Common Problems and Pitfalls in Fluid Inclusion Study: A Review and Discussion


Abstract
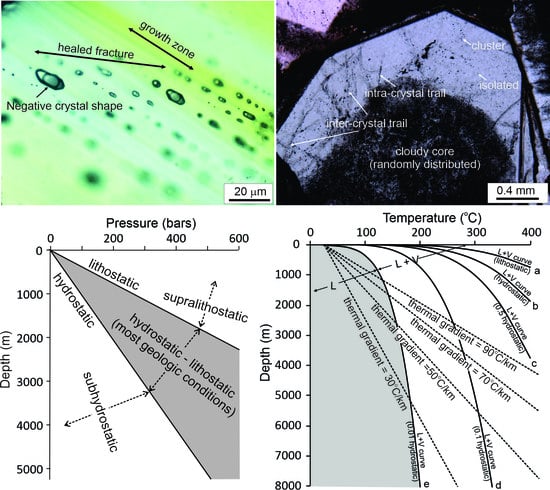
{kind=link}
{kind=link}
{kind=link}
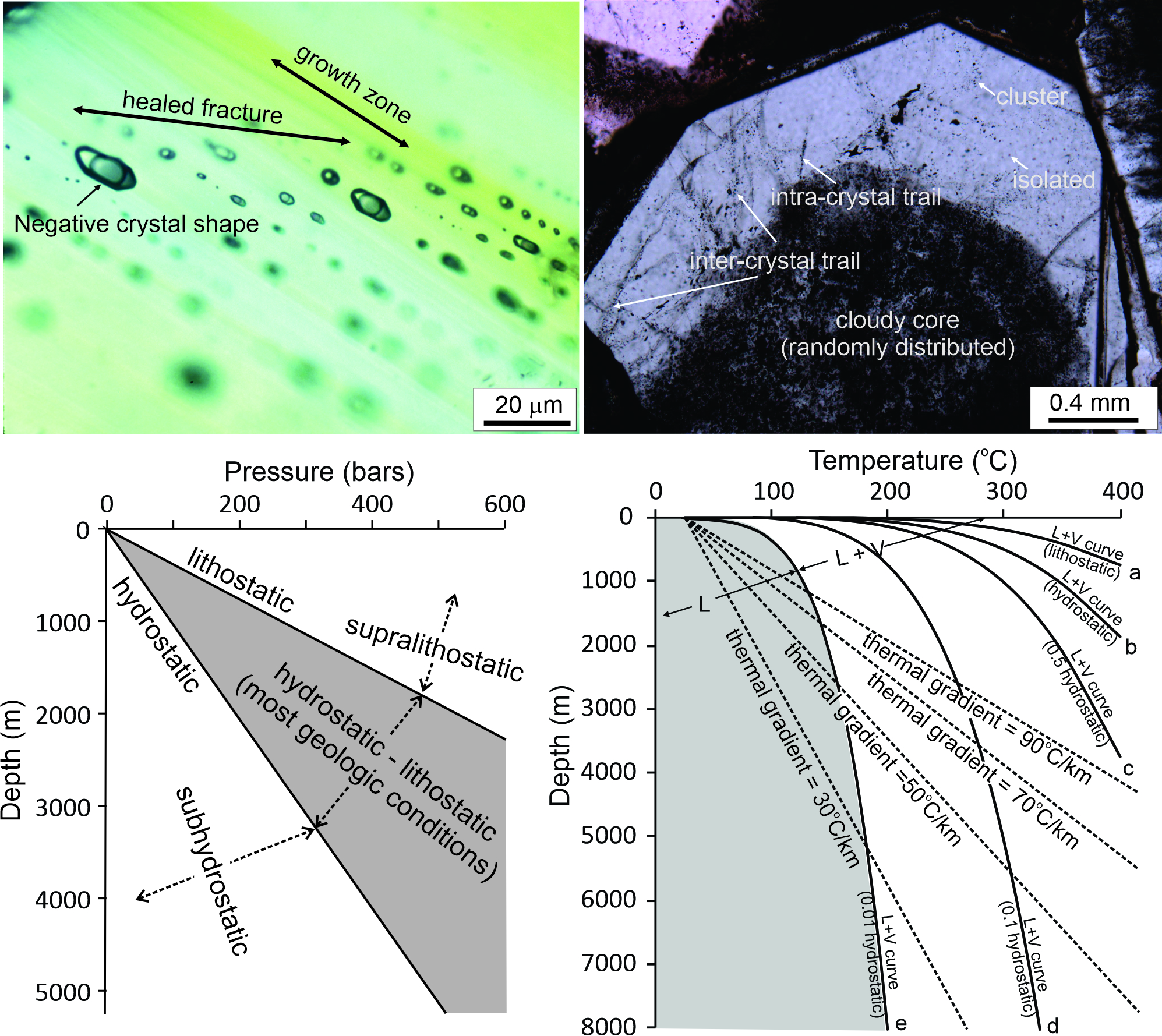
{kind=link}
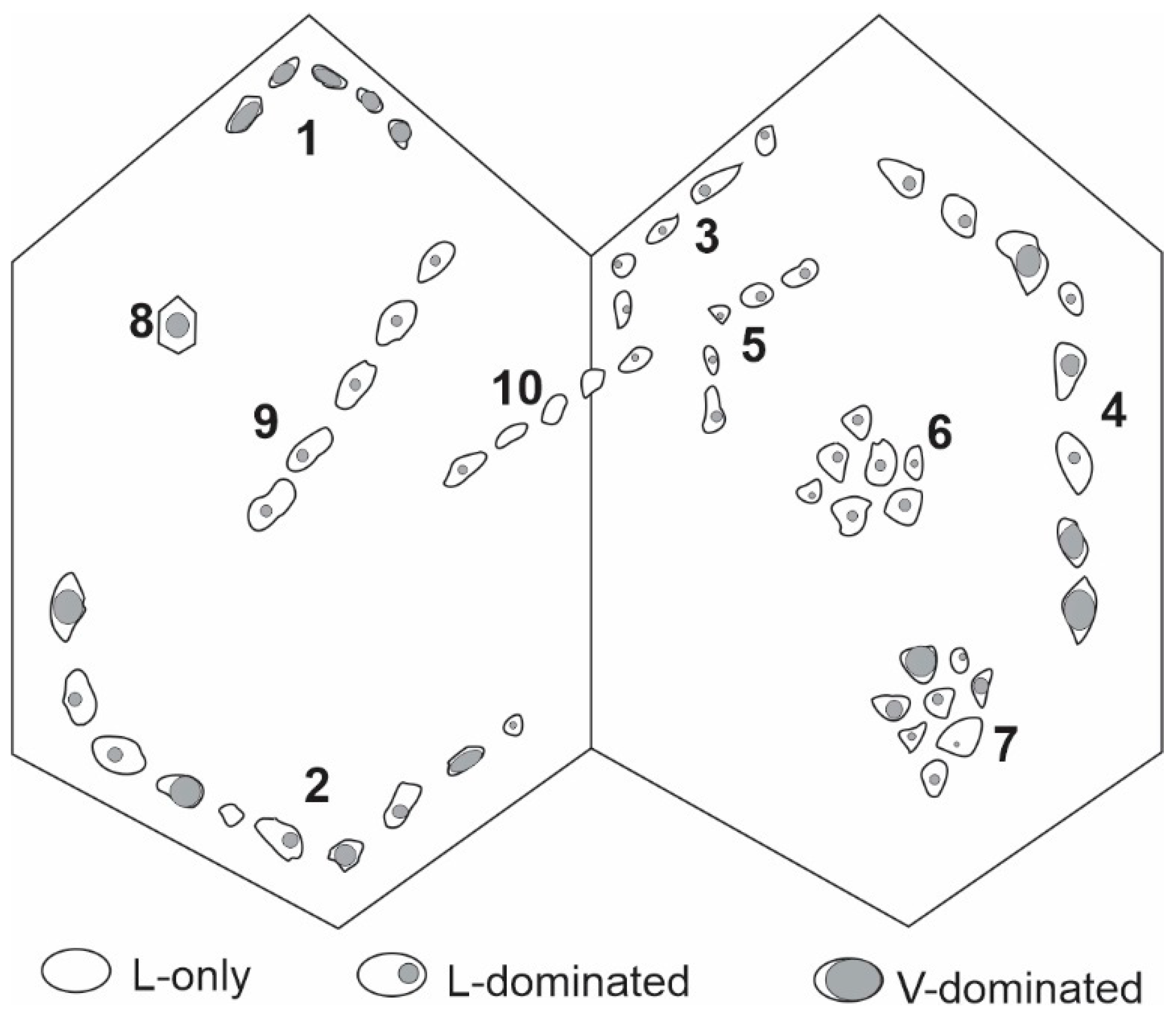
{kind=link}
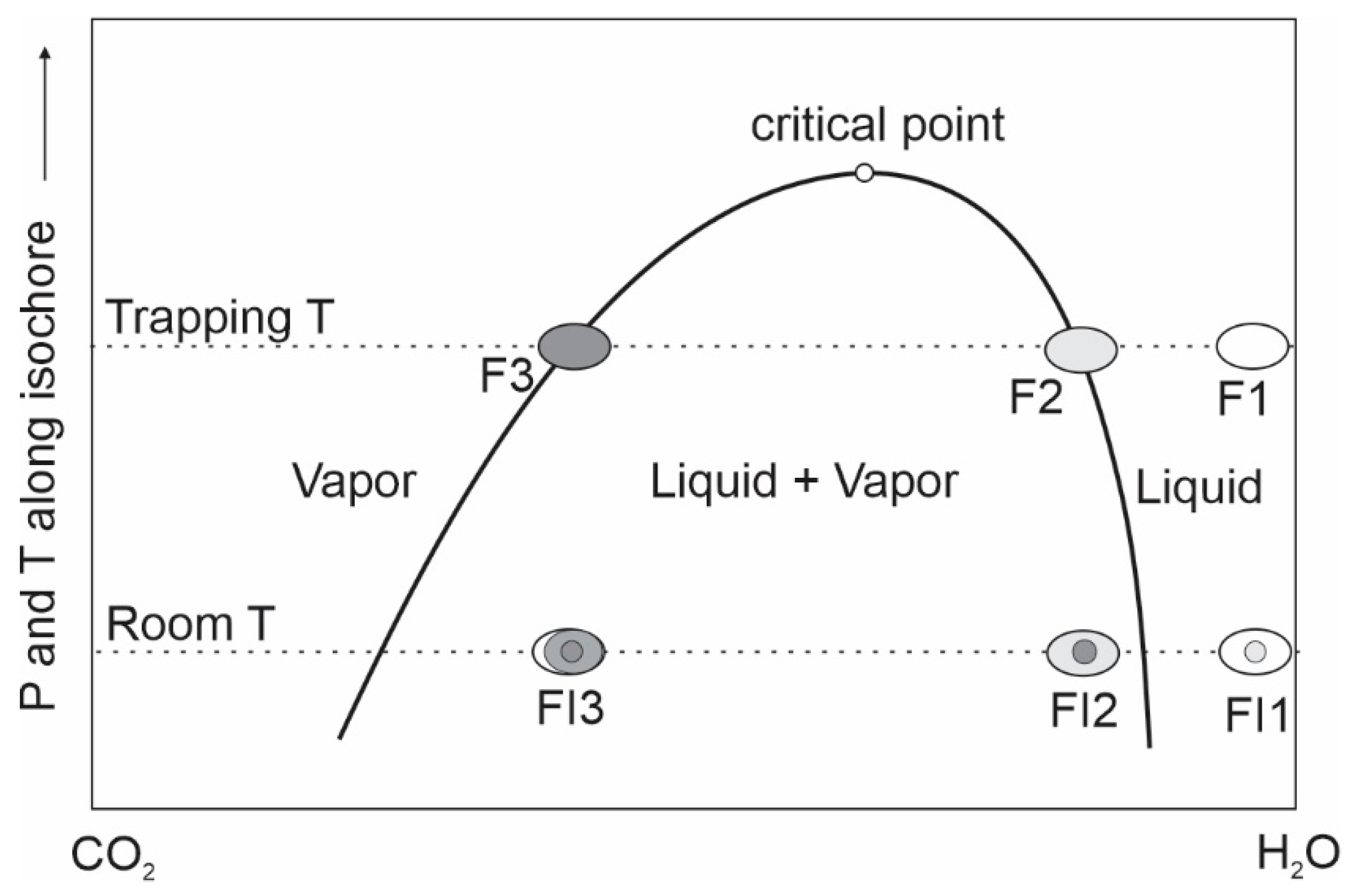
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Problems Related to Fluid Inclusion Petrography
2.1. Problem #1: Study of Fluid Inclusions without Determining the Paragenetic Position of the Host Mineral
2.2. Problem #2: Assigning Fluid Inclusions as “Primary” without Describing Their Actual Mode of Occurrence
2.3. Problem #3: Non-Use and Misuse of the ‘Fluid Inclusion Assemblage’ (FIA) Concept
3. Problems Related to Metastability
3.1. Problem #4: Misunderstanding of the Nature of First Melting and Misuse of ‘Eutectic Temperature’
3.2. Problem #5: Ignoring Liquid-Only Inclusions without Justification
3.3. Problem #6: Measurement of Ice Melting Temperature without the Presence of Vapor
4. Problems Related to Fluid Phase Relationships
4.1. Problem #7: Classifying Fluid Inclusions without Specifying If They Homogenize to Liquid or Vapor Phase
4.2. Problem #8: Treating Multiple Types of Fluid Inclusions as “Coexisting” without Considering If This Is Compatible with Fluid Phase Equilibria
4.3. Problem #9: Ambiguity Regarding Fluid Immiscibility and Its Recognition
4.4. Problem #10: Assigning Solid Phases in Fluid Inclusions as Daughter Minerals without Justification
5. Problems Related to Fluid Temperature and Pressure Calculation and Interpretation
5.1. Problem #11: Underestimation of the Complexity of the Wide Range of Homogenization Temperatures
5.2. Problem #12: Underestimation of Uncertainties of Fluid Pressure Calculation
5.3. Problem #13: Underestimation of Uncertainties in Depth Estimation
6. Problems Related to Bulk Fluid Inclusion Analysis
Problem #14: Lack of Petrographic and Microthermometric Evidence to Justify the Suitability of Bulk Fluid Inclusion Analysis and Interpretation of the Results
7. Problems Related to Data Presentations
7.1. Problem #15: Lack of Objective and Detailed Description of Raw Data
7.2. Problem #16: Skewed Representation of Fluid Inclusion Data
8. Concluding Remarks
- Paragenetic study is essential for any fluid inclusion study. Conducting a fluid inclusion study without knowing the paragenetic position of the host mineral may lead to serious problems in data interpretation.
- It is often difficult to unambiguously classify fluid inclusions as primary. Assigning fluid inclusions as “primary” without describing the supporting textural evidence may create problems in the interpretation of data.
- It is important to use the ‘fluid inclusion assemblage’ (FIA) approach as much as possible to select fluid inclusions for study. Even if individual FIAs cannot be determined unambiguously, the FIA concept should be applied to constrain the validity of the microthermometric data and their interpretations.
- The first melting temperature should not be described as the “eutectic temperature”, because in many cases, fluid inclusions do not freeze completely even at the temperature of liquid nitrogen. The terms “first melting” or “apparent eutectic” are descriptive and therefore preferred.
- Liquid-only inclusions should not be excluded from study simply because they cannot yield microthermometric data. In the case of vadose-zone fluid entrapment, liquid-only inclusions are the only inclusions that can provide valid P–T information about the paleofluids. In some other low-temperature environments such as early diagenesis, liquid-only inclusions may truly record the P–T conditions of trapping, whereas biphase inclusions may have resulted from post-entrapment modification.
- Final ice-melting temperatures measured without the presence of the vapor phase cannot be used to calculate fluid salinity due to metastability.
- It is important to specify if a fluid inclusion homogenizes into the liquid or vapor phase when classifying fluid inclusions and recording their phase transition temperatures. Homogenization into liquid or vapor has very different implications for fluid density and pressure.
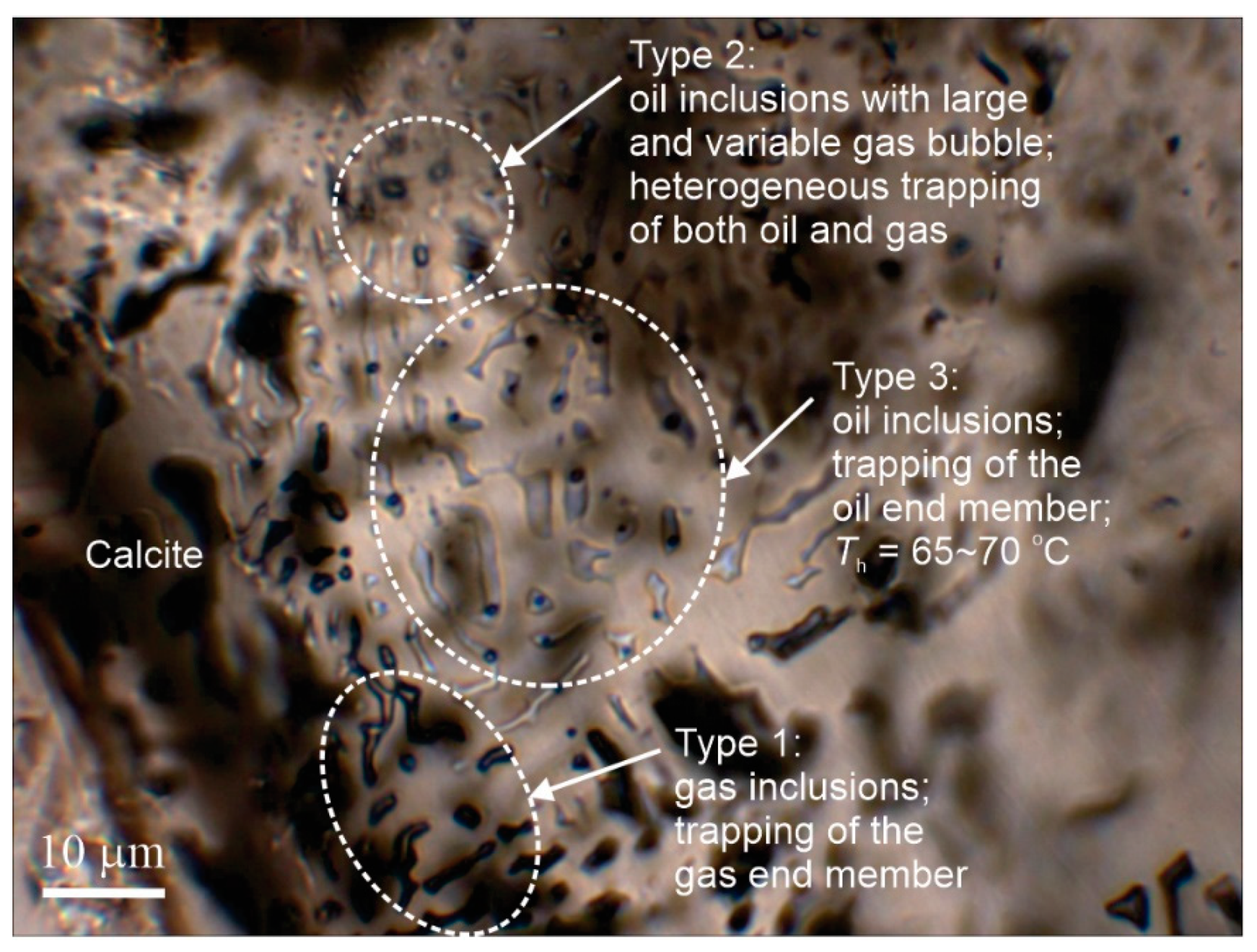
- The occurrence of multiple types of fluid inclusions in a mineral should not be simply described as “coexistence” of these types of inclusions even if they all appear to be “primary”. An examination of the compatibility of these types of fluid inclusions in terms of phase equilibria is required, and whether or not these different types of fluid inclusions can be all coeval should be evaluated accordingly.
- Fluid boiling is used to describe the process of phase change from liquid to liquid + vapor for single-volatile fluid systems, whereas fluid immiscibility describes the state of coexistence of liquid and vapor for both single-volatile and multi-volatile systems. Heterogenous trapping, which produces liquid-dominated and vapor-dominated inclusions with variable homogenization temperatures, is good evidence for fluid immiscibility, but in such FIAs, only inclusions that can be proven to have trapped end-member liquid or end-member vapor yield meaningful homogenization temperatures.
- Solid phases in fluid inclusions may be daughter minerals or accidently entrapped solids. Even if they are soluble salt crystals, they should not be automatically interpreted as daughter minerals. The best evidence for the presence of daughter minerals is if they have uniform phase proportions in all inclusions within an individual FIA (and hence they have very similar melting temperatures).
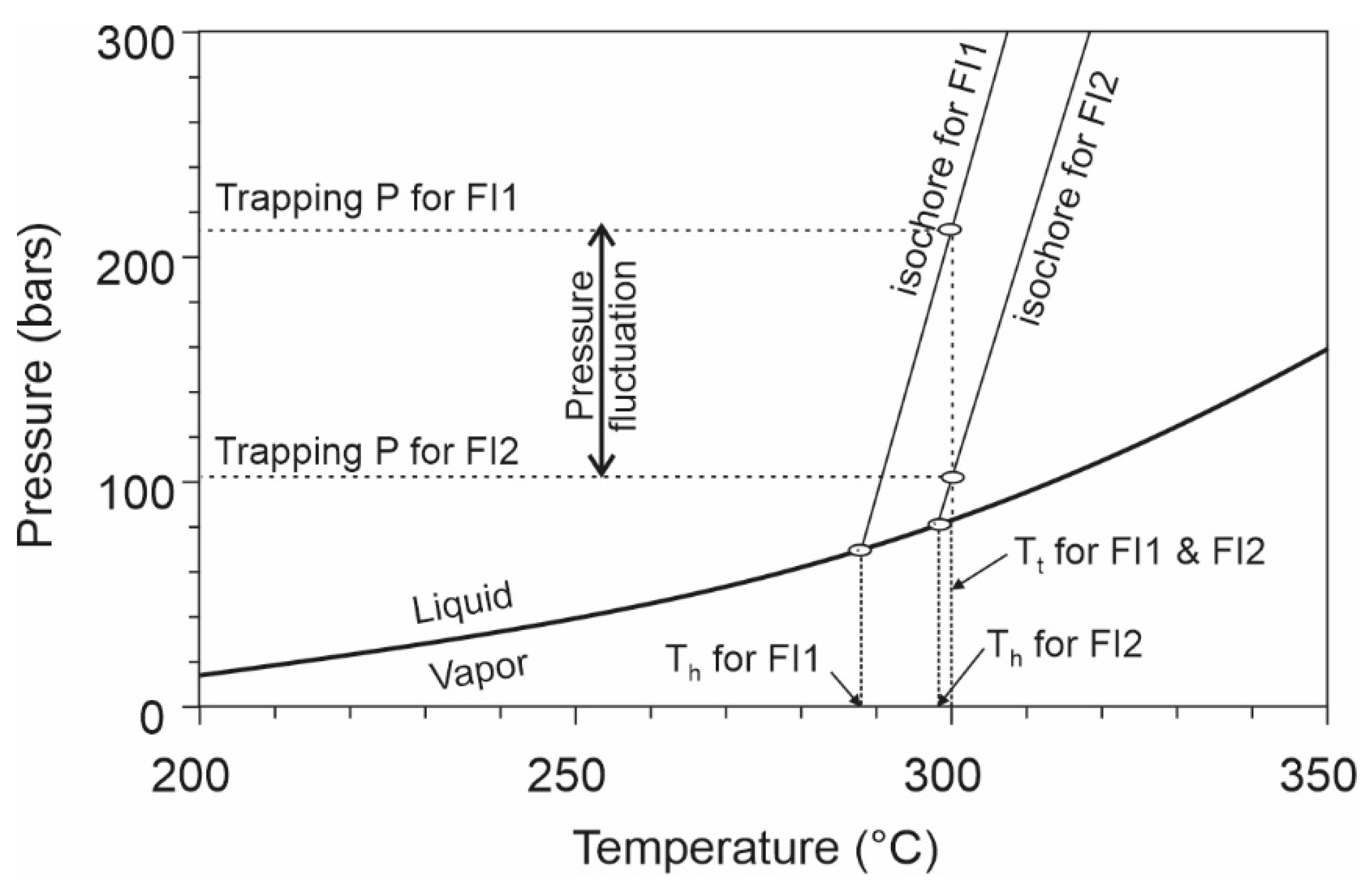
- The wide range of homogenization temperatures documented in many studies may be partly attributed to artifacts and partly to real P–T fluctuation. The common artifacts include failure to recognize different generations of fluid inclusions, heterogeneous trapping and post-entrapment modifications (e.g., necking down through a phase boundary, stretching, deformation of the host crystal). Even after the exclusion of the artifacts, the range of homogenization temperatures should not be simply considered as reflecting the variation of fluid temperature, as pressure fluctuation can also result in variation of homogenization temperatures.
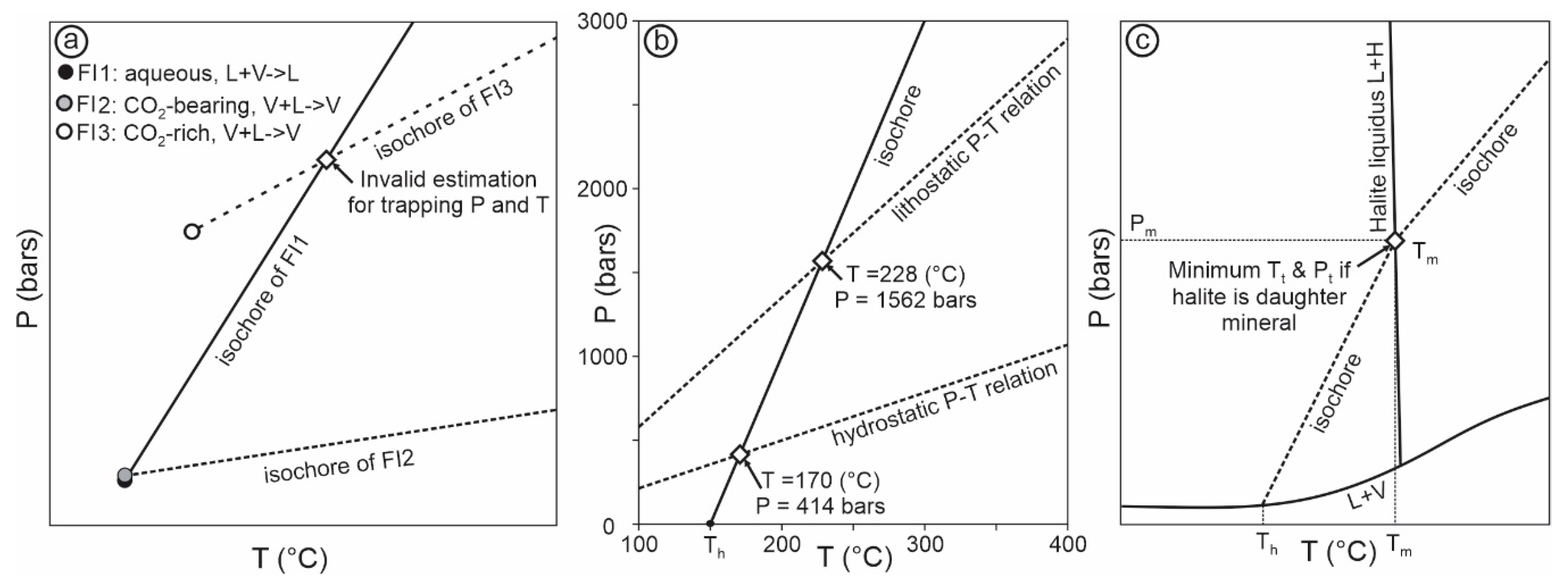
- Although fluid pressure is one of the most important parameters that one may aim to estimate from fluid inclusion studies, the uncertainties of fluid pressure calculation are generally higher than assumed. The uncertainties include those inherent to the chosen equation-of-state, the sensitivity of pressure to temperature as governed by the equations, and those involved in the estimation of the trapping temperatures. It is important to know the limit of the various fluid pressure calculation methods in order to avoid overinterpretation of the meaning of the fluid pressure values.
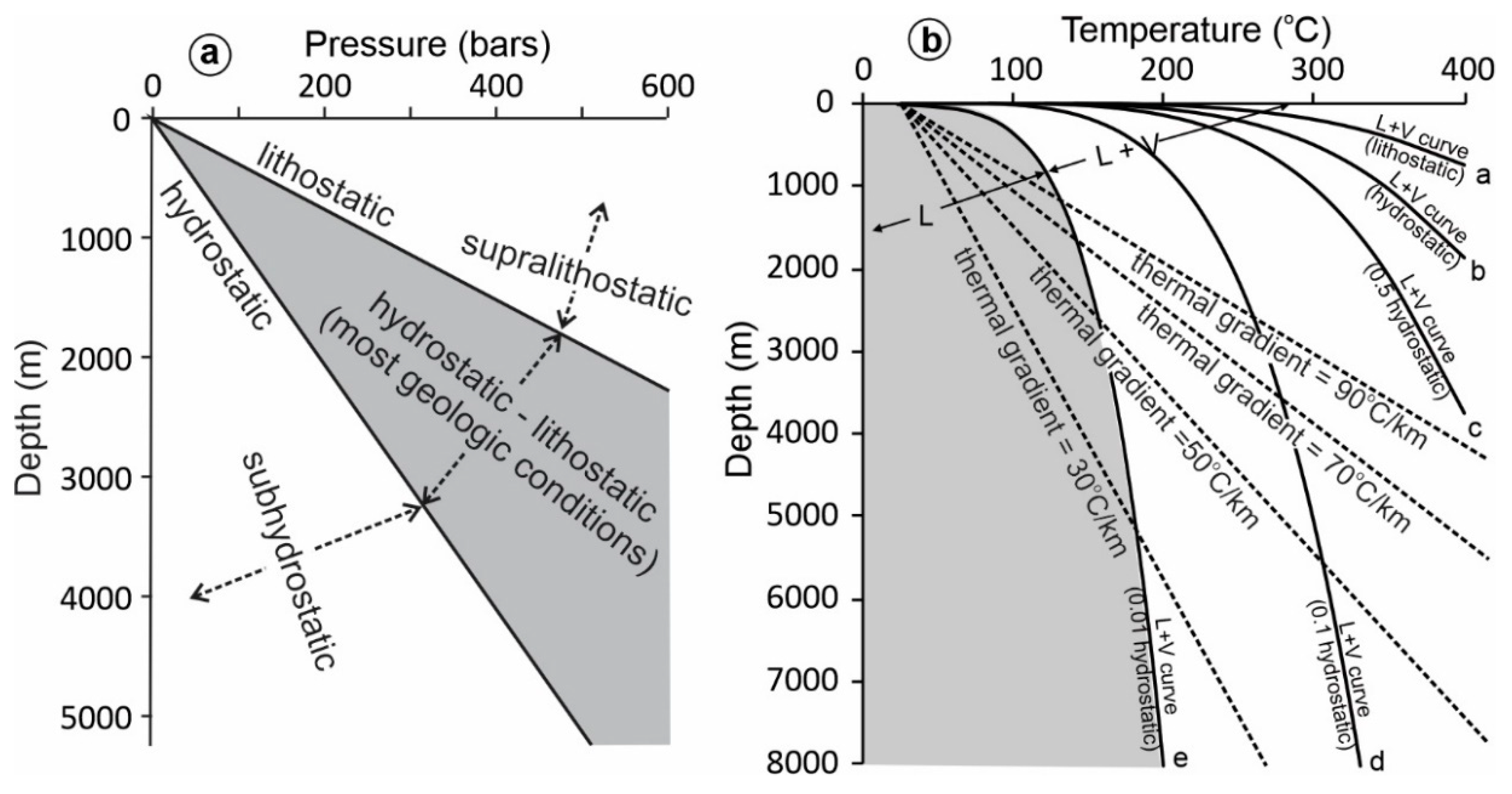
- The ultimate purpose of fluid pressure calculation is often to estimate the depth. However, it should be emphasized that even if fluid pressure has been constrained with confidence, it is not straightforward to calculate depth from fluid pressure. Much of the uncertainty comes from the difficulty in determining the fluid pressure regime, which may vary from subhydrostatic to supralithostatic.
- Bulk fluid inclusion analyses are useful methods for determining the compositions of paleofluids. However, the meaningful application of these methods requires that the sample contains a single or at least a dominant generation of fluid inclusions that are of interest. This requirement should not be neglected in sample preparation and data interpretation.
- Although raw data may not be accommodated in a publication, they should be provided in supplementary material. Detailed information about fluid inclusion occurrences and phase changes at homogenization should be described. The number of digits after the decimal point should reflect the true precision of the microthermometric data. Fluid inclusion data should not be simply treated as any geochemical data in a statistical approach. The quality of the data is more important than the quantity. The average value rather than that of each inclusion within an individual FIA should be used in diagrams in order to avoid overrepresentation (some FIA allow many more measurements than others), and the significance of the range of a parameter should not be masked by the average or peak in a diagram.
- Despite the many potential problems, fluid inclusion study remains an indispensable method for studying paleofluids. Knowing the potential problems and taking steps to avoid them or minimize their impact are critical for a successful fluid inclusion study.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sorby, H.C. On the microscopical structure of crystals, indicating the origin of minerals and rocks. Geol. Soc. London Quart. J. 1858, 14, 453–500. [Google Scholar] [CrossRef]
- Hollister, L.S.; Crawford, M.L. (Eds.) Fluid Inclusions-Applications in Petrology; Mineralogical Association of Canada Publications: Québec, QC, Canada, 1981; Volume 6, p. 304. [Google Scholar]
- Roedder, E. Fluid inclusions. Geol. Soc. Am. Bull. 1984, 12, 644. [Google Scholar]
- Shepherd, T.J.; Rankin, A.H.; Alderton, D.H.M. A Practical Guide to Fluid Inclusion Studies; Blackie: Glasgow, Scotland, UK, 1985; p. 239. [Google Scholar]
- Anderson, T.; Frezzotti, M.-L.; Burke, E.A.J. Editorial-A tribute to Jacques Touret. Lithos 2001, 55, ix–xi. [Google Scholar] [CrossRef]
- Samson, I.; Anderson, A.; Marshall, D. (Eds.) Fluid Inclusions. Analysis and Interpretation; Mineralogical Association of Canada Publications: Québec, QC, Canada, 2003; Volume 32, p. 374. [Google Scholar]
- Lu, H.; Fan, H.; Ni, P.; Ou, G.; Shen, K.; Zhang, W. Fluid Inclusions; Science Press: Beijing, China, 2004; p. 487. (In Chinese) [Google Scholar]
- Hurai, V.; Huraiová, M.; Slobodník, M.; Thomas, R. Geofluids: Developments in Microthermometry, Spectroscopy, Thermodynamics, and Stable Isotopes; Elsevier: Amsterdam, The Netherlands, 2015; p. 489. [Google Scholar]
- Roedder, E.; Bodnar, R.J. Fluid inclusion studies of hydrothermal deposits. In Geochemistry of Hydrothermal Ore Deposits, 3rd ed.; Barnes, H.L., Ed.; John Wiley & Sons: New York, NY, USA, 1997; pp. 657–698. [Google Scholar]
- Wilkinson, J.J. Fluid inclusions in hydrothermal ore deposits. Lithos 2001, 55, 229–272. [Google Scholar] [CrossRef]
- Bodnar, R.J.; Lecumberri-Sanchez, P.; Moncada, D.; Steele-MacInnis, M. Fluid inclusions in hydrothermal ore deposits. In Treatise on Geochemistry, 2nd ed.; Holland, H.D., Turekian, K.K., Eds.; Elsevier: Oxford, UK, 2014; Volume 13, pp. 119–142. [Google Scholar]
- Chi, G. Constraints from fluid inclusion studies on hydrodynamic models of mineralization. Acta. Petrol. Sin. 2015, 31, 907–917. [Google Scholar]
- Roedder, E.; Bodnar, R.J. Geologic pressure determinations from fluid inclusion studies. An. Rev. Earth Planet Sci. 1980, 8, 263–301. [Google Scholar] [CrossRef]
- Goldstein, R.H.; Reynolds, T.J. Systematics of Fluid Inclusions in Diagenetic Minerals. Soc. Sed. Geol. Short Course 1994, 31, 199. [Google Scholar]
- Goldstein, R.H. Fluid inclusions in sedimentary and diagenetic systems. Lithos 2001, 55, 159–193. [Google Scholar] [CrossRef]
- Gagnon, J.E.; Samson, I.M.; Fryer, B.J. LA-ICP-MS analysis of fluid inclusions. In Fluid Inclusions: Analysis and Interpretation; Samson, I., Anderson, A., Marshall, D., Eds.; Mineralogical Association of Canada Publications: Québec, QC, Canada, 2003; Volume 32, pp. 291–322. [Google Scholar]
- Frezzotti, M.L.; Tecce, F.; Casagli, A. Raman spectroscopy for fluid inclusion analysis. J. Geochem. Explor. 2012, 112, 1–20. [Google Scholar] [CrossRef]
- Van den Kerkhof, A.M.; Hein, U. Fluid inclusion petrography. Lithos 2001, 55, 27–47. [Google Scholar] [CrossRef]
- Bodnar, R.J. Introduction to fluid inclusions. In Fluid Inclusions: Analysis and Interpretation; Samson, I., Anderson, A., Marshall, D., Eds.; Mineralogical Association of Canada Publications: Québec, QC, Canada, 2003; Volume 32, pp. 1–8. [Google Scholar]
- Lambrecht, G.; Diamond, L.W. Morphological ripening of fluid inclusions and coupled zone-refining in quartz crystals revealed by cathodoluminescence imaging: Implications for CL-petrography, fluid inclusion analysis and trace-element geothermometry. Geochim. Cosmochim. Acta 2014, 141, 381–406. [Google Scholar] [CrossRef]
- Diamond, L.W.; Tarantola, A. Interpretation of fluid inclusions in quartz deformed by weak ductile shearing: Reconstruction of differential stress magnitudes and pre-deformation fluid properties. Earth Planet. Sci. Lett. 2015, 417, 107–119. [Google Scholar] [CrossRef]
- Diamond, L.W. Fluid inclusion evidence for P-V-T-X evolution of hydrothermal solution in late-Alpine gold-quartz veins at Brusson, Val d’Ayas, northwest Italian Alps. Am. J. Sci. 1990, 290, 912–958. [Google Scholar] [CrossRef]
- Rabiei, M.; Chi, G.; Normand, C.; Davis, W.J.; Fayek, M.; Blamey, N. Hydrothermal REE (xenotime) mineralization at Maw Zone, Athabasca Basin, Canada, and its relationship with unconformity-related uranium deposits. Econ. Geol. 2017, 112, 1483–1507. [Google Scholar] [CrossRef]
- Fall, A.; Bodnar, R.J. How precisely can the temperature of a fluid event be constrained using fluid inclusions? Econ. Geol. 2018, 113, 1817–1843. [Google Scholar] [CrossRef]
- Wang, K.; Chi, G.; Bethune, K.M.; Li, Z.; Blamey, N.; Card, C.; Potter, E.G.; Liu, Y. Fluid P-T-X characteristics and evidence for boiling in the formation of the Phoenix uranium deposit (Athabasca Basin, Canada): Implications for unconformity-related uranium mineralization mechanisms. Ore Geol. Rev. 2018, 101, 122–142. [Google Scholar] [CrossRef]
- Touret, J.L.R. Fluids in metamorphic rocks. Lithos 2001, 55, 1–25. [Google Scholar] [CrossRef]
- Chi, G.; Lu, H. Validation and representation of fluid inclusion microthermometric data using the fluid inclusion assemblage (FIA) concept. Acta. Petrol. Sin. 2008, 9, 1945–1953. [Google Scholar]
- Roedder, E. Changes in ore fluid with time, from fluid inclusion studies at Creede, Colorado. In Proceedings of the IAGOD 4th Symposium, Varna, Bulgaria, 19–25 September 1977; pp. 179–185. [Google Scholar]
- Haynes, F.M. Determination of fluid inclusion compositions by sequential freezing. Econ. Geol. 1985, 80, 1436–1439. [Google Scholar] [CrossRef]
- Diamond, L.W. Stability of CO2-clathrate-hydrate + CO2 liquid + CO2 vapour + aqueous KCl-NaCl solutions: Experimental determination and application to salinity estimates of fluid inclusions. Geochim. Cosmochim. Acta 1992, 56, 273–280. [Google Scholar] [CrossRef]
- Steele-MacInnis, M.; Bodnar, R.J.; Naden, J. Numerical mode to determine the composition of H2O-NaCl-CaCl2 fluid inclusions based on microthermometric and microanalytical data. Geochim. Cosmochim. Acta 2011, 75, 21–40. [Google Scholar] [CrossRef]
- Samson, I.M.; Walker, R.T. Cryogenic Raman spectroscopic studies in the system NaCl-CaCl2-H2O and implications for low-temperature phase behavior in aqueous fluid inclusions. Can. Mineral. 2000, 38, 35–43. [Google Scholar] [CrossRef]
- Baumgartner, M.; Bakker, R.J. CaCl2-hydrate nucleation in synthetic fluid inclusions. Chem. Geol. 2009, 265, 335–344. [Google Scholar] [CrossRef]
- Chu, H.; Chi, G.; Chou, I.-M. Freezing and melting behaviors of H2O-NaCl-CaCl2 solutions in fused silica capillaries and glass-sandwiched films: Implications for fluid inclusion studies. Geofluids 2016, 16, 518–532. [Google Scholar] [CrossRef]
- Davis, D.W.; Lowenstein, T.K.; Spencer, R.J. Melting behavior of fluid inclusions in laboratory-grown halite crystals in the systems NaCl-H2O, NaCl-KCl-H2O, NaCl-MgCl2-H2O, and NaCl-CaCl2-H2O. Geochim. Cosmochim. Acta 1990, 54, 591–601. [Google Scholar] [CrossRef]
- Diamond, L.W. Systematics of H2O inclusions. In Fluid Inclusions: Analysis and Interpretation; Samson, I., Anderson, A., Marshall, D., Eds.; Mineralogical Association of Canada Publications: Québec, QC, Canada, 2003; Volume 32, pp. 55–79. [Google Scholar]
- Kruger, Y.; Stoller, P.; Ricka, J.; Frenz, M. Femtosecond lasers in fluid-inclusion analysis: Overcoming metastable phase states. Eur. J. Miner. 2007, 19, 693–706. [Google Scholar] [CrossRef]
- Diamond, L.W. Review of the systematics of CO2–H2O fluid inclusions. Lithos 2001, 55, 69–99. [Google Scholar] [CrossRef]
- Pichavant, M.; Ramboz, C.; Weisbrod, A. Fluid immiscibility in natural processes: Use and misuse of fluid inclusion data. I. Phase equilibria analysis-A theoretical and geometrical approach. Chem. Geol. 1982, 37, 1–27. [Google Scholar] [CrossRef]
- Mernagh, T.P.; Leys, C.; Henley, R.W. Fluid inclusion systematics in porphyry copper deposits: The super-giant Grasberg deposit, Indonesia, as a case study. Ore Geol. Rev. 2020, 123, 103570. [Google Scholar] [CrossRef]
- Ramboz, C.; Pichavant, M.; Weisbrod, A. Fluid immiscibility in natural processes: Use and misuse of fluid inclusion data. II. Interpretation of fluid inclusion data in terms of immiscibility. Chem. Geol. 1982, 37, 29–48. [Google Scholar] [CrossRef]
- Belkin, H.; De Vivo, B.; Gianelli, G.; Lattanzi, P. Fluid inclusions in minerals from the geothermal fields of Tuscany, Italy. Geothermics 1985, 14, 59–72. [Google Scholar] [CrossRef]
- Bali, E.; Aradi, L.E.; Zierenberg, R.; Diamond, L.W.; Pettke, T.; Szabó, A.; Guðfinnsson, G.H.; Friðleifsson, G.O.; Szabó, C. Geothermal energy and ore-forming potential of 600 °C mid-ocean-ridge hydrothermal fluids. Geology 2020, 48, 1221–1225. [Google Scholar] [CrossRef]
- Mavrogenes, J.A.; Bodnar, R.J. Hydrogen movement into and out of fluid inclusions in quartz: Experimental evidence and geologic implications. Geochim. Cosmochim. Acta 1994, 58, 141–148. [Google Scholar] [CrossRef]
- Campbell, A.R.; Lundberg, S.A.W.; Dunbar, N.W. Solid inclusions of halite in quartz: Evidence for the halite trend. Chem. Geol. 2001, 173, 179–191. [Google Scholar] [CrossRef][Green Version]
- Becker, S.P.; Fall, A.; Bodnar, R.J. Synthetic fluid inclusions. XVII. PVTX properties of high salinity H2O-NaCl solutions (>30 wt. % NaCl): Application to fluid inclusions that homogenize by halite disappearance from porphyry copper and other hydrothermal ore deposits. Econ. Geol. 2008, 103, 539–554. [Google Scholar] [CrossRef]
- Lecumberri-Sanchez, P.; Steele MacInnis, M.; Bodnar, R.J. A numerical model to estimate trapping conditions of fluid inclusions that homogenize by halite disappearance. Geochim. Cosmochim. Acta 2012, 92, 14–22. [Google Scholar] [CrossRef]
- Bakker, R.J. Package FLUIDS 1. Computer programs for analysis of fluid inclusion data and for modelling bulk fluid properties. Chem. Geol. 2003, 194, 3–23. [Google Scholar] [CrossRef]
- Bodnar, R.J. Philosophy of fluid inclusion analysis. In Minerals, Methods and Applications; De Vivo, B., Frezzotti, M.L., Eds.; Virginia Tech: Blacksburg, VA, USA, 1994; pp. 1–6. [Google Scholar]
- Steele-MacInnis, M.; Lecumberri-Sanchez, P.; Bodnar, R.J. Hokieflincs_H2O-NACL: A Microsoft Excel spreadsheet for interpreting microthermometric data from fluid inclusions based on the PVTX properties of H2O–NaCl. Comput. Geosci. 2012, 49, 334–337. [Google Scholar] [CrossRef]
- Sibson, R.H.; Robert, F.; Poulsen, K.H. High angle reverse faults, fluid pressure cycling, and mesothermal gold-quartz deposits. Geology 1988, 16, 551–555. [Google Scholar] [CrossRef]
- Chi, G.; Guha, J.; Lu, H.-Z. Separation mechanism in the formation of proximal and distal tin-polymetallic deposits, Xinlu ore field, southern China-Evidence from fluid inclusion data. Econ. Geol. 1993, 88, 916–933. [Google Scholar] [CrossRef]
- Richter, L.; Diamond, L.W.; Atanasova, P.; Banks, D.A.; Gutzmer, J. Hydrothermal formation of heavy rare earth element (HREE)-xenotime deposits at 100oC in a sedimentary basin. Geology 2018, 46, 263–266. [Google Scholar] [CrossRef]
- Sibson, R.H.; Moore, J.M.M.; Rankin, A.H. Seismic pumping—A hydrothermal fluid transport mechanism. J. Geol. Soc. 1975, 131, 653–659. [Google Scholar] [CrossRef]
- Gleeson, S.A. Bulk analysis of electrolytes in fluid inclusions. In Fluid Inclusions: Analysis and Interpretation; Samson, I., Anderson, A., Marshall, D., Eds.; Mineralogical Association of Canada Publications: Québec, QC, Canada, 2003; Volume 32, pp. 233–246. [Google Scholar]
- Blamey, N.J.F. Compositional and evolution of crustal, geothermal, and hydrothermal fluids interpreted using quantitative fluid inclusion gas analysis. J. Geochem. Explor. 2012, 116–117, 17–27. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, G.; Diamond, L.W.; Lu, H.; Lai, J.; Chu, H. Common Problems and Pitfalls in Fluid Inclusion Study: A Review and Discussion. Minerals 2021, 11, 7. https://doi.org/10.3390/min11010007
Chi G, Diamond LW, Lu H, Lai J, Chu H. Common Problems and Pitfalls in Fluid Inclusion Study: A Review and Discussion. Minerals. 2021; 11(1):7. https://doi.org/10.3390/min11010007
Chicago/Turabian StyleChi, Guoxiang, Larryn W. Diamond, Huanzhang Lu, Jianqing Lai, and Haixia Chu. 2021. "Common Problems and Pitfalls in Fluid Inclusion Study: A Review and Discussion" Minerals 11, no. 1: 7. https://doi.org/10.3390/min11010007
APA StyleChi, G., Diamond, L. W., Lu, H., Lai, J., & Chu, H. (2021). Common Problems and Pitfalls in Fluid Inclusion Study: A Review and Discussion. Minerals, 11(1), 7. https://doi.org/10.3390/min11010007