Molecular Modeling of Ammonia Gas Adsorption onto the Kaolinite Surface with DFT Study


Abstract
1. Introduction
2. Calculational Methods and Models
2.1. Computational Methods
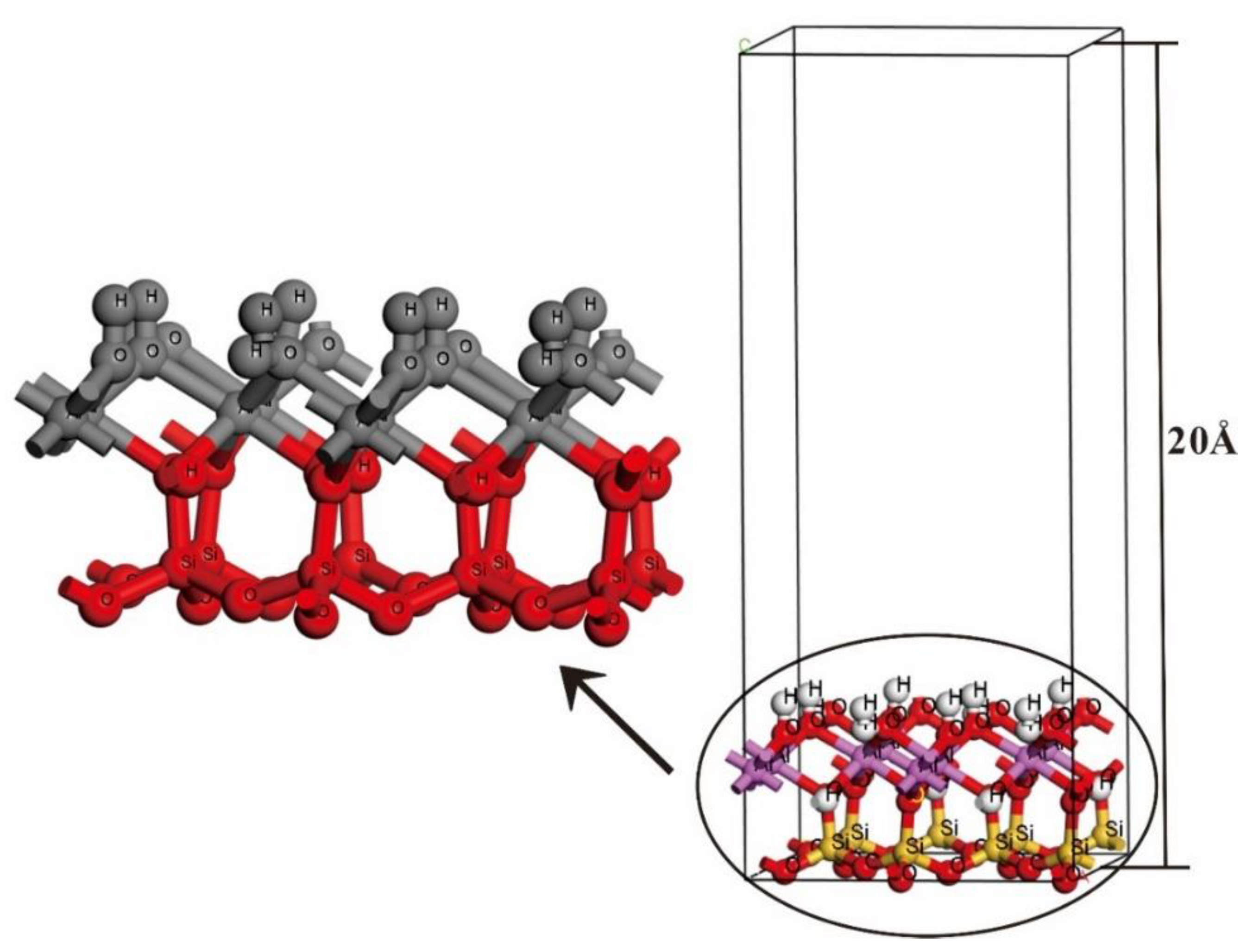
2.2. Computational Models
3. Results and Discussion
3.1. Adsorption Energy and Stable Adsorption Configuration
3.2. Bond and Mulliken Electric Charge
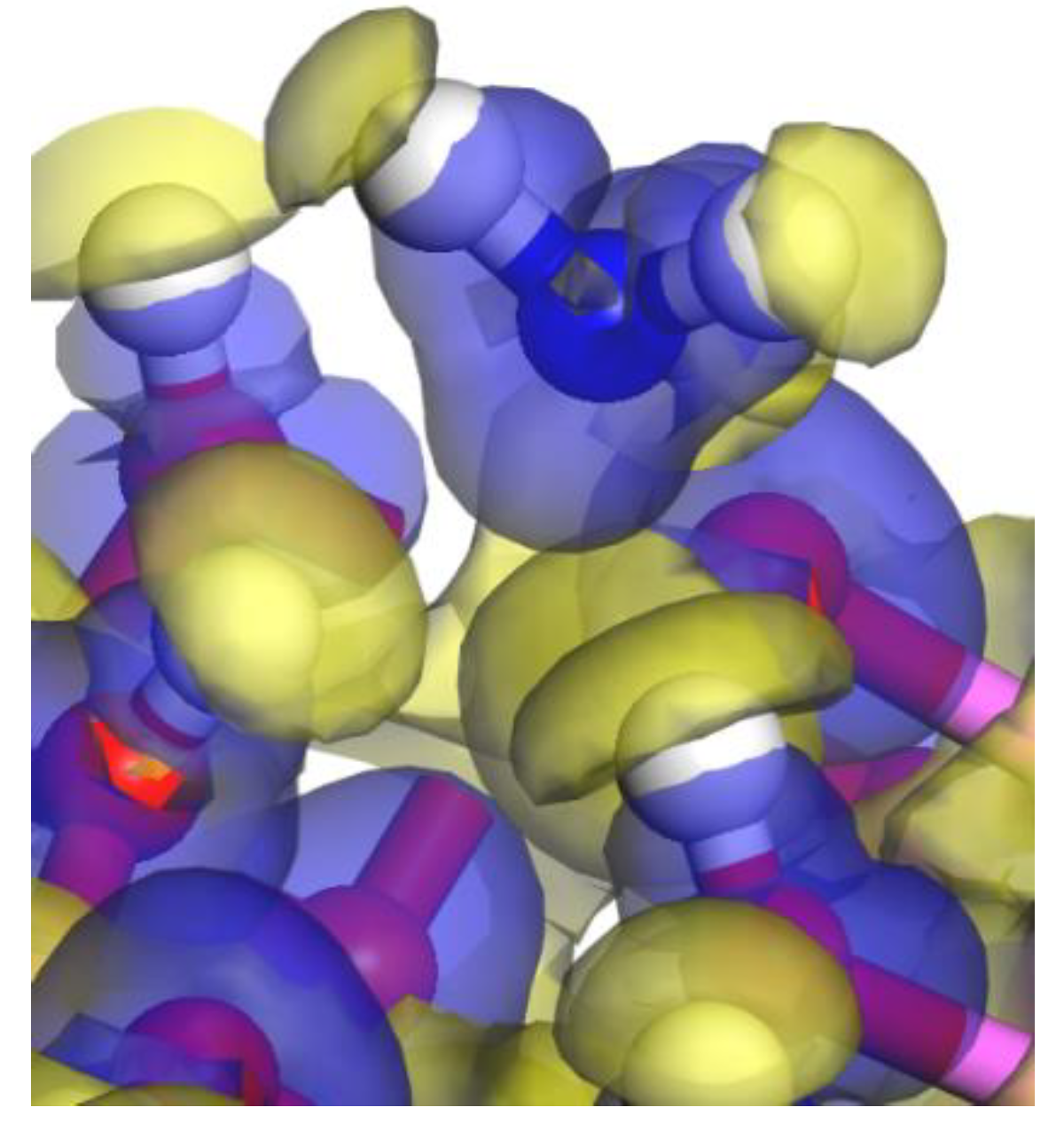
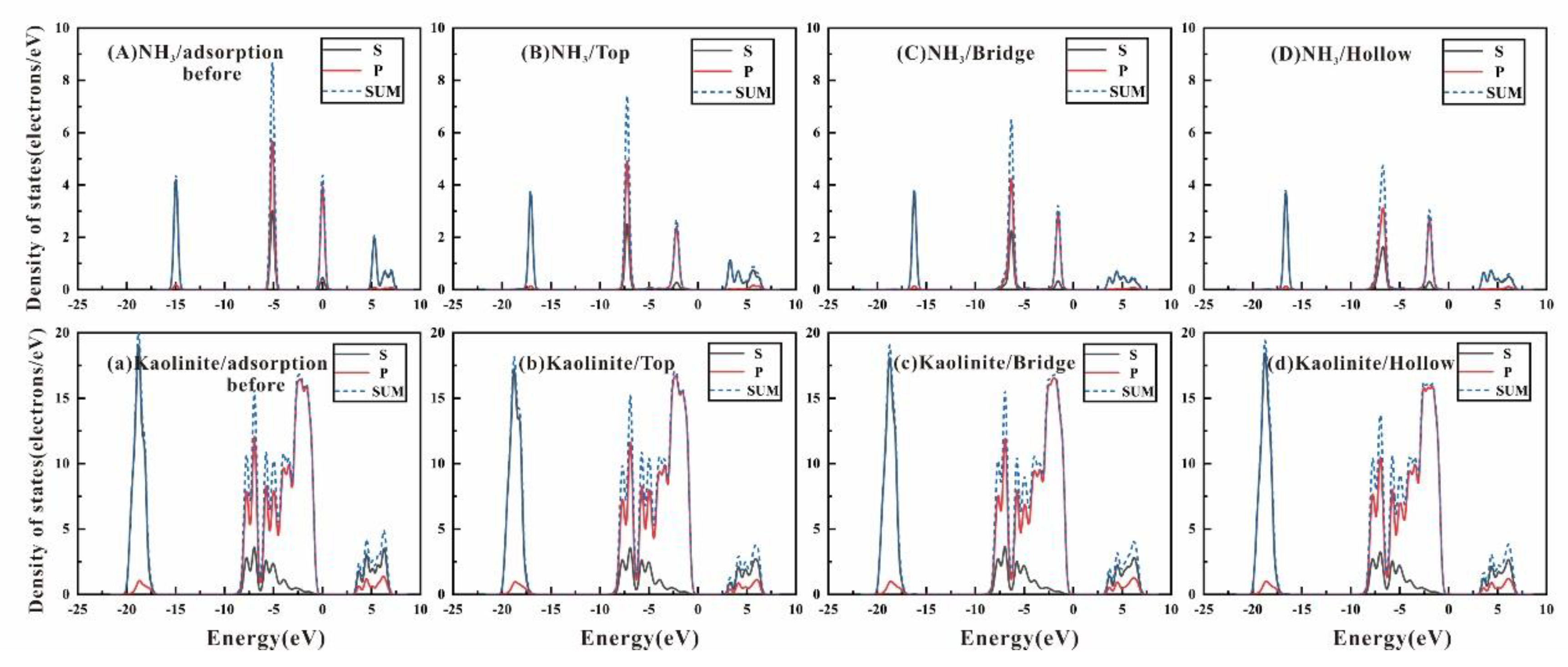
3.3. Density of State and Electron Transfer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Turpin, B.J.; Lim, H.-J. Species contributions to pm2.5 mass concentrations: Revisiting common assumptions for estimating organic mass. Aerosol Sci. Technol. 2001, 35, 602–610. [Google Scholar] [CrossRef]
- Bretschneider, B.; Kurfurst, J. Air Pollution Control Technology; Elsevier: Amsterdam, The Netherlands, 1987. [Google Scholar]
- Buijsman, E.; Maas, H.F.; Asman, W.A. Anthropogenic NH3 emissions in Europe. Atmos. Environ. (1967) 1987, 21, 1009–1022. [Google Scholar] [CrossRef]
- Bouwman, A.; Lee, D.; Asman, W.; Dentener, F.; Van Der Hoek, K.; Olivier, J. A global high-resolution emission inventory for ammonia. Glob. Biogeochem. Cycles 1997, 11, 561–587. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Dasgupta, P.K.; Dong, S. Solubility of ammonia in liquid water and generation of trace levels of standard gaseous ammonia. Atmos. Environ. (1967) 1986, 20, 565–570. [Google Scholar] [CrossRef]
- Behera, S.N.; Sharma, M.; Aneja, V.P.; Balasubramanian, R. Ammonia in the atmosphere: A review on emission sources, atmospheric chemistry and deposition on terrestrial bodies. Environ. Sci. Pollut. Res. 2013, 20, 8092–8131. [Google Scholar] [CrossRef]
- Søgaard, H.T.; Sommer, S.G.; Hutchings, N.; Huijsmans, J.; Bussink, D.; Nicholson, F. Ammonia volatilization from field-applied animal slurry—The alfam model. Atmos. Environ. 2002, 36, 3309–3319. [Google Scholar] [CrossRef]
- Heumann, W.L. Industrial Air Pollution Control Systems; Mc Graw-Hill: New York, NY, USA, 1997; Volume 620. [Google Scholar]
- Travlou, N.A.; Bandosz, T.J. N-doped polymeric resin-derived porous carbons as efficient ammonia removal and detection media. Carbon 2017, 117, 228–239. [Google Scholar] [CrossRef]
- Forzatti, P. Present status and perspectives in de-nox scr catalysis. Appl. Catal. A Gen. 2001, 222, 221–236. [Google Scholar] [CrossRef]
- Sharonov, V.E.; Aristov, Y.I. Ammonia adsorption by MgCl2, CaCl2 and BaCl2 confined to porous alumina: The fixed bed adsorber. React. Kinet. Catal. Lett. 2005, 85, 183–188. [Google Scholar] [CrossRef]
- Petit, C.; Karwacki, C.; Peterson, G.; Bandosz, T.J. Interactions of ammonia with the surface of microporous carbon impregnated with transition metal chlorides. J. Phys. Chem. C 2007, 111, 12705–12714. [Google Scholar] [CrossRef]
- Thang, H.V.; Grajciar, L.; Nachtigall, P.; Bludský, O.; Areán, C.O.; Frýdová, E.; Bulanek, R. Adsorption of CO2 in fau zeolites: Effect of zeolite composition. Catal. Today 2014, 227, 50–56. [Google Scholar] [CrossRef]
- Rodeghero, E.; Pasti, L.; Nunziante, G.; Chenet, T.; Gigli, L.; Plaisier, J.R.; Martucci, A. Highlighting the capability of zeolites for agro-chemicals contaminants removal from aqueous matrix: Evidence of 2-ethyl-6-methylaniline adsorption on zsm-12. Am. Miner. 2019, 104, 317–324. [Google Scholar] [CrossRef]
- Eberly, P.E., Jr. Adsorption properties of naturally occurring erionite and its cationic-exchanged forms. Am. Miner. 1964, 49, 30–40. [Google Scholar]
- Peng, C.; Min, F.; Liu, L.; Chen, J. A periodic dft study of adsorption of water on sodium-montmorillonite (001) basal and (010) edge surface. Appl. Surf. Sci. 2016, 387, 308–316. [Google Scholar] [CrossRef]
- Wungu, T.D.K.; Agusta, M.K.; Saputro, A.G.; Dipojono, H.K.; Kasai, H. First principles calculation on the adsorption of water on lithium–montmorillonite (Li-MMT). J. Phys. Condens. Matter Inst. Phys. J. 2012, 24, 475506. [Google Scholar] [CrossRef] [PubMed]
- Bergaya, F.; Lagaly, G. General introduction: Clays, clay minerals, and clay science. Dev. Clay Sci. 2006, 1, 1–18. [Google Scholar]
- Brigatti, M.F.; Galan, E.; Theng, B. Structures and mineralogy of clay minerals. Dev. Clay Sci. 2006, 1, 19–86. [Google Scholar]
- Kremleva, A.; Krüger, S.; Rösch, N. Density functional model studies of uranyl adsorption on (001) surfaces of kaolinite. Langmuir 2008, 24, 9515–9524. [Google Scholar] [CrossRef]
- Schoonheydt, R.; Johnston, C. Surface and interface chemistry of clay minerals. Dev. Clay Sci. 2006, 1, 87–113. [Google Scholar]
- Chen, J.; Min, F.F.; Liu, L.; Liu, C.; Lu, F. Experimental investigation and DFT calculation of different amine/ammonium salts adsorption on kaolinite. Appl. Surf. Sci. 2017, 419, 241–251. [Google Scholar] [CrossRef]
- Sato, H.; Ono, K.; Johnston, C.T.; Yamagishi, A. First-principle study of polytype structures of 1: 1 dioctahedral phyllosilicates. Am. Miner. 2004, 89, 1581–1585. [Google Scholar] [CrossRef]
- Schaef, H.T.; Glezakou, V.-A.; Owen, A.T.; Ramprasad, S.; Martin, P.F.; McGrail, B.P. Surface condensation of CO2 onto kaolinite. Environ. Sci. Technol. Lett. 2013, 1, 142–145. [Google Scholar] [CrossRef]
- Zhao, J.; He, M.C. Theoretical study of heavy metal Cd, Cu, Hg, and Ni(II) adsorption on the kaolinite (001) surface. Appl. Surf. Sci. 2014, 317, 718–723. [Google Scholar] [CrossRef]
- Zhang, B.; Kang, J.; Kang, T. Effect of water on methane adsorption on the kaolinite (001) surface based on molecular simulations. Appl. Surf. Sci. 2018, 439, 792–800. [Google Scholar] [CrossRef]
- Jun, C.; Fan-fei, M.; Ling-yun, L.; Chen-liang, P. DFT calculations of different amine/ammonium cations adsorption on kaolinite (001) surface. J. China Coal Soc. 2016, 41, 3115–3121. [Google Scholar]
- Segall, M.; Shah, R.; Pickard, C.; Payne, M. Population analysis of plane-wave electronic structure calculations of bulk materials. Phys. Rev. B 1996, 54, 16317. [Google Scholar] [CrossRef]
- Segall, M.; Pickard, C.; Shah, R.; Payne, M. Population analysis in plane wave electronic structure calculations. Mol. Phys. 1996, 89, 571–577. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Ireta, J.; Neugebauer, J.; Scheffler, M. On the accuracy of DFT for describing hydrogen bonds: Dependence on the bond directionality. J. Phys. Chem. A 2004, 108, 5692–5698. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using castep. Zeitschrift für Kristallographie-Crystalline Materials 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.a.; Pickard, C.J.; Hasnip, P.J.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the castep code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.; Joannopoulos, A.J. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- McLachlan, A. Self-consistent field theory of the electron spin distribution in π-electron radicals. Mol. Phys. 1960, 3, 233–252. [Google Scholar] [CrossRef]
- Bish, D.L. Rietveld refinement of the kaolinite structure at 1.5 k. Clays Clay Miner. 1993, 41, 738–744. [Google Scholar] [CrossRef]
- Xia, L.-Y.; Zhong, H.; Liu, G.-Y.; Li, X.-G. Electron bandstructure of kaolinite and its mechanism of flotation using dodecylamine as collector. J. Cent. South Univ. Technol. 2009, 16, 73–79. [Google Scholar] [CrossRef]
- Bickmore, B.R.; Bosbach, D.; Hochella, M.F.; Charlet, L.; Rufe, E. In situ atomic force microscopy study of hectorite and nontronite dissolution: Implications for phyllosilicate edge surface structures and dissolution mechanisms. Am. Miner. 2001, 86, 411–423. [Google Scholar] [CrossRef]
- Hu, Y.; Wei, S.; Hao, J.; Miller, J.; Fa, K. The anomalous behavior of kaolinite flotation with dodecyl amine collector as explained from crystal structure considerations. Int. J. Miner. Process. 2005, 76, 163–172. [Google Scholar] [CrossRef]
- Xiao, L.H.; Michaelides, A.; Xiao, L.H.; Michaelides, A. Water on the hydroxylated (001) surface of kaolinite: From monomer adsorption to a flat 2D wetting layer. Surf. Sci. 2008, 602, 960–974. [Google Scholar]
- Gray, P.V.; Brown, D.M. Density of SiO2–Si interface states. Appl. Phys. Lett. 1966, 8, 31–33. [Google Scholar] [CrossRef]
- Shirley, D.A. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 1972, 5, 4709. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exchange-Correlation Functions/eV | Lattice Parameter (Å) | Cell Angles (°) | Total Energy/eV | ||||
|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | ||
| LDA + CA-PZ/400 | 5.082 | 8.820 | 7.155 | 91.734 | 105.078 | 89.839 | −8689.188 |
| LDA + CA-PZ (DFT-D)/400 | 5.045 | 8.752 | 7.099 | 91.966 | 105.389 | 89.885 | −8689.142 |
| GGA + PBE/400 | 5.209 | 9.068 | 7.485 | 91.675 | 104.836 | 89.726 | −8682.448 |
| GGA + PBE (DFT-D)/400 | 5.176 | 8.977 | 7.339 | 91.724 | 105.037 | 89.962 | −8682.408 |
| GGA + PW91 | 5.176 | 8.984 | 7.371 | 91.757 | 105.445 | 89.813 | −8693.178 |
| GGA + WC | 5.183 | 9.024 | 7.347 | 91.652 | 105.077 | 89.740 | −8667.929 |
| GGA-PBESOL | 5.182 | 9.030 | 7.335 | 91.634 | 104.928 | 89.743 | −8656.095 |
| Experimental results [45] | 5.154 | 8.942 | 7.391 | 91.926 | 105.046 | 89.797 | |
| Surface | Adsorption Position | NH3 on Kaolinite Final Enthalpy (eV) | Adsorption Energy (eV) |
|---|---|---|---|
| (001) | Top1 | −17,689.03 | −0.19 |
| Top2 | −17,689.29 | −0.45 | |
| Top3 | −17,689.12 | −0.27 | |
| Bridge1 | −17,689.27 | −0.43 | |
| Bridge2 | −17,689.33 | −0.48 | |
| Bridge3 | −17,689.36 | −0.52 | |
| Hollow1 | −17,689.38 | −0.54 | |
| Hollow2 | −17,689.37 | −0.53 | |
| Hollow3 | −17,689.35 | −0.51 | |
| Hollow4 | −17,689.11 | −0.27 |
| Bond Length and Band Angle | Adsorption Before | Top2 | Bridge3 | Hollow1 |
|---|---|---|---|---|
| Longest N–H bond length (Å) | 1.110 | 1.030 | 1.033 | 1.036 |
| Shortest N–H bond length (Å) | 1.028 | 1.029 | 1.030 | |
| Largest H–N–H angle (°) | 109.471 | 107.885 | 108.112 | 108.550 |
| Smallest H–N–H angle (°) | 107.268 | 106.222 | 106.506 | |
| Adsorption energy (eV) | - | 0.45 | 0.52 | 0.54 |
| Adsorption Site | Bond | Population (bulk/after) | Bond Length (bulk/after)/Å |
|---|---|---|---|
| adsorption before | N–HN1 | 0.64/0.65 | 1.039/1.036 |
| N–HN2 | 0.64/0.65 | 1.035/1.030 | |
| N–HN3 | 0.64/0.66 | 1.039/1.030 | |
| Top2 | N–H1 | 0.09 | 1.887 |
| N–O1 | −0.10 | 2.869 | |
| Bridge3 | N–H1 | 0.00 | 2.960 |
| N–H2 | 0.06 | 2.037 | |
| N–H3 | −0.01 | 2.599 | |
| N–O2 | −0.06 | 2.979 | |
| Hollow1 | N–H1 | 0.06 | 2.016 |
| N–H2 | 0.02 | 2.326 | |
| N–H3 | −0.02 | 2.552 | |
| HN1–O3 | 0.03 | 2.094 |
| Adsorption Site | Atom | Adsorption State | s | p | Total | Charge (e) |
|---|---|---|---|---|---|---|
| Hollow1 | HN1 | Before | 0.59 | 0 | 0.59 | 0.41 |
| After | 0.61 | 0 | 0.61 | 0.39 | ||
| ∆(After-Before) | 0.02 | 0 | 0.02 | −0.02 | ||
| HN2 | Before | 0.59 | 0 | 0.59 | 0.41 | |
| After | 0.58 | 0 | 0.58 | 0.42 | ||
| ∆(After-Before) | −0.01 | 0 | −0.01 | 0.01 | ||
| HN3 | Before | 0.59 | 0 | 0.59 | 0.41 | |
| After | 0.59 | 0 | 0.59 | 0.41 | ||
| ∆(After-Before) | 0.00 | 0 | 0.00 | 0.00 | ||
| N | Before | 1.74 | 4.49 | 6.23 | −1.23 | |
| After | 1.72 | 4.46 | 6.18 | −1.18 | ||
| ∆(After-Before) | −0.02 | −0.03 | −0.05 | 0.05 | ||
| H1 | Before | 0.55 | 0 | 0.55 | 0.45 | |
| After | 0.57 | 0 | 0.57 | 0.43 | ||
| ∆(After-Before) | 0.02 | 0 | 0.02 | −0.02 | ||
| H2 | Before | 0.54 | 0 | 0.54 | 0.46 | |
| After | 0.55 | 0 | 0.55 | 0.45 | ||
| ∆(After-Before) | 0.01 | 0 | 0.01 | −0.01 | ||
| O3 | Before | 1.85 | 5.20 | 7.05 | −1.05 | |
| After | 1.86 | 5.16 | 7.02 | −1.02 | ||
| ∆(After-Before) | 0.01 | −0.04 | −0.03 | 0.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Q.; Li, Y.; Qiao, X.; Guo, Y.; Zhao, Y.; Zhang, Q.; Ju, Y.; Shi, Y. Molecular Modeling of Ammonia Gas Adsorption onto the Kaolinite Surface with DFT Study. Minerals 2020, 10, 46. https://doi.org/10.3390/min10010046
Cheng Q, Li Y, Qiao X, Guo Y, Zhao Y, Zhang Q, Ju Y, Shi Y. Molecular Modeling of Ammonia Gas Adsorption onto the Kaolinite Surface with DFT Study. Minerals. 2020; 10(1):46. https://doi.org/10.3390/min10010046
Chicago/Turabian StyleCheng, Qi, Yongbing Li, Xiaojuan Qiao, Yang Guo, Yang Zhao, Qiuyuan Zhang, Yiwen Ju, and Yaolin Shi. 2020. "Molecular Modeling of Ammonia Gas Adsorption onto the Kaolinite Surface with DFT Study" Minerals 10, no. 1: 46. https://doi.org/10.3390/min10010046
APA StyleCheng, Q., Li, Y., Qiao, X., Guo, Y., Zhao, Y., Zhang, Q., Ju, Y., & Shi, Y. (2020). Molecular Modeling of Ammonia Gas Adsorption onto the Kaolinite Surface with DFT Study. Minerals, 10(1), 46. https://doi.org/10.3390/min10010046