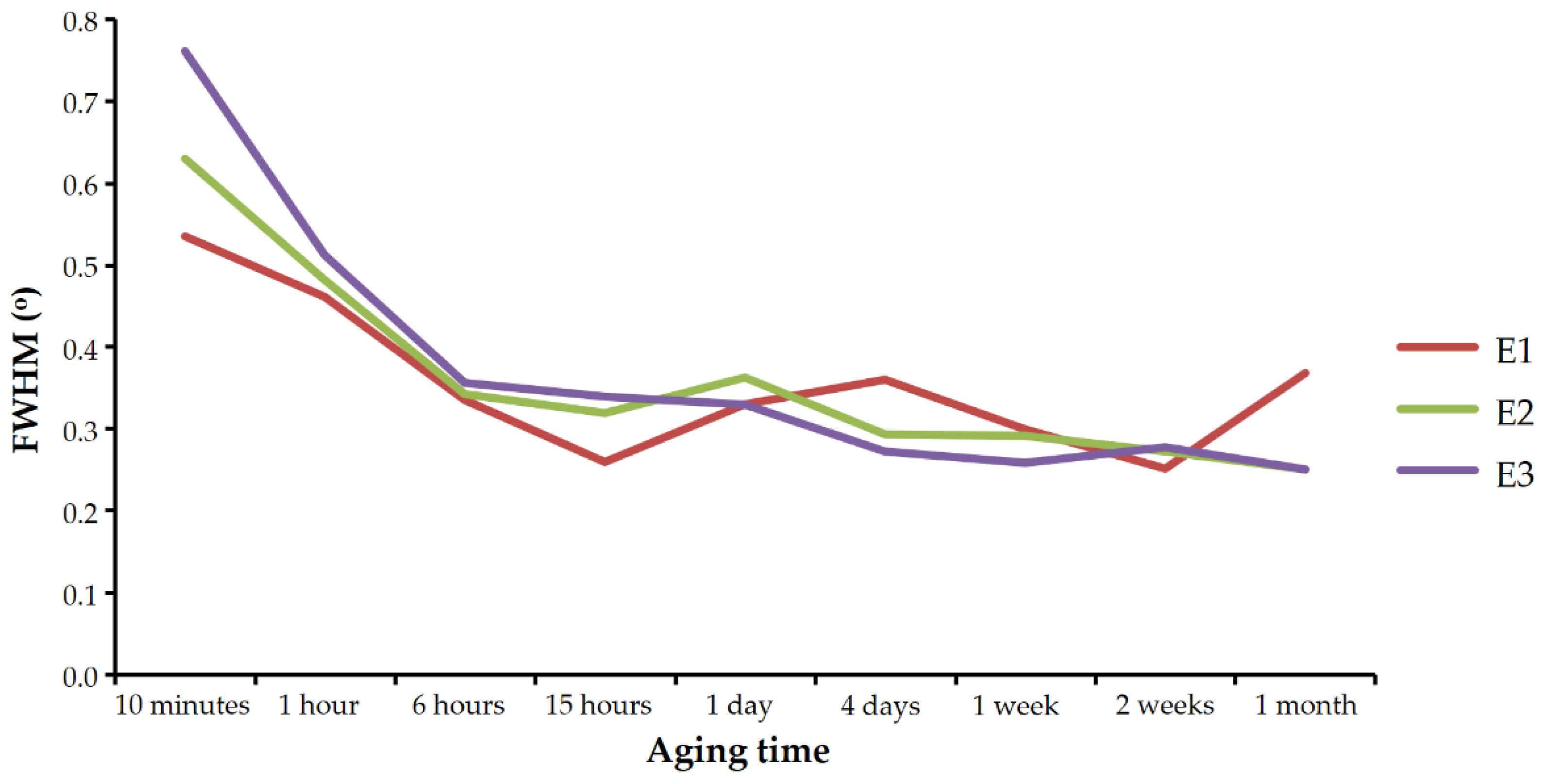
Figure 4 shows the evolution of the FWHM of the 112 vaterite reflection in the diffractograms of sets E1, E2, and E3 with aging time. As can be seen, the reflection became narrower with time in all three experiments. As discussed below, this behavior cannot be explained by compositional heterogeneities of the crystals and has to be interpreted as an increase in vaterite crystallinity with aging time.
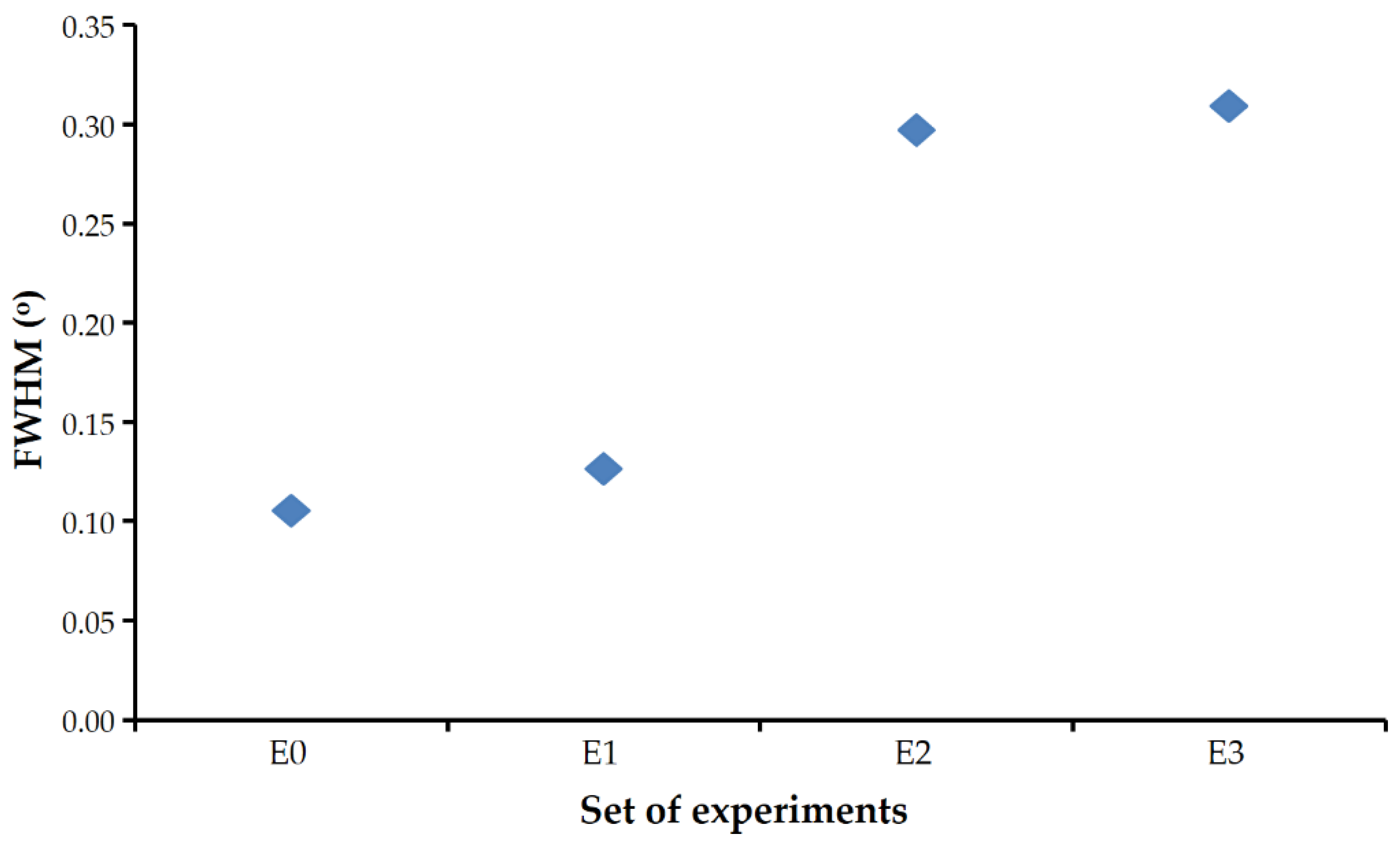
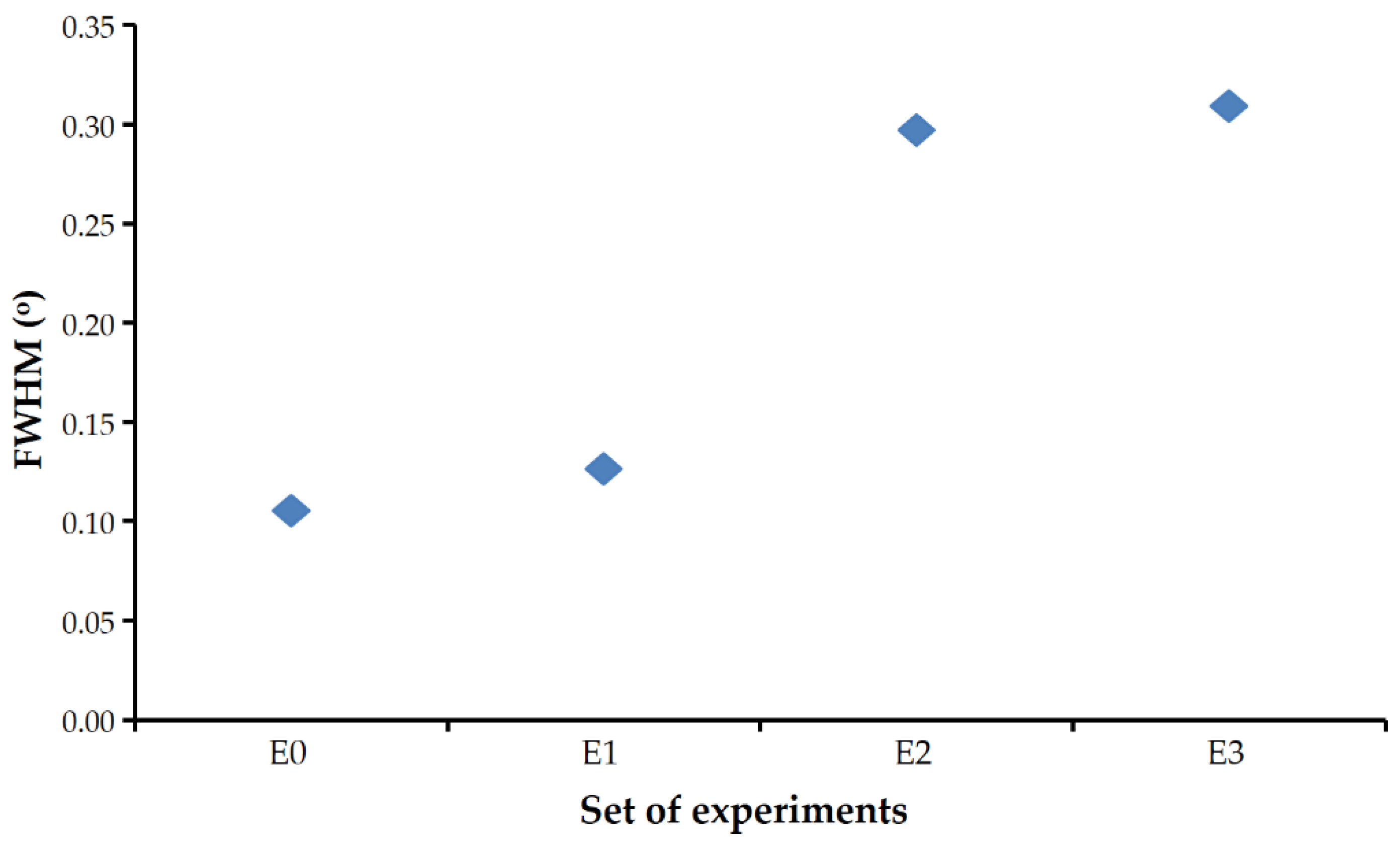
FWHM of the calcite reflections found in the experiments carried out in the presence of Se(IV) were significantly larger than those obtained in the control experiment.
Figure 5 shows these values for the 104 reflections (the most intense) of the most crystalline calcite that could be obtained in each set. The presence of selenium prevented the calcite from reaching a degree of crystallinity as high as that observed in similar experiments free of this element (set E0).
3.3. Cell Parameters of Vaterite Precipitated in the Presence of Se(IV)
The incorporation of foreign ions into a crystalline structure can lead to a variation in cell parameters with respect to the pure phase. The effect of selenite incorporation in the calcite structure has been studied in the scientific literature [
12]. According to these studies, the incorporation of selenium replacing carbonate causes the calcite cell volume to increase. The maximum amount of Se(IV) that these authors have found in calcite is 27.6 mmol/kg, and with this concentration of selenite, the cell volume had increased by 0.11%. The effect that the eventual incorporation of selenite into the vaterite structure may have on its cell parameters is unknown at this time.
For each one of the diffractograms, Miller indexes were assigned to a minimum of 15 reflections of vaterite in the P63/mmc space group. Once the assignment was made, the cell parameters and their volume were obtained and refined by the Rietveld method, using the X’pert High Score Plus program. The obtained values are shown in
Table 3.
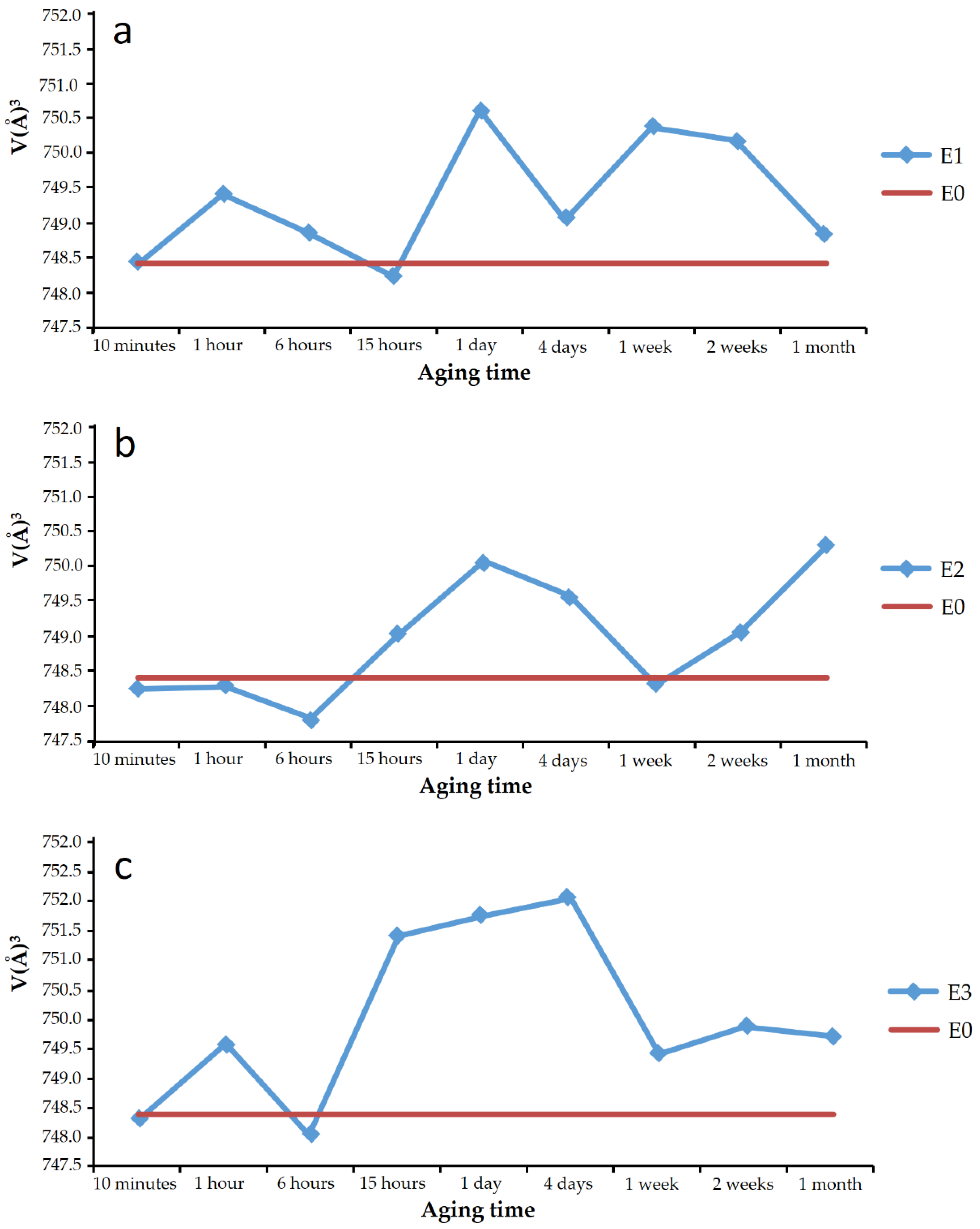
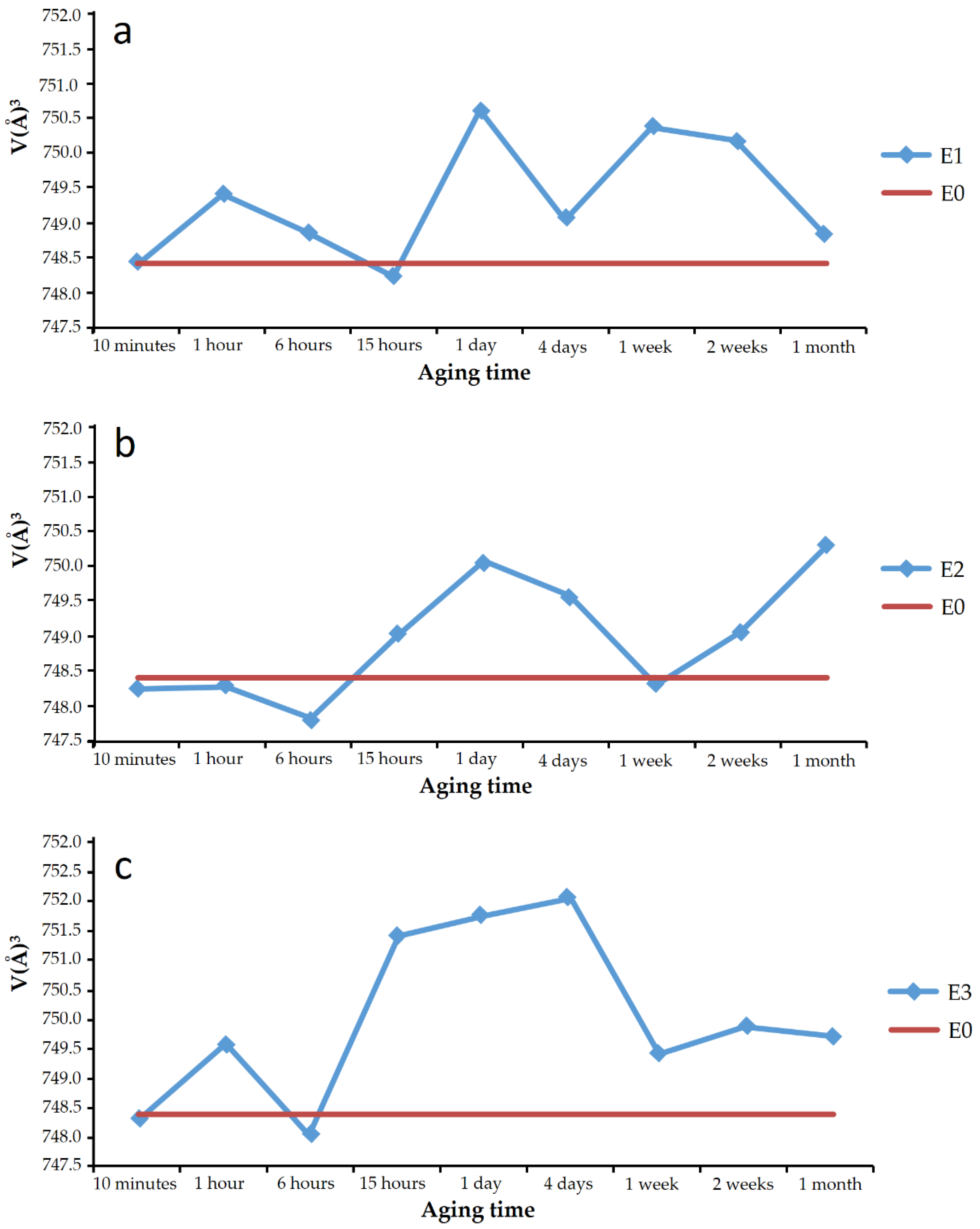
Plots a, b, and c in
Figure 6 show, respectively, the cell volume of the vaterite from experiments E1, E2, and E3 for the different aging times. In the plots, the cell volume of the vaterite shown in the JCPDS database record is marked with a horizontal line, and it is coincident with that calculated for the vaterite of experiments E0. In addition, superimposed on each graph, the phases that were present in the sample, together with the vaterite, are indicated.
Virtually, all the vaterite solids precipitated in the presence of SeO
32− showed larger cell volume than pure vaterite. The difference was small, less than 1% in all cases, but it is almost an order of magnitude higher than the difference found in the literature [
27] for calcite doped with 27.6 mmol/kg Se(IV).
Observations did not show that the variation of the cell volume followed a regular trend with aging time, nor could the trend be directly related to the appearance of other phases, such as calcite or CaSeO3·H2O in the solids. It can be pointed out that vaterite with higher cell volumes was obtained at aging times of around 1 day. The discussion of these results should be combined with the chemical analysis of vaterite precipitates, described in a following subsection of this work.
3.4. Morphology of the Solid Phase Subsection
Under scanning electron microscope, the identification of phases in the solids precipitated in the absence of Se(IV) was immediate. The morphology was evident and the results agreed with those obtained by X-ray diffraction.
Figure 7 shows precipitates of the set E0 at different aging times. Calcite precipitated as rhombohedral crystals and vaterite as spherulites formed by aggregation of tiny individual crystals [
27]. The size of calcite crystals was different according to the aging time. In the early stages, when it coexisted with vaterite, the edge of the rhombohedron could reach 10 µm. Later, smaller crystals precipitated, giving rise to an important size variety in the sample, ranging from 1 to 10 µm of edge. The vaterite spherulites were quite homogeneous in size and had a diameter between 4 and 7 µm.
The morphology of the vaterite in the experiments performed in the presence of Se(IV) was also spherulitic and the size was similar to that observed in the control experiment.
Figure 8 shows some of these spherulites in precipitates from experiments E1, E2, and E3.
CaSeO
3·H
2O crystals showed a characteristic lamellar morphology with a leafy appearance. In many cases, the platelets were embedding vaterite spherulites, as shown in
Figure 9.
The lamellar habit of CaSeO
3·H
2O precipitated from aqueous solutions was previously reported in the scientific literature and agreed with predictions of the Donnay–Harker model [
32].
3.5. Chemical Composition of Precipitated Solid Phases
Once the phases were morphologically identified, they were analyzed by EDX. The chemical analysis of calcite and CaSeO
3·H
2O were coherent with their stoichiometric formula. However, vaterite incorporated certain proportions of Se.
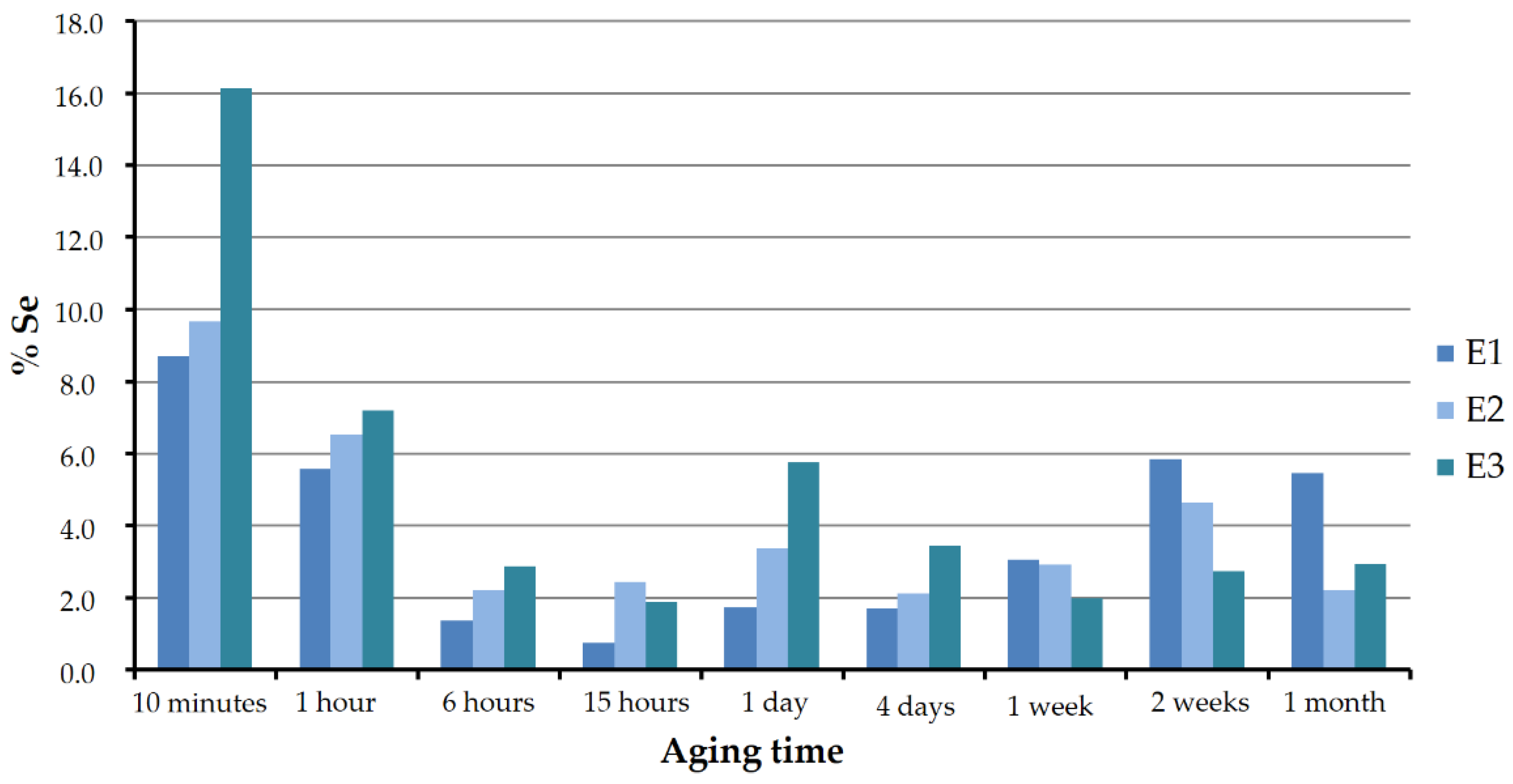
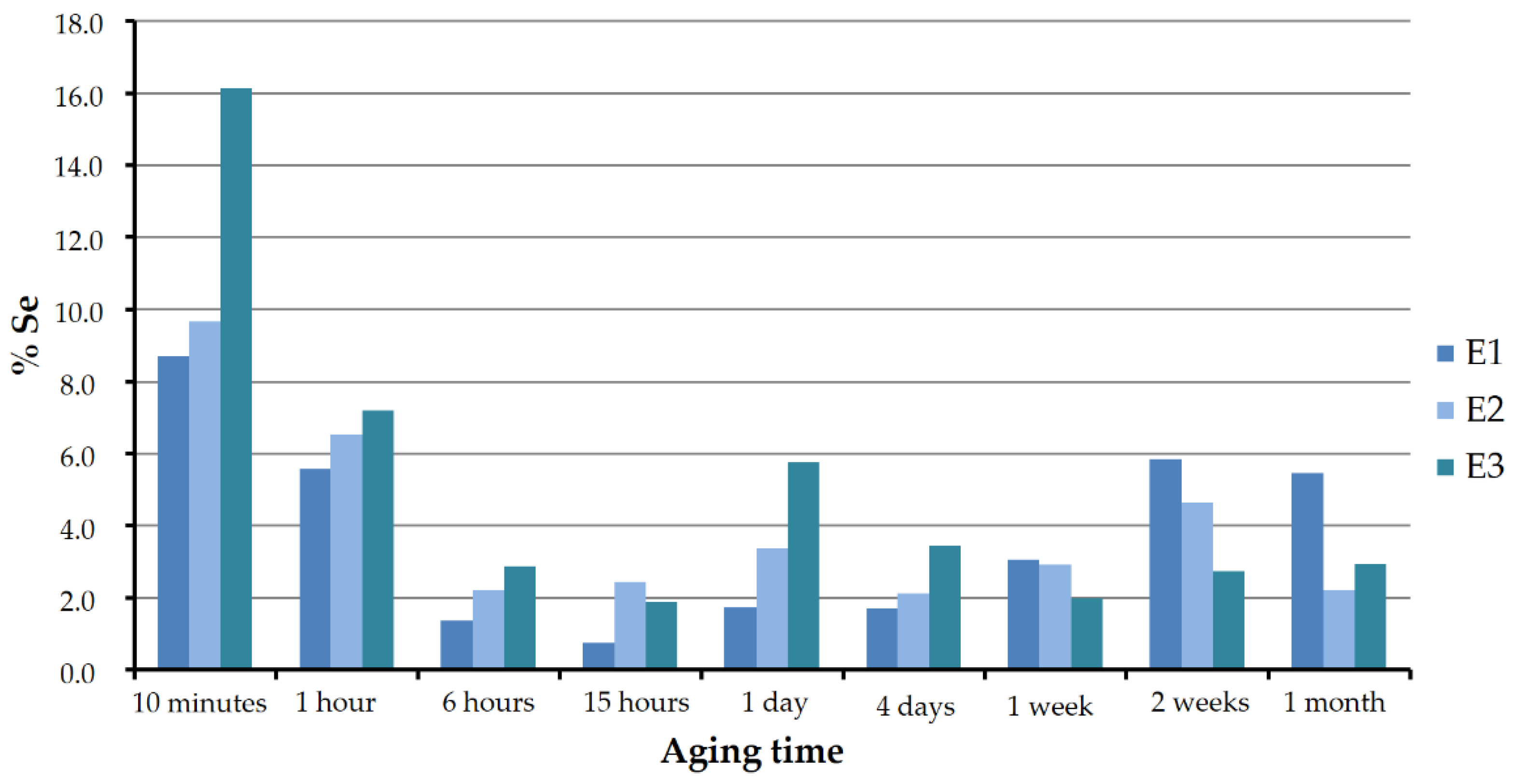
Figure 10 shows the Se:Ca atomic ratio as a percentage in the analyzed vaterite of sets E1, E2, and E3 for the different aging periods. As can be seen, in all the three cases the pattern was similar. In the first stages, the solids were found to be richer in selenium. The atomic ratio reached values around 16, 10, and 9, respectively, in experiments E3, E2, and E1. With increasing aging time, vaterite lost selenium until it reached a minimum between approximately 0.7% and 2.5%, at around 15 to 24 h. This minimum coincided with the precipitation of CaSeO
3·H
2O. From that moment on, the selenium–calcium proportions slightly increased or decreased, but without exceeding 6%.
In the early stages, the highest selenium concentrations in vaterite corresponded to the highest selenium concentrations in the starting solutions. However, this trend was less clear and even seemed to be reversed for longer aging times, starting from one week.
3.6. Initial Aqueous Solutions Characterization and Modelization
The aqueous solutions from which precipitation occurred were modeled with the Phreeq software. The sit.dat database, developed by Amphos21, BRGM, and ANDRA, was used in all simulations. According to these calculations, the initial solution was oversaturated with respect to the three main polymorphs of CaCO
3 (calcite, aragonite, and vaterite) and also, with respect to monohydorcalcite (CaCO
3·H
2O) and the CaSeO
3·H
2O phase, as shown in
Table 4.
3.7. Aqueous Solutions Evolution with Aging Time
Once precipitation takes place, the evolution of the solids with aging time may also have an effect on the aqueous solution, both on its overall chemical composition and on the distribution of ionic species. In general, these changes imply that the remaining solution is less concentrated in the elements that are incorporated into the solid and that the supersaturation with respect to the precipitating phases decreases.
It is well known that the pH value is of great importance in carbonates precipitation because of its influence on the distribution of the carbonate species and therefore on the supersaturation. In addition, pH also has an important influence on the distribution of Se(IV)-containing species, although this influence is not as clearly established in the literature as it is for carbonates. In general, at very low pH values, the dominant species is H
2SeO
3, while HSeO
3− is dominant at intermediate values and SeO
32− at high pH.
Figure 11 shows the distribution of species in a solution of similar concentration to that of the experiments developed in this work.
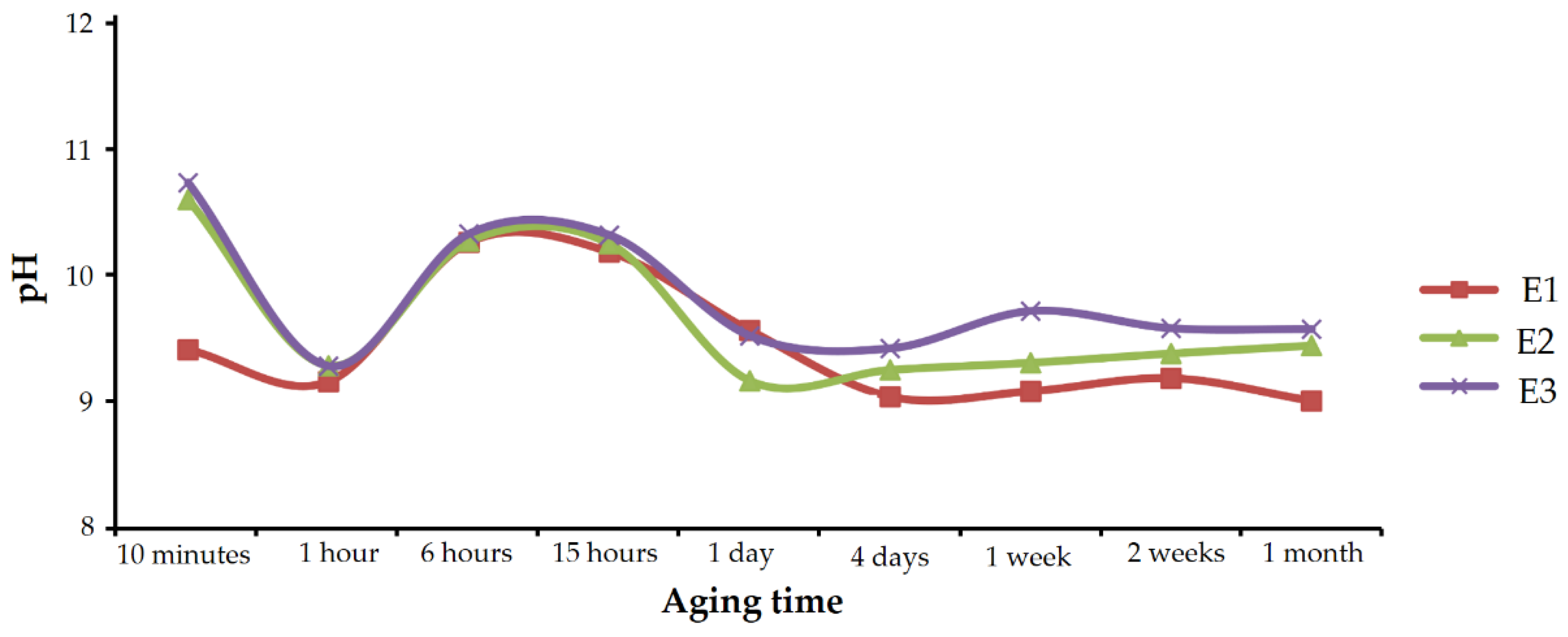
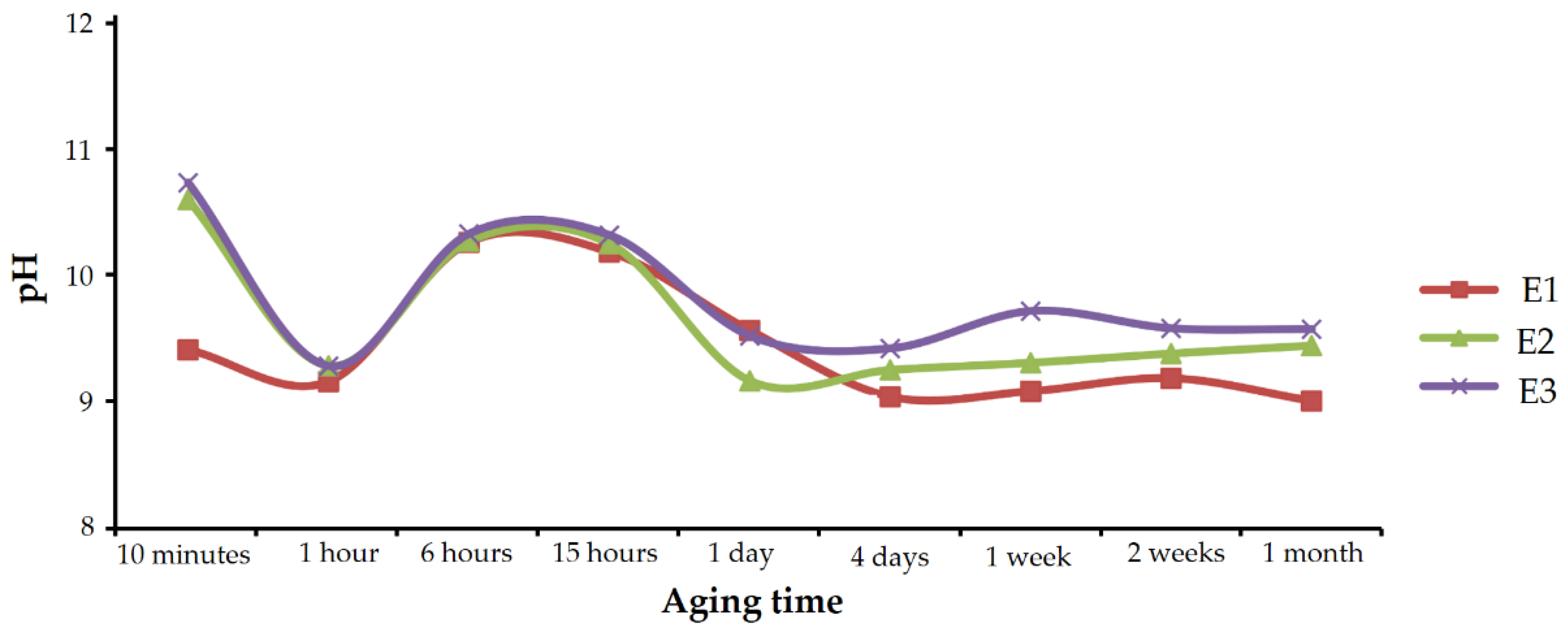
The pH of the solutions in each set of experiments was monitored during aging, and the measured values are shown in
Figure 12.
In all the three cases, the evolution was similar. The pH value sharply decreased in the first hour; from values close to 11, it quickly approached 9. After 1 h, pH went up to reach maximums above 10 at 6 to 15 h of aging time. From then on, pH dropped again, approaching 9, and it remained practically stable until the end of the experiments.
According to the results described above, the decrease observed in the first hour was caused by the rapid precipitation of calcium carbonate. This decrease was smaller in experiments containing Se(IV) than in experiments performed under similar conditions in the absence of selenium, where values below 8 were reached [
27]. The oscillating pH observed after 1 h of aging, has a more complex explanation which is described in the Discussion section. It should be noted that the pH evolution correlated very well with the composition of the precipitating solids. If the above pH evolution graph of
Figure 12 is compared with
Figure 10, which shows the chemical composition of vaterite precipitates, it can be seen that higher enrichments in Se(IV) correspond to higher pH values. Although there are several possible interpretations for this good correlation, one of the clearest would be that the increase in pH indicates an enrichment in free CO
32- and therefore higher supersaturation in calcium carbonate. This high supersaturation would imply a higher precipitation rate that favors the incorporation of impurities, i.e., a higher amount of selenite in the solid.
On the other hand, the stabilization of the pH in prolonged aging times was coincident with the precipitation of CaSeO3·H2O. The decrease in calcium and carbonate concentration in the aqueous solution, due to the precipitation of vaterite, must have brought the system closer to equilibrium with respect to the calcium carbonates, and the precipitation was slower than in the first stages.
4. Discussion
The above results, allow proposing the following “sequence of events”. Once the parent aqueous solutions were mixed, highly supersaturated solutions with respect to the three main calcium carbonate phases, and rather less with respect to CaSeO3·H2O, were produced. The instantaneous precipitation of solid phases caused a sharp drop in pH, from about 11 at the beginning of the experiment, to approaching 9 after 1 h in all experiments. Following the normal crystallization sequence of calcium carbonates, vaterite precipitated instantaneously as a calcite precursor phase. In the absence of Se(IV), the transformation of vaterite into calcite by a dissolution–recrystallization process that has been discussed in several previous works, progressed rapidly, so that after 6 h the whole solid was calcite and showed a good degree of crystallinity that was manifested in narrow and very sharp peaks in the corresponding diffraction diagram. The morphology of these crystals was rhombohedral. However, in experiments E1, E2, and E3, carried out in the presence of Se(IV), vaterite remained for much longer, and the aging history was different.
In these cases, the width of the reflections indicates that the early vaterite precipitates had low crystallinity. This low crystallinity and the high precipitation rate in a highly supersaturated system, both favored the incorporation of a significant amount of selenite into the crystal structure. This incorporation was verified by EDX analyses, that also have shown quantities of Se in vaterite of up to 16% (Se:Ca). All vaterite precipitates showed the characteristic spherulitic morphology. Such a significant incorporation of selenium does not seem to imply a large change in the cell parameters of the vaterite.
Although the initial solutions were also supersaturated with respect to CaSeO3·H2O, the saturation index for this phase was appreciably lower than for calcium carbonates, so it did not form as rapidly as vaterite. In addition, the precipitation of selenium-rich vaterite caused the aqueous solution to be depleted in calcium and selenium, further lowering the saturation index with respect to calcium selenite.
Selenite-rich vaterite persisted in the fluid as the only solid phase for a certain period of time. The higher the selenite concentration, the longer the persistence: 6 h in set E1, 15 h in E2, and 1 day in E3. This indicates that Se(IV)-doped vaterite was more stable than Se(IV)-doped calcite. According to the literature [
12,
29], the selenite ion can partially substitute the carbonate ion in the calcite structure. The morphology and dimensions of the selenite ion made its incorporation in the calcite structure difficult and implies a certain modification in the cell parameters. Moreover, although there are no experimental data on the solubility of Se(IV)-doped calcite, it should be more soluble than pure calcite as confirmed by molecular modeling [
12]. In the case of vaterite, the partial substitution of carbonate ions by selenite ions would be more favorable since this CaCO
3 phase is less dense than calcite. In vaterite, the anionic positions are particularly flexible, so it could better accommodate foreign anions. This fact would also explain the little effect of selenium on the dimensions of the vaterite cell parameters found in the present work. For a certain range of composition, the solubility of vaterite with selenium can approach that of pure calcite and can even be lower than that of calcite with the same amount of selenium. This influence of Se(IV) on the solubility of calcite and vaterite hinders the vaterite–calcite transformation.
The progressive depletion in selenium of vaterite finally permitted the transformation into calcite to begin. This happened after 6 h of aging in set E1, 15 h in E2, and nearly 1 day in E3. At this time, the vaterite had a low selenium concentration. Calcite formed in this transformation was free of selenium, as evidenced by the EDX analysis and position of the reflections in the diffractograms that perfectly matched that of the standard JCPS database records. However, the morphology of calcite was affected by the presence of selenium in the aqueous solution. No rhombohedron morphologies as shown by crystals formed from vaterite in selenite-free solutions were found. On the contrary, calcite crystals showed a spherulitic morphology that was difficult to distinguish from that of vaterite. This influence of selenite on calcite growth was in agreement with molecular-scale observations in the literature [
30].
In experiments of set E3, the beginning of the transformation of vaterite to calcite occurred simultaneously with the precipitation of CaSeO3·H2O. The release of a significant amount of SeO32- by the dissolution of Se-doped vaterite, which calcite is unable to incorporate into its structure, increased the oversaturation of the system with respect to this phase. However, in experiments of sets E1 and E2, the amount of selenite contributed was not so important, and this phase still did not precipitate.
Under the explored conditions, calcite was always a minority phase with respect to vaterite, and it even completely disappeared in a short period of time. In sets E1 and E2 calcite, disappeared completely a few hours after formation. In the case of E3, it persisted for a period between 1 and 4 days. This result contradicts the normal sequence of carbonate crystallization. In light of the results, a plausible explanation would be as follows: vaterite doped with a certain amount of selenite had lower solubility than pure vaterite, but higher than calcite. Therefore, impure vaterite–calcite transformation took place, although more slowly than the pure vaterite–calcite transformation of experiment E0 that took place in the absence of Se. As the transformation took place, vaterite released selenium to the aqueous solution, which hindered the growth of calcite and promoted morphologies with a larger specific surface area than the rhombohedron. This morphology favored calcite dissolution in a medium in which its growth was already difficult because of the presence of selenium. Thus, calcite dissolved to form vaterite with selenite impurities.
After calcite dissolution, the remaining vaterite precipitates in the aqueous solution showed lower selenium content and higher crystallinity than the vaterite precipitates of the initial stages. Both factors favored their stability. The slow and progressive increase in SeO32- in the aqueous solution caused the final precipitation of CaSeO3·H2O observed. The relationship between the appearance of this phase and the dissolution–recrystallization of the vaterite spherulites was clearly observed in the scanning electron microscopy images that showed CaSeO3·H2O crystals embedding vaterite spherulites. The morphology of CaSeO3·H2O crystals was consistent with that predicted according to the Donnay–Harker model for this phase.
5. Conclusions
The presence of dissolved Se(IV) plays an important role in the precipitation of calcium carbonate from aqueous solution at room temperature. It has a great influence on the composition of the precipitated phases and especially on the crystallization sequence of the different CaCO3 polymorphs.
In the range of the explored concentration, the presence of the SeO32− anion increased the stability of the vaterite by delaying its transformation into calcite.
Vaterite synthesized at high supersaturation with respect to calcium carbonate, contained up to 16% atomic Se:Ca ratio in the initial stages of precipitation. However, this ratio decreased after a few hours of aging in the remaining aqueous solution.
In contrast with what has been observed in the scientific literature for calcite [
12,
29,
30], the incorporation of Se(IV) into the vaterite crystal structure was not observed to influence its cell parameters. This may be explained by the lower density of vaterite compared with calcite. In the crystal structure of vaterite, the positions in which anions were accommodated were wider and would allow not only the structural disorder of the carbonates but also the entry of foreign anions such as SeO
32−.
In aqueous media in which the calcium carbonate precipitation took place, the simultaneous precipitation of CaSeO3·H2O was conditioned not only by the availability of Ca2+ that was not incorporated into the carbonates but also by the availability of free SeO32−, which had not been incorporated into the vaterite crystal structure.
Under the conditions in which the experiments were carried out, CaSeO3·H2O grew in close association with the vaterite and showed a leafy or lamellar morphology that was in agreement with what would be predicted by the Donnay–Harker theory.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}