
Determination of Trace Metal (Mn, Fe, Ni, Cu, Zn, Co, Cd and Pb) Concentrations in Seawater Using Single Quadrupole ICP-MS: A Comparison between Offline and Online Preconcentration Setups

 and
and 
Abstract
:1. Introduction
2. Analytical Methods
2.1. Reagents and Materials
2.2. Automated Preconcentration Using seaFAST Module
2.3. ICP-MS
2.3.1. Calibration
2.3.2. Monitoring Instrument Drift
2.4. Procedural Blanks and Limit of Detection (LOD)
2.5. Reference Materials—NASS-7 and GEOTRACES (GSP and GSC)
2.6. Recovery
2.7. Quantifying Polyatomic Interferences
2.8. Data Verification
2.8.1. Precision
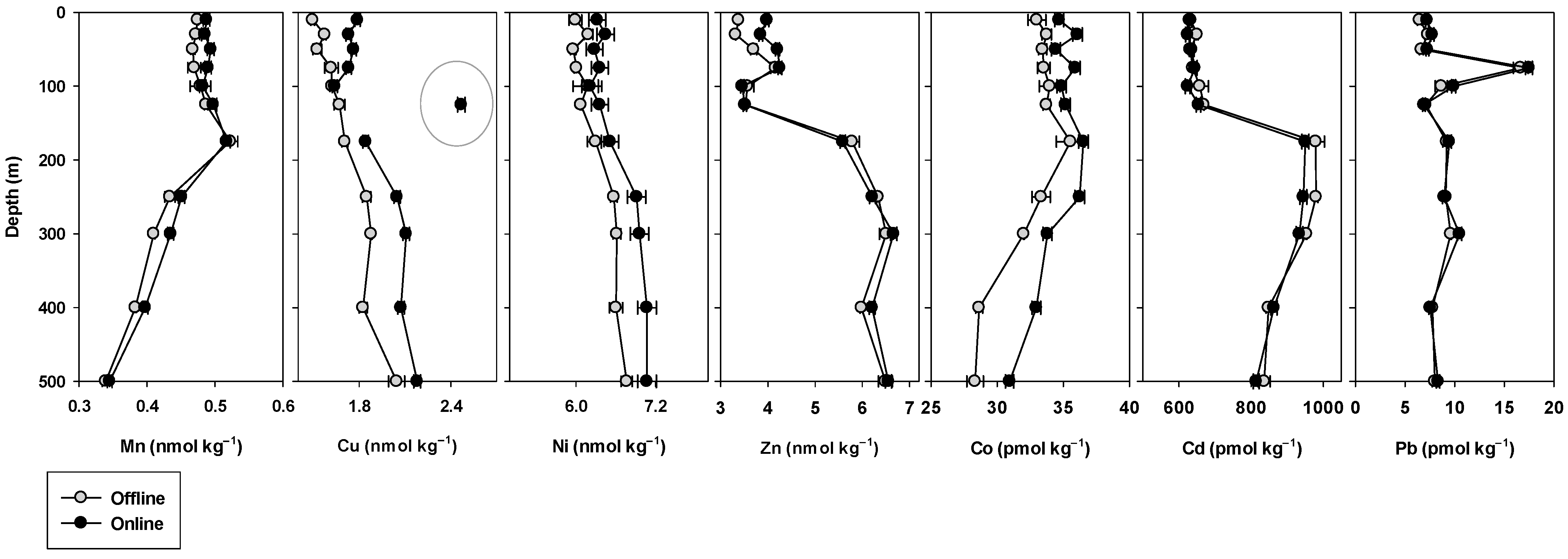
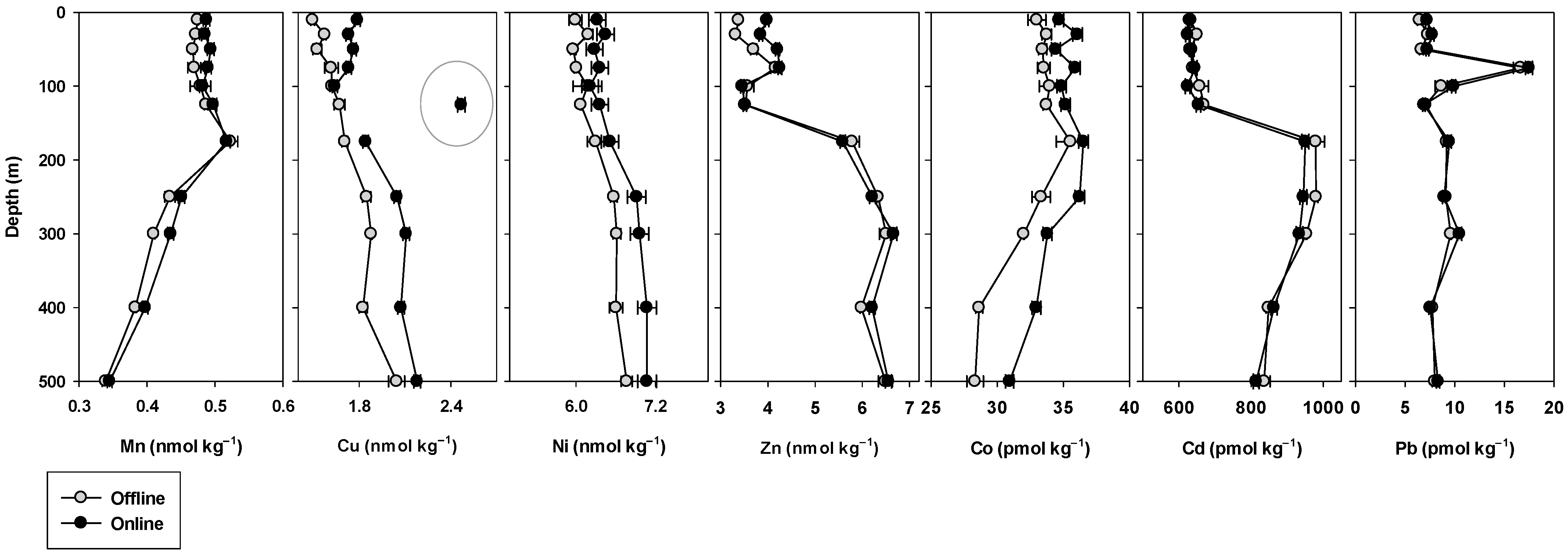
2.8.2. Crossover Station
2.8.3. External Intercalibration
3. Results
3.1. Blanks and Limits of Detection
3.2. Method Accuracy and Precision
3.3. Recovery
3.4. Polyatomic Interferences
3.5. Crossover and Intercalibration Stations
4. Discussion
4.1. Method Performance
4.2. Polyatomic Interference Removal
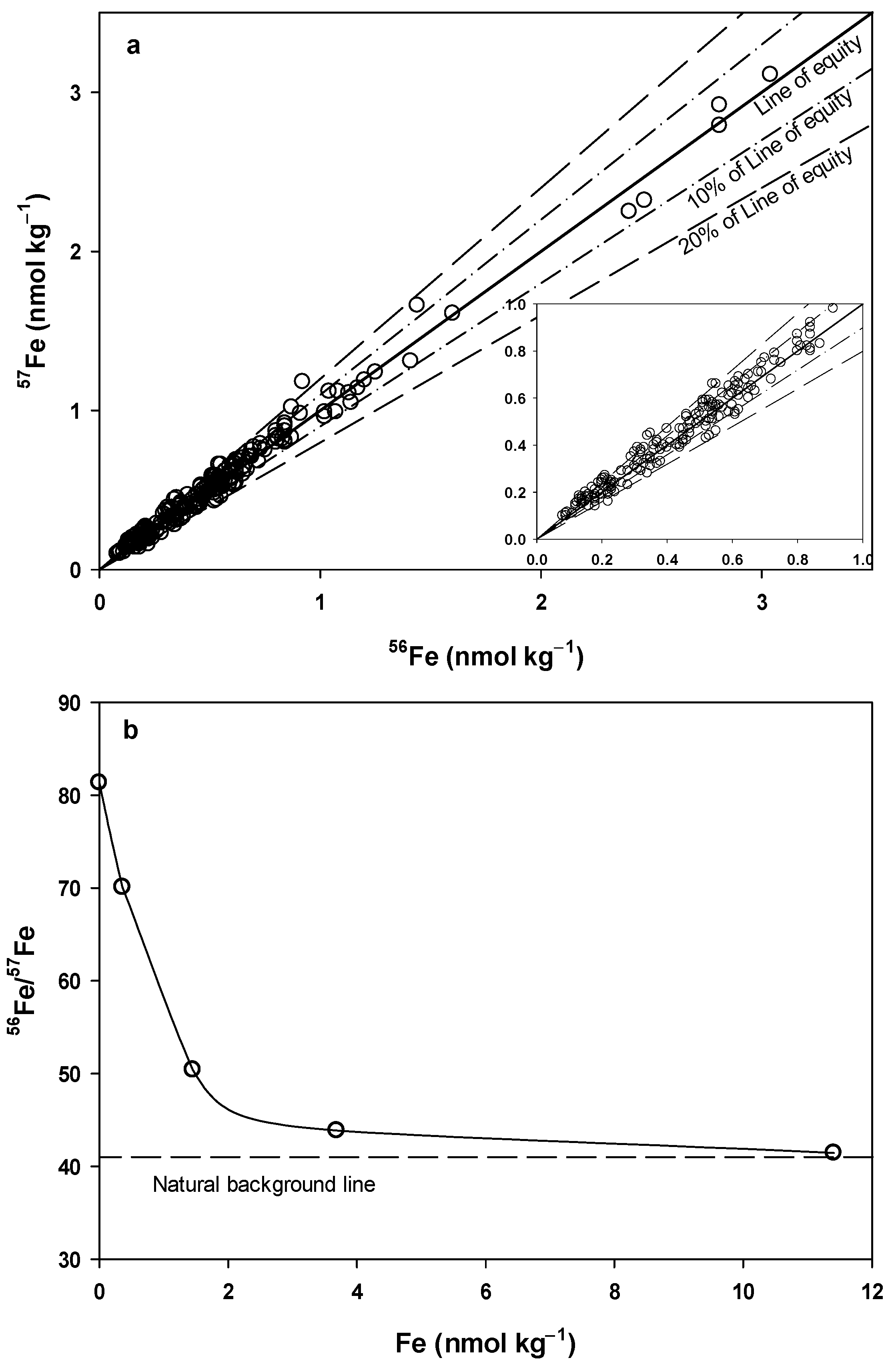
4.2.1. Recalculation of Fe
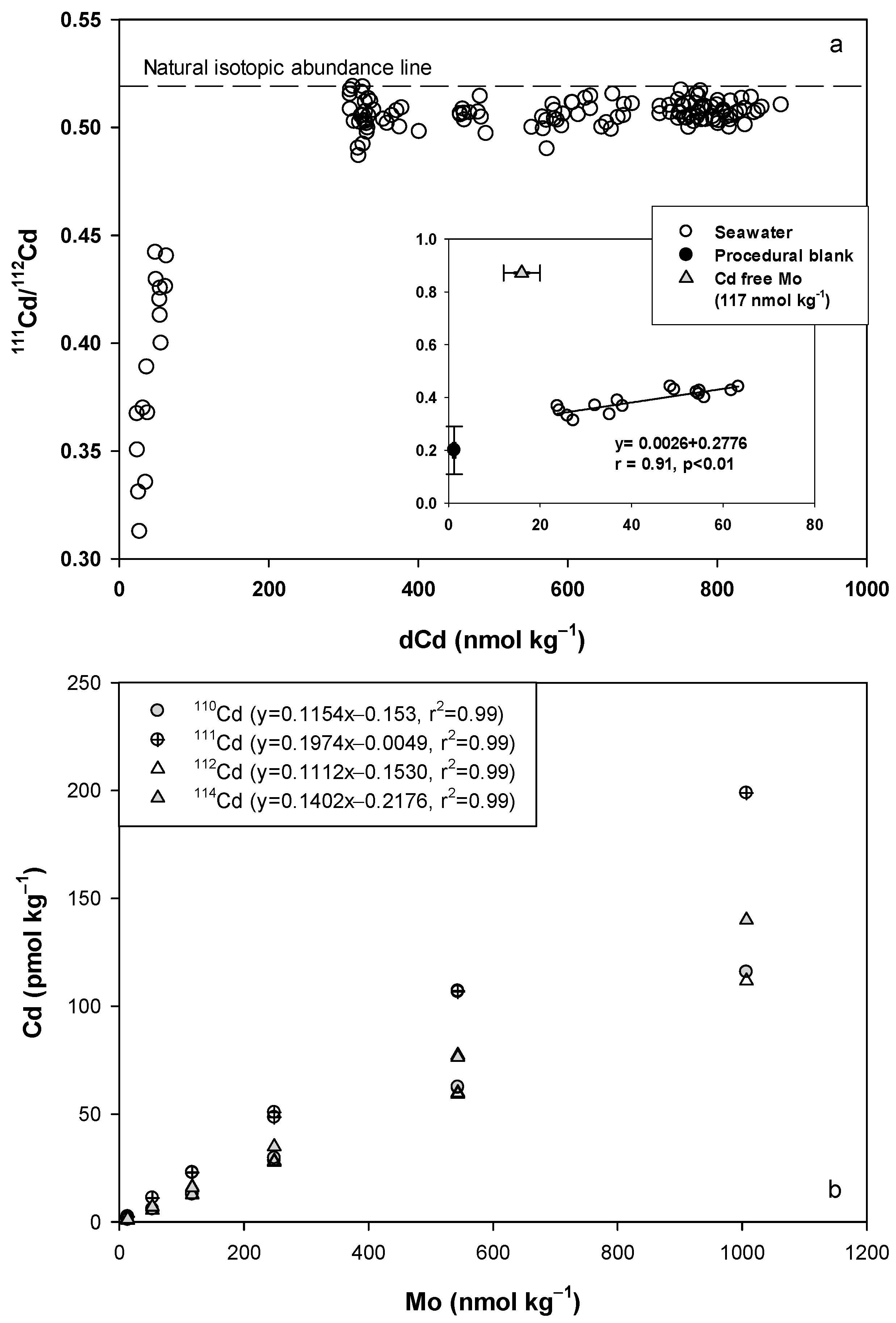
4.2.2. Recalculation of Cd
4.3. Crossover Stations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, R.F.; Francois, R.H.G.M.; Frank, M.; Henderson, G.M.; Jeandel, C.; Sharma, M. GEOSECS to GEOTRACES: Lessons Learned from Large Programs Studying Ocean Chemistry. In AGU Fall Meeting Abstracts; American Geophysical Union: Washington, DC, USA, 2019; p. OS41A-05. [Google Scholar]
- Bruland, K.W.; Franks, R.P. Sampling and analytical methods for the determination of copper, cadmium, zinc and nickel at the nanogram per litre level in seawater. Anal. Chim. Acta 1979, 28, 367–376. [Google Scholar] [CrossRef]
- Wu, J.; Boyle, E.A. Low Blank Preconcentration Technique for the Determination of Lead, Copper, and Cadmium in Small-Volume Seawater Samples by Isotope Dilution ICPMS. Anal. Chem. 1997, 69, 2464–2470. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.L.; Bruland, K.W. An improved method for rapid preconcentration and determination of bioactive trace metals in seawater using solid phase extraction and high resolution inductively coupled plasma mass spectrometry. Mar. Chem. 1998, 63, 145–153. [Google Scholar] [CrossRef]
- Bowie, A.R.; Achterberg, E.P.; Mantoura, R.F.C.; Worsfold, P.J. Determination of sub-nanomolar levels of iron in seawater using flow injection with chemiluminescence detection. Anal. Chim. Acta 1998, 361, 189–200. [Google Scholar] [CrossRef]
- Colombo, C.; van den Berg, C.M.G. Simultaneous determination of several trace metals in seawater using cathodic stripping voltammetry with mixed ligands. Anal. Chim. Acta 1997, 337, 29–40. [Google Scholar] [CrossRef]
- Kingston, H.M.; Barnes, I.L.; Brady, T.J.; Rains, T.C.; Champ, M.A. Separation of eight transition elements from alkali and alkaline earth elements in estuarine and seawater with chelating resin and their determination by graphite furnace atomic absorption spectrometry. Anal. Chem. 1978, 50, 2064–2070. [Google Scholar] [CrossRef]
- King, D.W.; Lin, J.; Kester, D.R. Spectrophotometric determination of iron (II) in seawater at nanomolar concentrations. Anal. Chim. Acta 1991, 247, 125–132. [Google Scholar] [CrossRef]
- Rapp, I.; Schlosser, C.; Rusiecka, D.; Gledhill, M.; Achterberg, E.P. Automated preconcentration of Fe, Zn, Cu, Ni, Cd, Pb, Co, and Mn in seawater with analysis using high-resolution sector field inductively-coupled plasma mass spectrometry. Anal. Chim. Acta 2017, 976, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.L.; Spence, J.; Janssen, D.J.; Ross, A.R.S.; Cullen, J.T. Determination of Mn, Fe, Ni, Cu, Zn, Cd and Pb in seawater using offline extraction and triple quadrupole ICP-MS/MS. J. Anal. At. Spectrom. 2018, 33, 304–313. [Google Scholar] [CrossRef]
- Wuttig, K.; Townsend, A.T.; van der Merwe, P.; Gault-Ringold, M.; Holmes, T.; Schallenberg, C.; Latour, P.; Tonnard, M.; Rijkenberg, M.J.A.; Lannuzel, D.; et al. Critical evaluation of a SeaFAST system for the analysis of trace metals in a wide range of marine samples. Talanta 2019, in press. [Google Scholar] [CrossRef]
- Strivens, J.E.; Brandenberger, J.M.; Johnston, R.K. Data trend shifts induced by method of concentration for trace metals in seawater: Automated online preconcentration vs. borohydride reductive coprecipitation of nearshore seawater samples for analysis of Ni, Cu, Zn, Cd, and Pb via ICP-MS. Limnol. Oceanogr. Methods 2019, 17, 266–276. [Google Scholar] [CrossRef]
- Vassileva, E.; Wysocka, I.; Orani, A.M.; Quétel, C. Off-line preconcentration and inductively coupled plasma sector field mass spectrometry simultaneous determination of Cd, Co, Cu, Mn, Ni, Pb and Zn mass fractions in seawater: Procedure validation. Spectrochim. Acta Part B At. Spectrosc. 2019, 153, 19–27. [Google Scholar] [CrossRef]
- Wysocka, I.; Vassileva, E. Method validation for high resolution sector field inductively coupled plasma mass spectrometry determination of the emerging contaminants in the open ocean: Rare earth elements as a case study. Spectrochim. Acta Part B At. Spectrosc. 2017, 128, 1–10. [Google Scholar] [CrossRef]
- Behrens, M.K.; Muratli, J.; Pradoux, C.; Wu, Y.; Böning, P.; Brumsack, H.J.; Goldstein, S.L.; Haley, B.; Jeandel, C.; Paffrath, R.; et al. Rapid and precise analysis of rare earth elements in small volumes of seawater—Method and intercomparison. Mar. Chem. 2016, 186, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Vassileva, E.; Wysocka, I. Development of procedure for measurement of Pb isotope ratios in seawater by application of seaFAST sample pre-treatment system and SF ICP-MS. Spectrochim. Acta Part B At. Spectrosc. 2016, 126, 93–100. [Google Scholar] [CrossRef]
- Poehle, S.; Schmidt, K.; Koschinsky, A. Determination of Ti, Zr, Nb, V, W and Mo in seawater by a new online-preconcentration method and subsequent ICP-MS analysis. Deep. Res. Part I Oceanogr. Res. Pap. 2015, 98, 83–93. [Google Scholar] [CrossRef]
- Tanner, S.D.; Baranov, V.I. Theory, Design, and Operation of a Dynamic Reaction Cell for ICP-MS. At. Spectrosc. 1999, 20, 45–78. [Google Scholar] [CrossRef]
- Cloete, R.; Loock, J.C.; Mtshali, T.; Fietz, S.; Roychoudhury, A.N. Winter and summer distributions of Copper, Zinc and Nickel along the International GEOTRACES Section GIPY05: Insights into deep winter mixing. Chem. Geol. 2019, 511, 342–357. [Google Scholar] [CrossRef]
- Cloete, R.; Loock, J.C.; van Horsten, N.R.; Fietz, S.; Mtshali, T.N.; Planquette, H.; Roychoudhury, A.N. Winter biogeochemical cycling of dissolved and particulate cadmium in the Indian sector of the Southern Ocean (GEOTRACES GIpr07 transect). Front. Mar. Sci. 2021, 8, 1014. [Google Scholar] [CrossRef]
- Cloete, R.; Loock, J.C.; van Horsten, N.R.; Menzel Barraqueta, J.-L.; Fietz, S.; Mtshali, T.N.; Planquette, H.; García-Ibáñez, M.I.; Roychoudhury, A.N. Winter dissolved and particulate zinc in the Indian Sector of the Southern Ocean: Distribution and relation to major nutrients (GEOTRACES GIpr07 transect). Mar. Chem. 2021, 236, 104031. [Google Scholar] [CrossRef]
- Cutter, G.; Casciotti, K.; Croot, P.; Geibert, W.; Heimbürger, L.-E.; Lohan, M.; Planquette, H.; van de Flierdt, T. Sampling and Sample-Handling Protocols for GEOTRACES Cruises; GEOTRACES International Project Office: Toulouse, France, 2017. [Google Scholar]
- Byrd, J.T.; Andreae, M.O. Dissolved and particulate tin in North Atlantic seawater. Mar. Chem. 1986, 19, 193–200. [Google Scholar] [CrossRef]
- McKelvey, B.A.; Orians, K.J. Dissolved zirconium in the North Pacific Ocean. Geochim. Cosmochim. Acta 1993, 57, 3801–3805. [Google Scholar] [CrossRef]
- Bekov, G.I.; Letokhov, V.S.; Radaev, V.N.; Baturin, G.N.; Egorov, A.S.; Kursky, A.N.; Narseyev, V.A. Ruthenium in the ocean. Nature 1984, 312, 748–750. [Google Scholar] [CrossRef]
- Liu, K.; Gao, X.; Li, L.; Chen, C.T.A.; Xing, Q. Determination of ultra-trace Pt, Pd and Rh in seawater using an off-line pre-concentration method and inductively coupled plasma mass spectrometry. Chemosphere 2018, 212, 429–437. [Google Scholar] [CrossRef]
- Feldmann, I.; Jakubowski, N.; Stuewer, D. Application of a hexapole collision and reaction cell in ICP-MS Part I: Instrumental aspects and operational optimization. Fresenius J. Anal. Chem. 1999, 365, 415–421. [Google Scholar] [CrossRef]
- Yamada, N. Kinetic energy discrimination in collision/reaction cell ICP-MS: Theoretical review of principles and limitations. Spectrochim. Acta Part B At. Spectrosc. 2015, 110, 31–44. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G. Molybdenum in natural waters: A review of occurrence, distributions and controls. Appl. Geochem. 2017, 84, 387–432. [Google Scholar] [CrossRef] [Green Version]
- Arnold, T.; Harvey, J.N.; Weiss, D.J. An experimental and theoretical investigation into the use of H2 for the simultaneous removal of ArO+ and ArOH+ isobaric interferences during Fe isotope ratio analysis with collision cell based Multi-Collector Inductively Coupled Plasma Mass Spectrometry. Spectrochim. Acta Part B 2008, 63, 666–672. [Google Scholar] [CrossRef]
- Sadagopa Ramanujam, V.M.; Yokoi, K.; Egger, N.G.; Dayal, H.H.; Alcock, N.W.; Sandstead, H.H. Polyatomics in zinc isotope ratio analysis of plasma samples by inductively coupled plasma-mass spectrometry and applicability of nonextracted samples for zinc kinetics. Biol. Trace Elem. Res. 1999, 68, 143–158. [Google Scholar] [CrossRef]
- Biller, D.V.; Bruland, K.W. Analysis of Mn, Fe, Co, Ni, Cu, Zn, Cd, and Pb in seawater using the Nobias-chelate PA1 resin and magnetic sector inductively coupled plasma mass spectrometry (ICP-MS). Mar. Chem. 2012, 130–131, 12–20. [Google Scholar] [CrossRef]
- Schlitzer, R.; Anderson, R.F.; Dodas, E.M.; Lohan, M.; Geibert, W.; Tagliabue, A.; Bowie, A.; Jeandel, C.; Maldonado, M.T.; Landing, W.M.; et al. The GEOTRACES Intermediate Data Product 2017. Chem. Geol. 2018, 493, 210–223. [Google Scholar] [CrossRef]
- Collier, R.W. Molybdenum in the Northeast Pacific Ocean. Limnol. Oceanogr. 1985, 30, 1351–1354. [Google Scholar] [CrossRef]
- Ho, P.; Lee, J.-M.; Heller, M.I.; Lam, P.J.; Shiller, A.M. The distribution of dissolved and particulate Mo and V along the U.S. GEOTRACES East Pacific Zonal Transect (GP16): The roles of oxides and biogenic particles in their distributions in the oxygen deficient zone and the hydrothermal plume. Mar. Chem. 2018, 201, 242–255. [Google Scholar] [CrossRef]
- Posacka, A.M.; Semeniuk, D.M.; Whitby, H.; Van Den Berg, C.M.; Cullen, J.T.; Orians, K.; Maldonado, M.T. Dissolved copper (dCu) biogeochemical cycling in the subarctic Northeast Pacific and a call for improving methodologies. Mar. Chem. 2017, 196, 47–61. [Google Scholar] [CrossRef]
- Saito, M.A.; Moffett, W. Cobalt and nickel in the Peru upwelling region: A major flux of labile cobalt utilized as a micronutrient. Glob. Biogeochem. Cycles 2004, 18, GB4030. [Google Scholar] [CrossRef] [Green Version]
- Morel, F.M.M.; Milligan, A.J.; Saito, M.A. Marine Bioinorganic Chemistry: The Role of Trace Metals in the Oceanic Cycles of Major Nutrients. In Treatise on Geochemistry, 2nd ed.; Holland, H.D., Turekian, K.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 123–150. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Mode of Analysis | Offline | Online |
|---|---|---|
| Preconcentration module | SC-4 DX seaFAST-pico | SC-4 DX seaFAST S3 |
| Column resin | Nobias (EDTriA and IDA) | Nobias (EDTriA and IDA) |
| Sample pH | 1.7 | 1.7 |
| Buffer | Ammonium acetate (pH: 6.0 ± 0.2) | Ammonium acetate (pH: 6.0 ± 0.2) |
| Eluent | 5% HNO3 | 5% HNO3 |
| Internal Standard | - | 10 µg kg−1 In |
| Sample loop (mL) | 10 | 3 |
| Elution cycles | 4 | 1 |
| Elution volume (mL) | 0.25 | 0.1 |
| Preconcentration factor | 40 | 30 |
| Sample throughput (min sample−1) | 20 | 11 |
| Mode of Analysis | Offline | Online |
|---|---|---|
| Instrument | Agilent 7900 | Agilent 7900 |
| Nebulizer | 200 µL PFA | PFA-ST microflow (400 μL min−1) |
| Cones | Ni plated (sample and skimmer) | Ni plated (sample and skimmer) |
| Sensitivity | >109 cps/ppm at <2% CeO | >109 cps/ppm at <2% CeO |
| RF power (W) | 1600 | 1450 |
| Spray chamber | Double pass | Cyclonic |
| Torch depth (mm) | 10 | 8 |
| Make-up gas (L/min) | 0.25 | 0 |
| He gas flow (mL/min) | 4.8 | 4.8 |
| H2 gas flow (mL/min) | 6 | 0 |
| Cool gas flow (L/min | 15 | 15 |
| Auxiliary gas flow (L/min) | 0.9 | 0.9 |
| Sample gas flow (L/min) | 0.95 | 1.07 |
| Sample uptake (s) | 18 | 100 |
| Sensitivity for 1 ppb Y (cps) | 45,000 | 32,000 |
| Oxide ratio | <0.3% | <0.3% |
| Isotope | Interference |
|---|---|
| 56Fe | 40Ar16O+ |
| 110Cd | 94Mo16O+; 110Pd |
| 111Cd | 95Mo16O+; 94Zr16O1H+ |
| 112Cd | 96Mo16O+; 40Ar216O2+; 96Ru16O+; 112Sn+ |
| 114Cd | 98Mo16O+; 98Ru16O+; 114Sn+ |
| Procedural Blank | Mn (nmol kg−1) | Fe (nmol kg−1) | Ni (nmol kg−1) | Cu (nmol kg−1) | Zn (nmol kg−1) | Co (pmol kg−1) | Cd (pmol kg−1) | Pb (pmol kg−1) |
|---|---|---|---|---|---|---|---|---|
| Online a (n = 5) | 0.007 ± 0.001 | 0.025 ± 0.005 | 0.021 ± 0.006 | 0.021 ± 0.005 | 0.067 ± 0.051 | 0.791 ± 0.122 | 0.451 ± 0.085 | 0.083 ± 0.032 |
| Online b (n = 77) | 0.018 ± 0.008 | 0.050 ± 0.020 | 0.052 ± 0.017 | 0.026 ± 0.017 | 0.260 ± 0.102 | 1.730 ± 0.910 | 0.671 ± 0.136 | 0.670 ± 0.440 |
| Offline a (n = 5) | 0.001 ± 0.001 | 0.023 ± 0.006 | 0.033 ± 0.004 | 0.086 ± 0.007 | 0.090 ± 0.008 | 0.687 ± 0.162 | 1.218 ± 0.296 | 0.218 ± 0.074 |
| Limit of detection | ||||||||
| Online a,c | 0.003 | 0.015 | 0.018 | 0.015 | 0.151 | 0.366 | 0.255 | 0.096 |
| Online b,c | 0.008 | 0.081 | 0.030 | 0.020 | 0.090 | 0.590 | 1.200 | 0.900 |
| Online b,d | 0.015 | 0.072 | 0.050 | 0.050 | 0.150 | 1.210 | 5.660 | 1.410 |
| Offline a,c | 0.001 | 0.019 | 0.011 | 0.020 | 0.024 | 0.485 | 0.888 | 0.221 |
| NASS-7 | Mn (nmol kg−1) | Fe (nmol kg−1) | Ni (nmol kg−1) | Cu (nmol kg−1) | Zn (nmol kg−1) | Co (pmol kg−1) | Cd (pmol kg−1) | Pb (pmol kg−1) |
|---|---|---|---|---|---|---|---|---|
| Certified | 13.47 ± 1.09 | 6.16 ± 0.47 | 4.14 ± 0.31 | 3.07 ± 0.22 | 6.27 ± 1.22 | 243.00 ± 24.00 | 142.00 ± 14.00 | 12.07 ± 3.86 |
| Online (n = 6) | 13.07 ± 0.07 | 5.98 ± 0.10 | 3.84 ± 0.13 | 2.90 ± 0.14 | 6.65 ± 0.44 | 228.00 ± 12.00 | 130.00 ± 2.10 | 11.35 ± 0.33 |
| Offline (n = 5) | 13.43 ± 0.78 | 5.77 ± 0.28 | 3.85 ± 0.10 | 3.11 ± 0.08 | 6.59 ± 0.07 | 261.42 ± 5.43 | 132.69 ± 3.08 | 15.19 ± 1.86 |
| GSP-62 | ||||||||
| Consensus | 0.78 ± 0.03 | 0.16 ± 0.05 | 2.60 ± 0.10 | 0.57 ± 0.05 | 0.03 ± 0.05 | 5.00 ± 0.70 | 2.00 ± 2.00 | 62.00 ± 5.00 |
| Online (n = 5) | 0.76 ± 0.04 | 0.21 ± 0.03 | 2.72 ± 0.15 | 0.63 ± 0.04 | b.d.l. | 6.00 ± 1.10 | 0.99 ± 0.35 a | 62.00 ± 8.00 |
| Offline (n = 5) | 0.73 ± 0.07 | 0.31 ± 0.08 | 2.44 ± 0.12 | 0.57 ± 0.02 | 0.10 ± 0.02 | 8.77 ± 4.75 | 4.07 ± 0.48 | 67.13 ± 3.91 |
| GSC 1-19 | ||||||||
| Consensus | 2.18 ± 0.08 | 1.54 ± 0.12 | 4.39 ± 0.21 | 1.10 ± 0.15 | 1.43 ± 0.10 | 81.70 ± 4.06 | 364.00 ± 22.00 | 39.00 ± 4.00 |
| Online (n = 5) | 2.17 ± 0.11 | 1.63 ± 0.10 | 4.59 ± 0.41 | 1.16 ± 0.11 | 1.46 ± 0.07 | 87.00 ± 6.80 | 366.00 ± 27.00 | 40.00 ± 5.00 |
| Offline (n = 5) | 2.01 ± 0.17 | 1.50 ± 0.07 | 3.93 ± 0.14 | 1.14 ± 0.04 | 1.41 ± 0.10 | 81.71 ± 4.06 | 345.00 ± 21.00 | 40.05 ± 1.79 |
| Offline | Mn (ng kg−1) | Fe (ng kg−1) | Ni (ng kg−1) | Cu (ng kg−1) | Zn (ng kg−1) | Co (ng kg−1) | Cd (ng kg−1) | Pb (ng kg−1) |
|---|---|---|---|---|---|---|---|---|
| SOSW | 23.21 ± 1.11 | 17.79 ± 0.07 | 379.59 ± 0.21 | 92.32 ± 0.21 | 543.66 ± 5.74 | 0.54 ± 0.03 | 98.39 ± 1.75 | 1.91 ± 0.01 |
| SOSW + spike (n = 5) | 232.70 ± 7.24 | 228.21 ± 5.15 | 598.46 ± 15.47 | 309.47 ± 6.53 | 746.84 ± 11.54 | 206.64 ± 4.73 | 299.13 ± 4.99 | 207.20 ± 2.99 |
| Spike recovery | 209.49 | 210.41 | 218.88 | 217.15 | 203.18 | 206.1 | 200.75 | 205.28 |
| Spike recovery (%) | 105 | 105 | 109 | 108 | 101 | 103 | 100 | 102 |
| Online | ||||||||
| SOSW | 11.50 ± 0.55 | 10.10 ± 0.97 | 258.90 ± 2.21 | 18.31 ± 1.02 | 10.28 ± 2.22 | 0.81 ± 0.02 | 34.90 ± 1.22 | 1.54 ± 0.01 |
| SOSW + spike (n = 5) | 174.50 ± 2.32 | 169.00 ± 3.52 | 424.10 ± 9.32 | 180.21 ± 8.55 | 178.12 ± 5.52 | 66.07 ± 2.21 | 99.10 ± 2.23 | 159.50 ± 3.22 |
| Spike recovery | 163.00 | 158.90 | 165.20 | 161.90 | 167.84 | 65.27 | 64.21 | 157.96 |
| Spike recovery (%) | 101 | 98 | 102 | 100 | 104 | 101 | 100 | 98 |
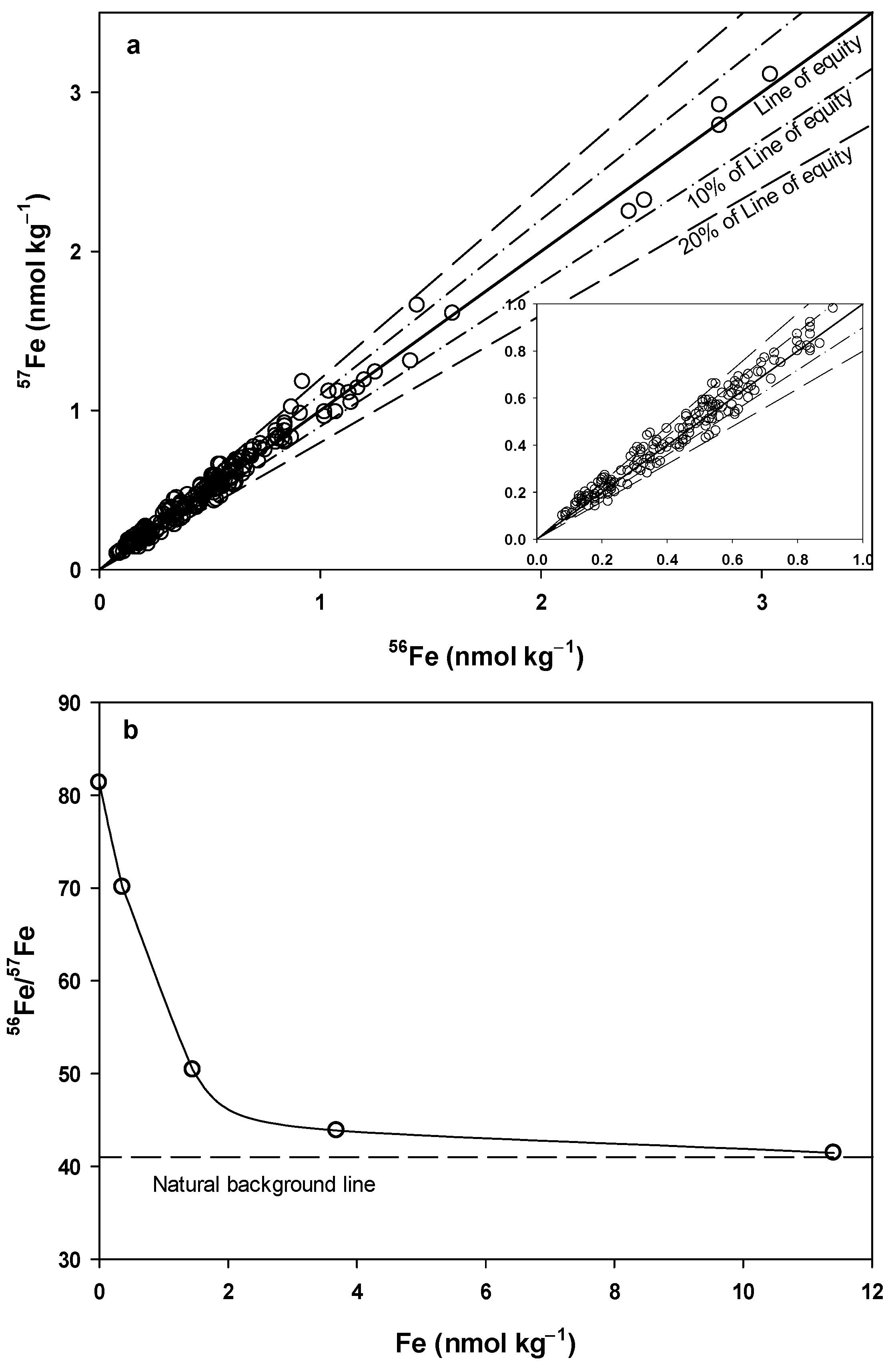
| Ratio of Instrumental Signal | Procedural Blank | Seawater | Seawater (Blank Subtracted) | Natural Abundance |
|---|---|---|---|---|
| 56Fe/57Fe | 84.5 ± 19.9 | 52.0 ± 4.5 | 42.9 ± 4.1 | 43.2 |
| (n = 77) | (n > 200) | (n > 200) | ||
| 111Cd/112Cd | 0.20 ± 0.09 | 0.51 ± 0.01 | 0.52 ± 0.03 | 0.52 |
| (n = 77) | (n > 200) a | (n > 200) a | ||
| 0.40 ± 0.08 | 0.43 ± 0.04 | |||
| (n > 20) b | (n > 20) b |
| Material | 110Cd/111Cd | 110Cd/112Cd | 111Cd/112Cd | 114Cd/112Cd |
|---|---|---|---|---|
| Natural abundance | 0.97 | 0.52 | 0.53 | 1.19 |
| Solution | ||||
| 1% HCl | 1.66 ± 0.45 | 0.33 ± 0.06 | 0.23 ± 0.05 | 0.98 ± 0.08 |
| dCd-free solution | ||||
| Mo (13.5 nmol kg−1) | 0.63 ± 0.12 | 0.52 ± 0.10 | 0.83 ± 0.01 | 1.49 ± 0.11 |
| Mo (53.3 nmol kg−1) a | 0.53 | 0.49 | 0.83 | 1.56 |
| Mo (117.2 nmol kg−1) | 0.56 ± 0.03 | 0.52 ± 0.10 | 0.83 ± 0.01 | 1.49 ± 0.11 |
| Mo (248.9 nmol kg−1) | 0.56 ± 0.03 | 0.50 ± 0.01 | 0.88 ± 0.04 | 1.62 ± 0.03 |
| Mo (543.5 nmol kg−1) | 0.56 ± 0.01 | 0.50 ± 0.01 | 0.90 ± 0.01 | 1.68 ± 0.01 |
| Mo (1006.7 nmol kg−1) a | 0.56 | 0.50 | 0.90 | 1.63 |
| Reference standard | ||||
| GSP (dCd: 2 ± 2 pmol kg−1) | 0.55 ± 0.01 | 0.48 ± 0.01 | 0.83 ± 0.03 | 1.57 ± 0.06 |
| GSC (dCd: 364 ± 22 pmol kg−1) | 0.90 | 0.47 ± 0.01 | 0.52 ± 0.01 | 1.30 ± 0.01 |
| Instrumental parameter/Element | This Study | Rapp et al. | Wuttig et al. | Jackson et al. | Strivens et al. | Vassileva et al. | |||
|---|---|---|---|---|---|---|---|---|---|
| Instrument | Q-ICPMS | Q-ICPMS | SF-ICPMS | SF-ICPMS | QQQ-ICPMS | iCapQ ICP-MS | ICP-SFMS | ||
| Precon. module | seaFAST pico | seaFAST S3 | seaFAST pico | seaFAST S2 | seaFAST pico | seaFAST 2 | seaFAST pico | ||
| Configuration | offline | online | offline | Offline | Offline | Online | Offline | ||
| Blank sol. | 1% HCl | 2% HNO3 | HCl | 0.2% HNO3 | 0.2% HCl | 2%HNO3 | |||
| Precon. factor | 40 | 30 | 10 | 40 | 8 and 16 | 25 | |||
| Short-term (n = 5) | Short-term (n = 5) | Long-term (n = 77) | Long-term (n = 116) | Mid-term (n = 20) | |||||
| Mn | Blank | 0.001 ± 0.001 | 0.007 ± 0.001 | 0.018 ± 0.008 | 0.014 ± 0.003 | - | 0.006 | - | 0.009 ± 0.002 |
| LOD | 0.001 | 0.003 | 0.008 | 0.016 | 0.007 | 0.002 | - | 0.018 | |
| Fe | Blank | 0.023 ± 0.006 | 0.025 ± 0.005 | 0.05 ± 0.020 | 0.068 ± 0.010 | - | 0.14 | - | - |
| LOD | 0.019 | 0.015 | 0.081 | 0.029 | 0.060 | 0.290 | - | - | |
| Ni | Blank | 0.033 ± 0.004 | 0.021 ± 0.006 | 0.052 ± 0.017 | 0.111 ± 0.020 | - | 0.053 | - | 0.034 ± 0.014 |
| LOD | 0.011 | 0.018 | 0.030 | 0.059 | 0.090 | 0.030 | 0.249 | 0.068 | |
| Cu | Blank | 0.086 ± 0.007 | 0.021 ± 0.005 | 0.026 ± 0.017 | 0.014 ± 0.006 | - | 0.030 | - | 0.034 ± 0.008 |
| LOD | 0.020 | 0.015 | 0.020 | 0.009 | 0.040 | 0.008 | 0.122 | 0.047 | |
| Zn | Blank | 0.090 ± 0.008 | 0.067 ± 0.051 | 0.260 ± 0.102 | 0.030 ± 0.009 | - | 0.025 | - | 0.107 ± 0.030 |
| LOD | 0.024 | 0.151 | 0.090 | 0.028 | 0.120 | 0.017 | 0.194 | 0.061 | |
| Co | Blank | 0.687 ± 0.162 | 0.791 ± 0.122 | 1.730 ± 0.910 | 2.700 ± 0.800 | - | - | - | 0.509 ± 0.051 |
| LOD | 0.485 | 0.366 | 0.590 | 2.500 | 1.000 | - | - | 1.697 | |
| Cd | Blank | 1.218 ± 0.296 | 0.451 ± 0.085 | 0.671 ± 0.136 | 2.200 ± 0.300 | - | 0.34 | - | 0.361 ± 0.090 |
| LOD | 0.888 | 0.255 | 1.200 | 0.800 | 1.000 | 0.600 | 8.797 | 0.541 | |
| Pb | Blank | 0.218 ± 0.074 | 0.083 ± 0.032 | 0.670 ± 0.440 | 0.400 ± 0.200 | - | 0.74 | - | 0.003 ± 0.001 |
| LOD | 0.221 | 0.096 | 0.900 | 0.600 | 1.000 | 0.300 | 1.883 | 4.826 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samanta, S.; Cloete, R.; Loock, J.; Rossouw, R.; Roychoudhury, A.N. Determination of Trace Metal (Mn, Fe, Ni, Cu, Zn, Co, Cd and Pb) Concentrations in Seawater Using Single Quadrupole ICP-MS: A Comparison between Offline and Online Preconcentration Setups. Minerals 2021, 11, 1289. https://doi.org/10.3390/min11111289
Samanta S, Cloete R, Loock J, Rossouw R, Roychoudhury AN. Determination of Trace Metal (Mn, Fe, Ni, Cu, Zn, Co, Cd and Pb) Concentrations in Seawater Using Single Quadrupole ICP-MS: A Comparison between Offline and Online Preconcentration Setups. Minerals. 2021; 11(11):1289. https://doi.org/10.3390/min11111289
Chicago/Turabian StyleSamanta, Saumik, Ryan Cloete, Jean Loock, Riana Rossouw, and Alakendra N. Roychoudhury. 2021. "Determination of Trace Metal (Mn, Fe, Ni, Cu, Zn, Co, Cd and Pb) Concentrations in Seawater Using Single Quadrupole ICP-MS: A Comparison between Offline and Online Preconcentration Setups" Minerals 11, no. 11: 1289. https://doi.org/10.3390/min11111289
APA StyleSamanta, S., Cloete, R., Loock, J., Rossouw, R., & Roychoudhury, A. N. (2021). Determination of Trace Metal (Mn, Fe, Ni, Cu, Zn, Co, Cd and Pb) Concentrations in Seawater Using Single Quadrupole ICP-MS: A Comparison between Offline and Online Preconcentration Setups. Minerals, 11(11), 1289. https://doi.org/10.3390/min11111289