
Symmetry and Structure in the POT Family of Proton Coupled Peptide Transporters


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
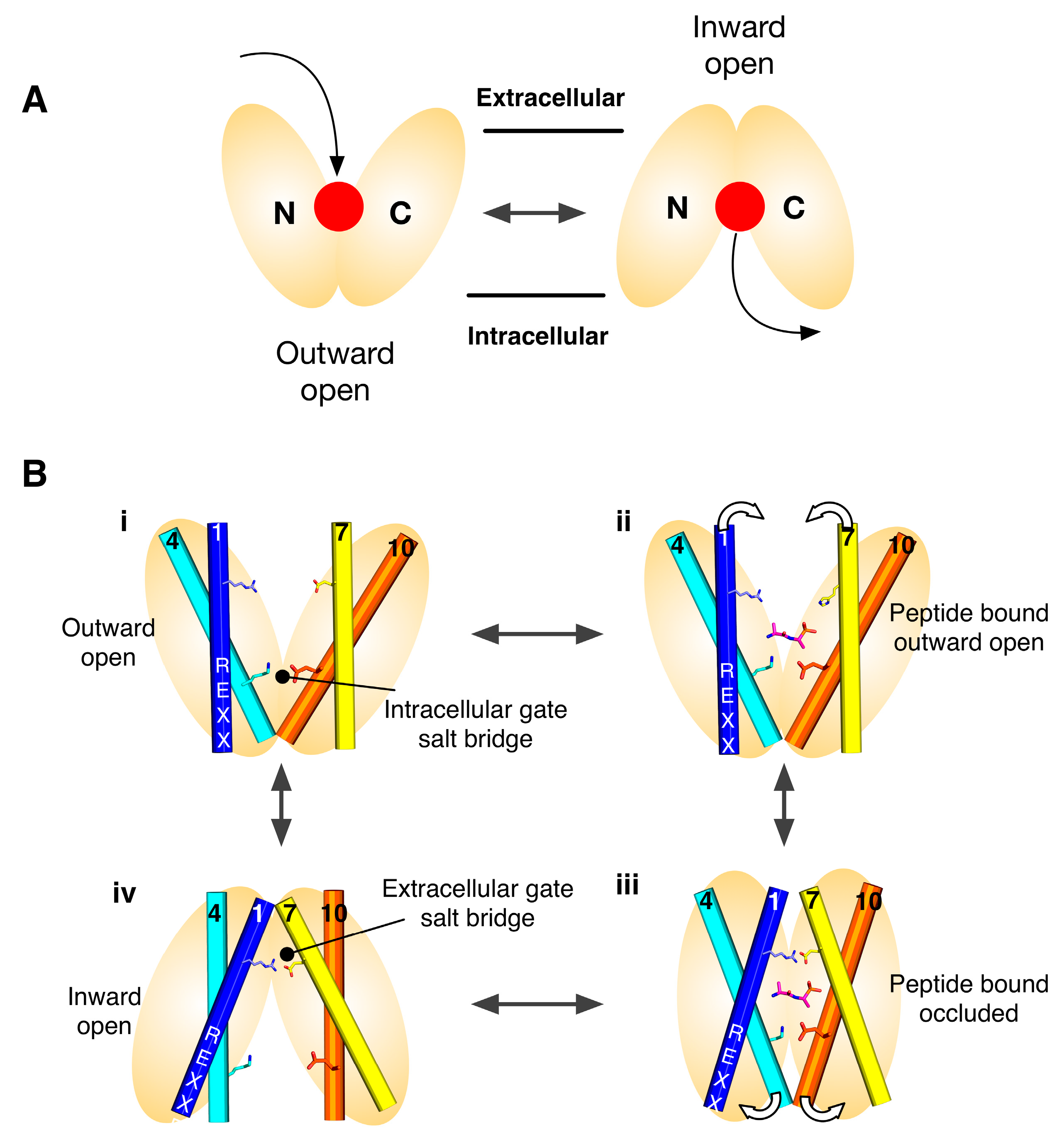
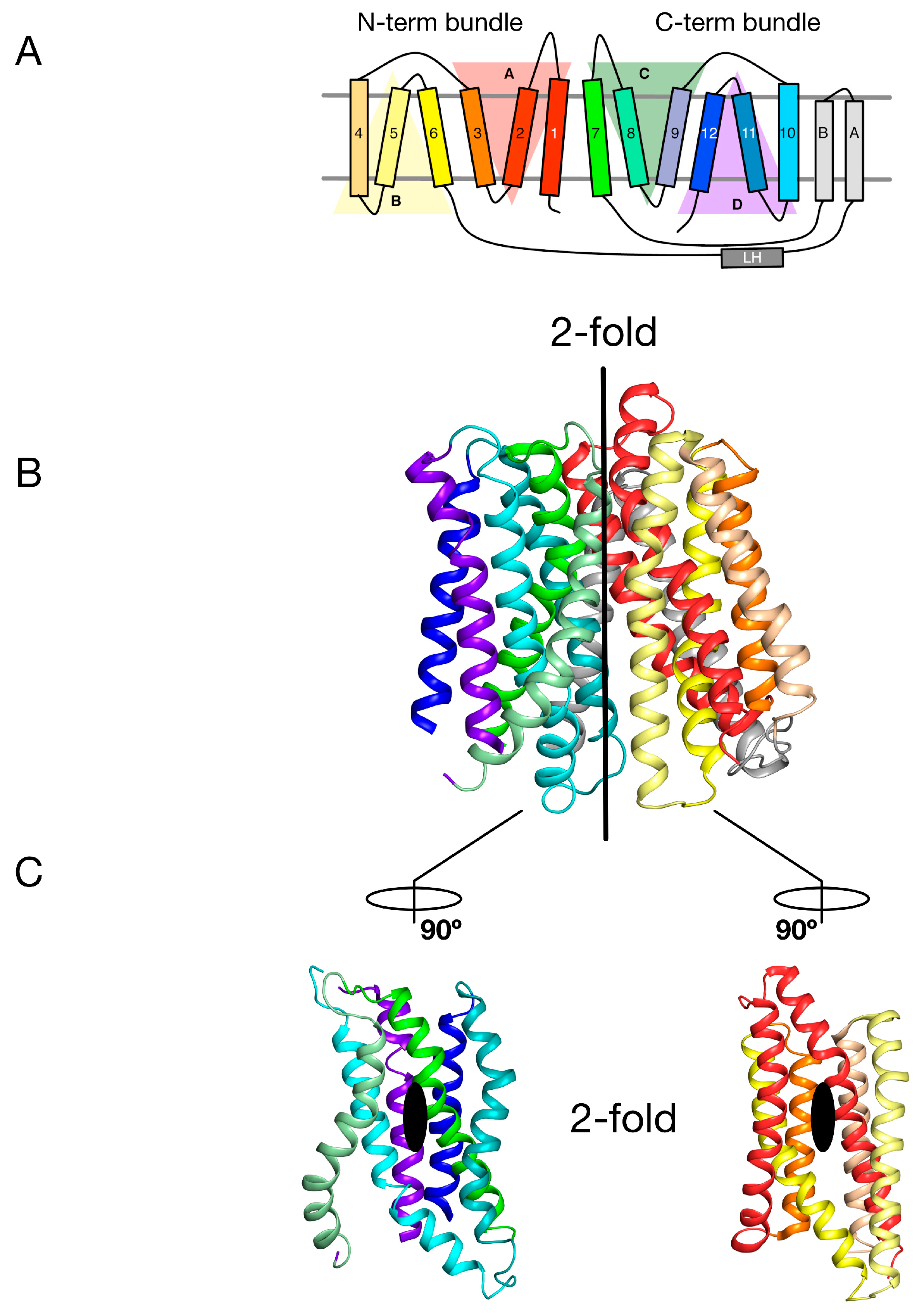
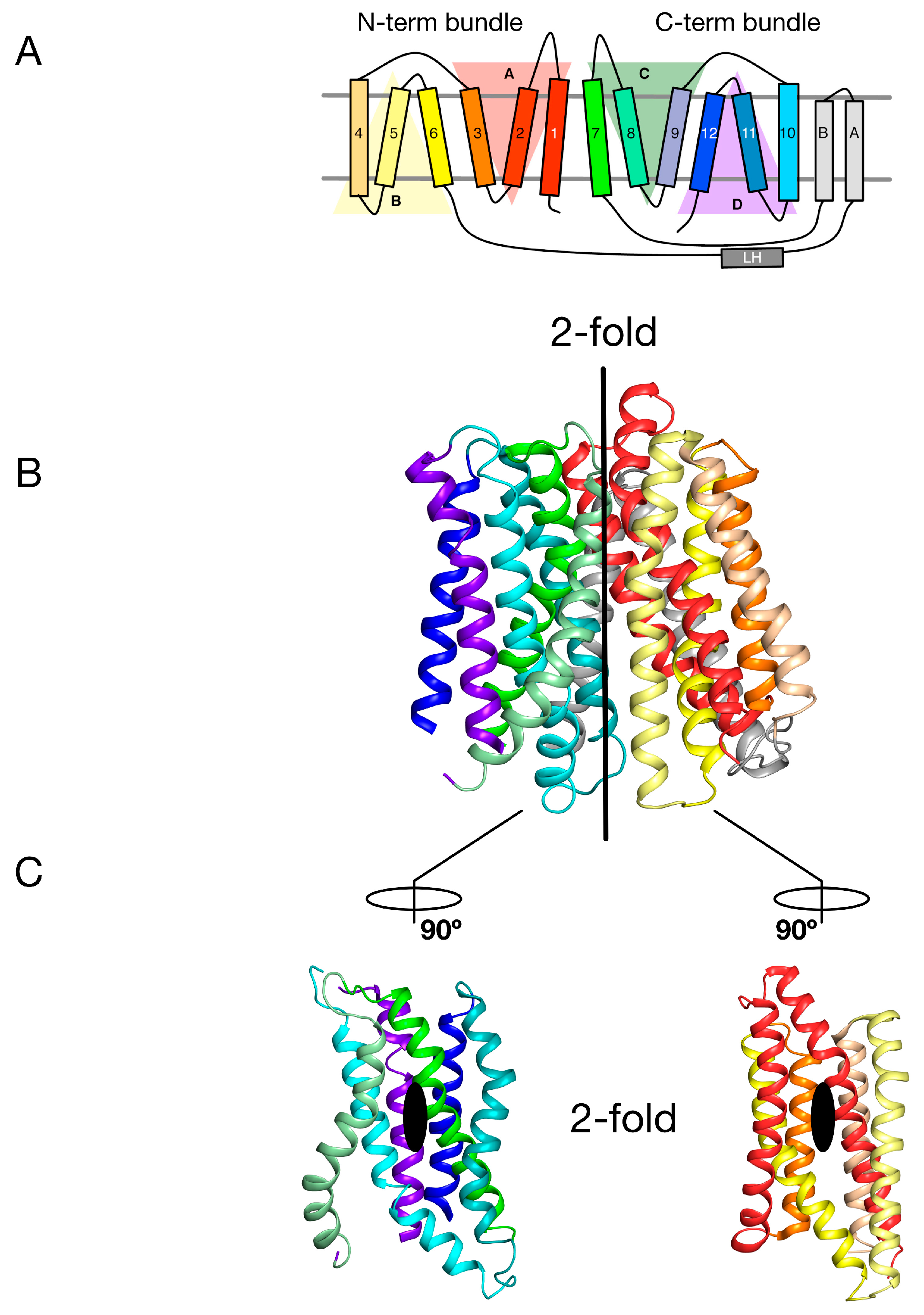
2. Symmetry-Exchange Underlies the Overall Conformational Change with the Major Facilitator Superfamily
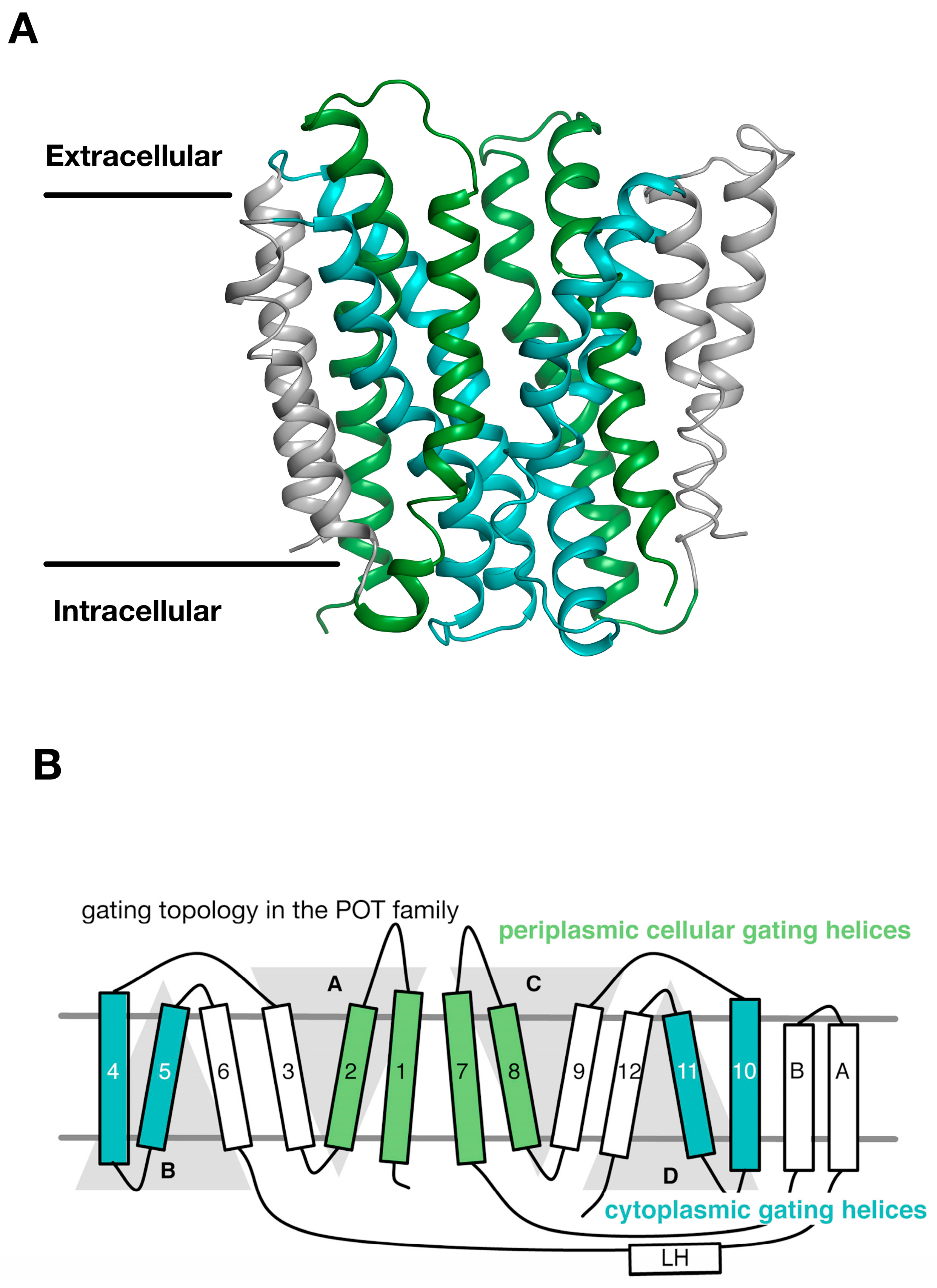
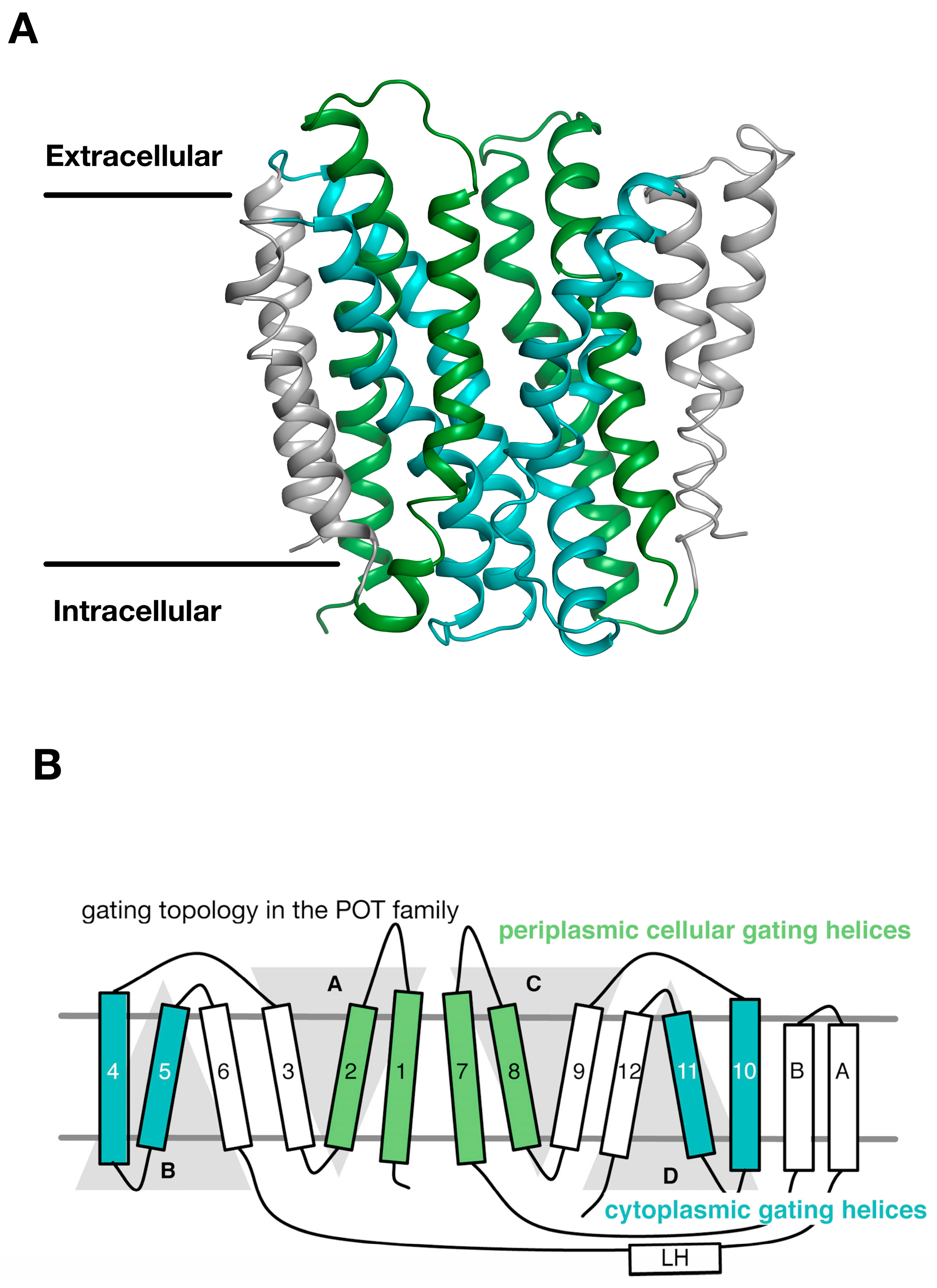
3. Gating Topology with the POT Family
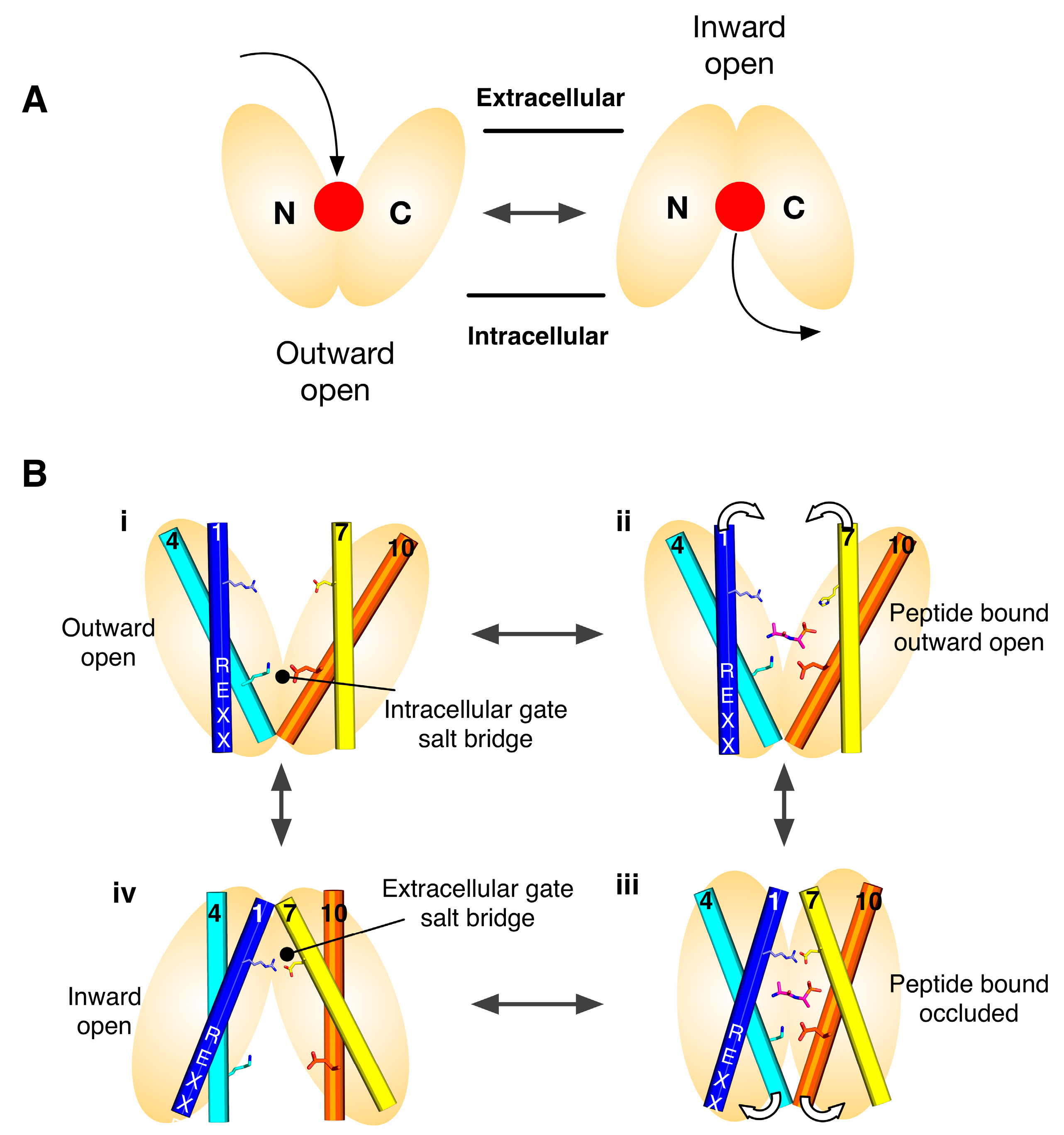
4. Salt Bridge Networks and Proton Binding
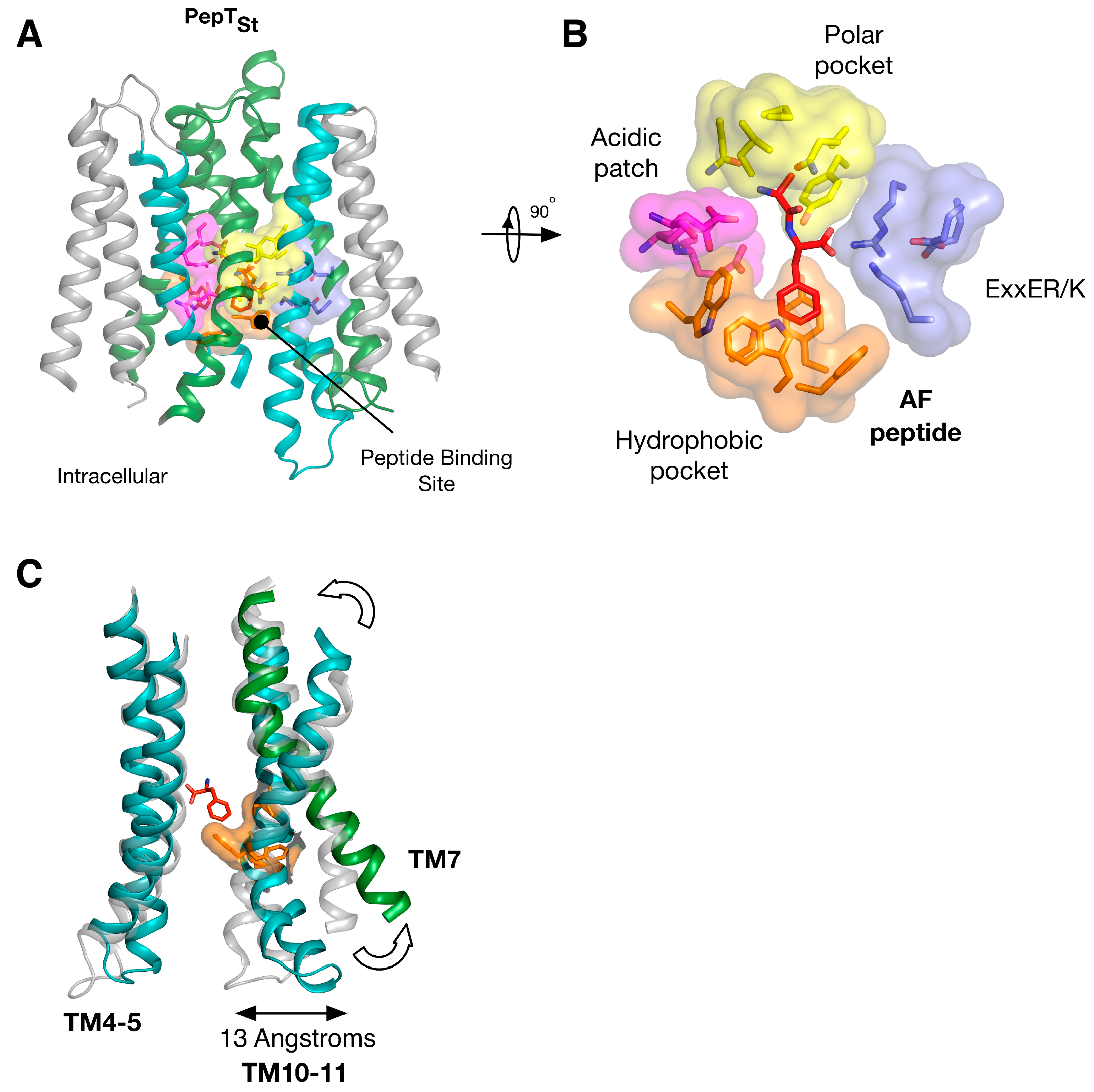
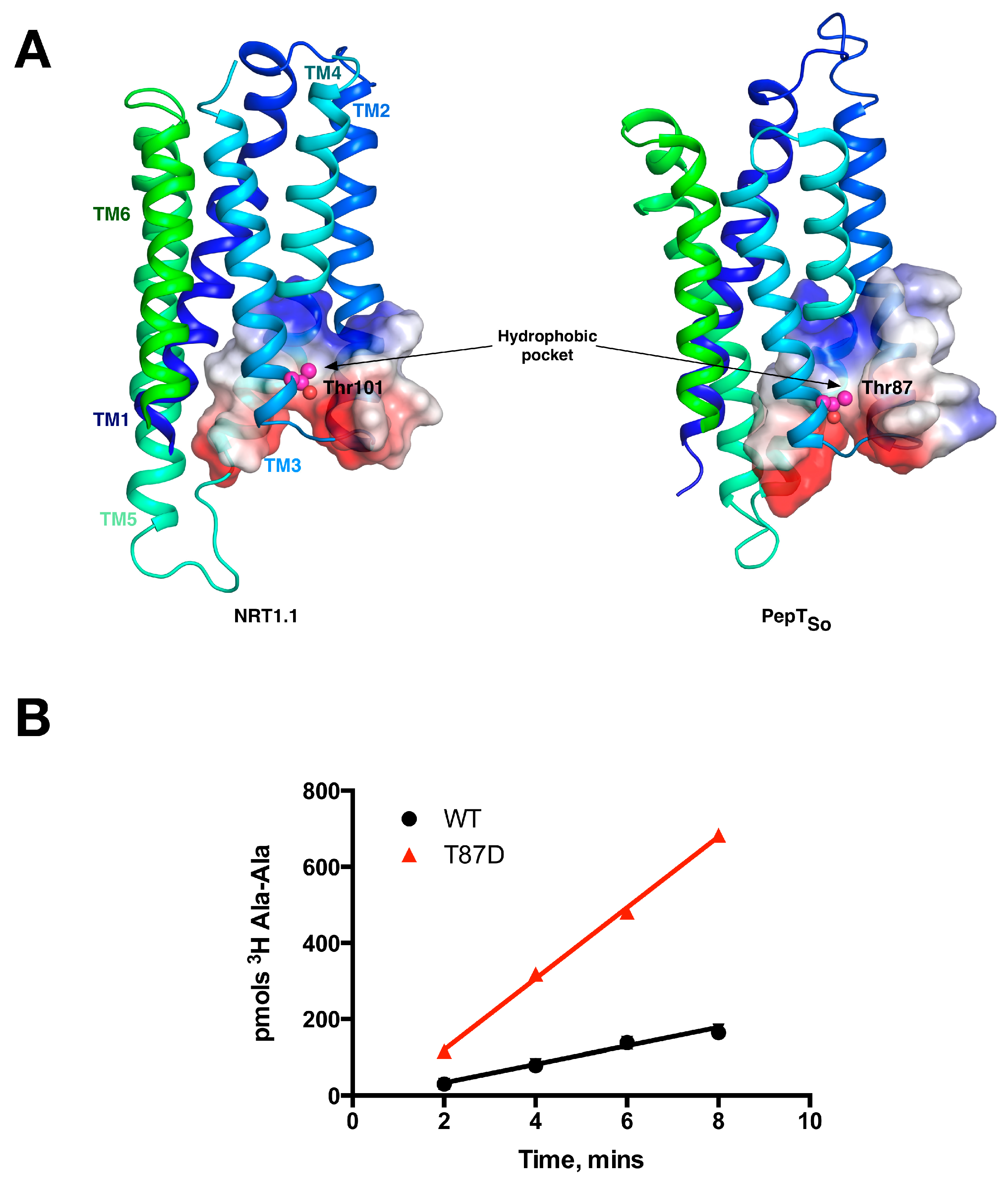
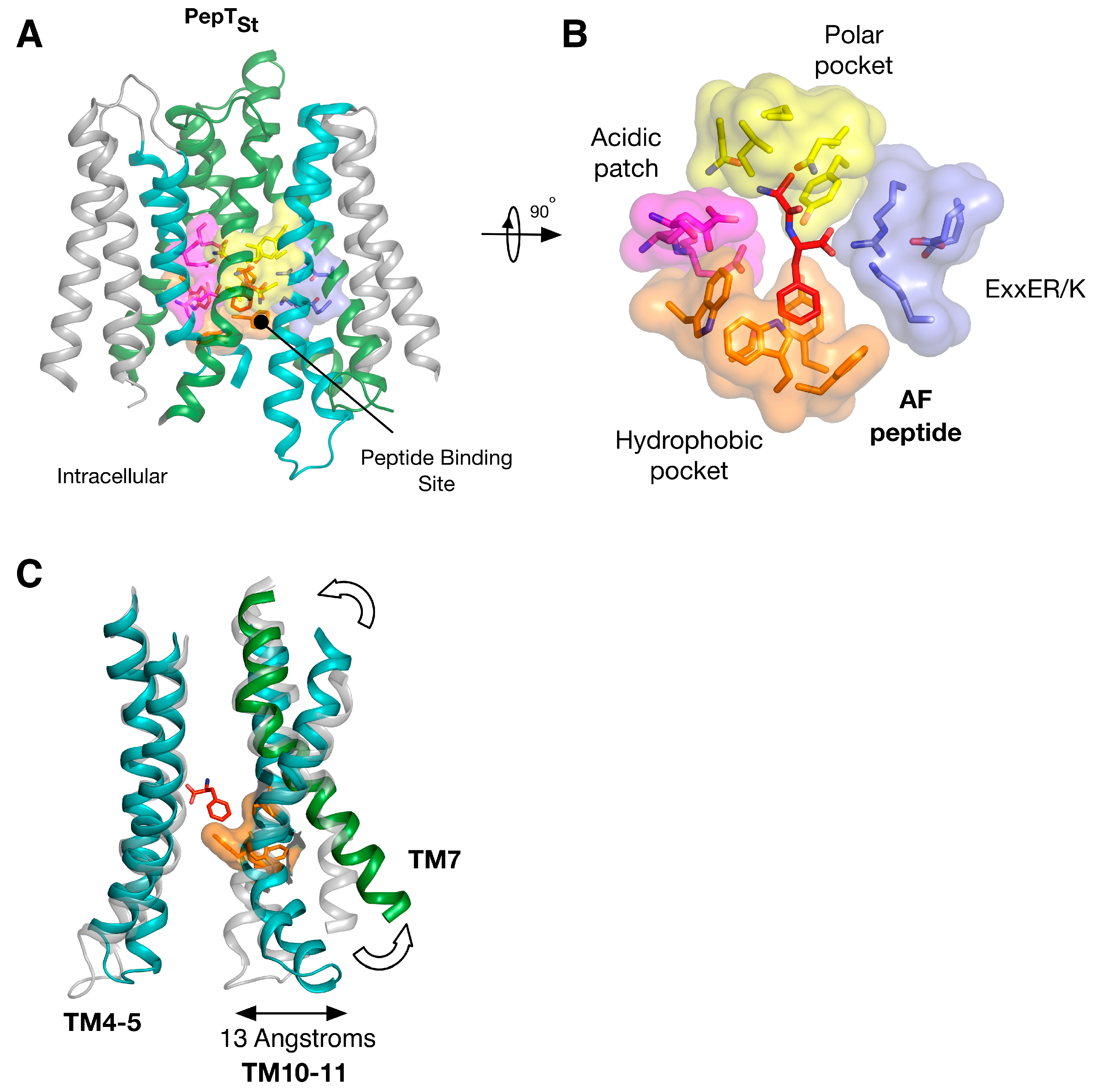
5. Peptide Binding and the Role of Specificity Pockets in Ligand Recognition
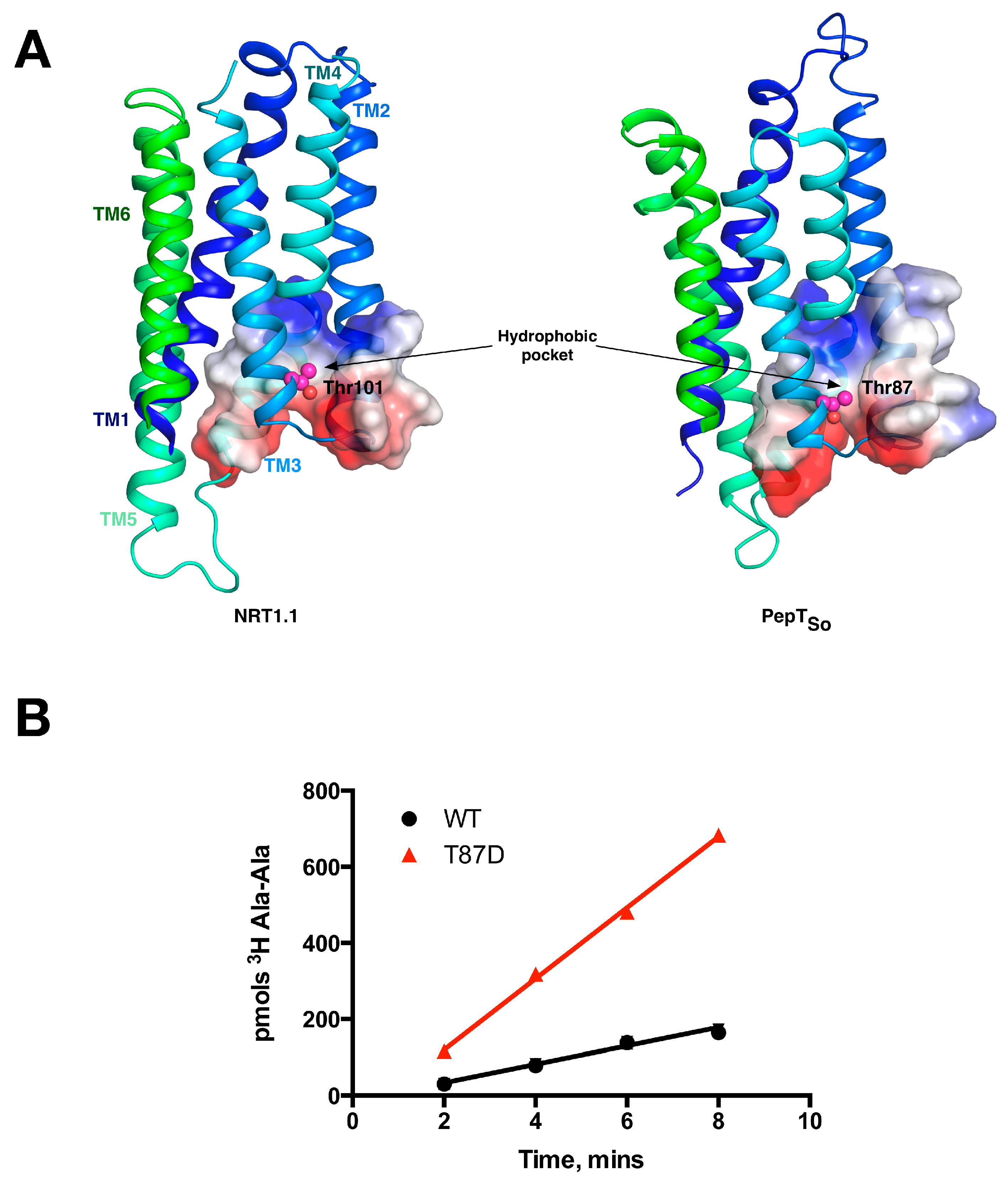
6. Regulation and Structural Adaptation
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Adibi, S.A. The oligopeptide transporter (Pept-1) in human intestine: Biology and function. Gastroenterology 1997, 113, 332–340. [Google Scholar] [CrossRef]
- Matthews, D.M. Protein Absorption: Development and Present State of the Subject; Wiley-Liss: New York, NY, USA, 1991; p. 414. [Google Scholar]
- Fei, Y.J.; Kanai, Y.; Nussberger, S.; Ganapathy, V.; Leibach, F.H.; Romero, M.F.; Singh, S.K.; Boron, W.F.; Hediger, M.A. Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature 1994, 368, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Daniel, H.; Rubio-Aliaga, I. An update on renal peptide transporters. Am. J. Physiol. Ren. Physiol. 2003, 284, F885–F892. [Google Scholar] [CrossRef] [PubMed]
- Brandsch, M. Drug transport via the intestinal peptide transporter PepT1. Curr. Opin. Pharmacol. 2013, 13, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Clemencon, B.; Hediger, M.A. Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol. Asp. Med. 2013, 34, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Daniel, H.; Spanier, B.; Kottra, G.; Weitz, D. From bacteria to man: Archaic proton-dependent peptide transporters at work. Physiology 2006, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hikida, A.; Kawai, S.; Lan, V.T.; Motoyama, T.; Kitagawa, S.; Yoshikawa, Y.; Kato, R.; Kawarasaki, Y. Analysing the substrate multispecificity of a proton-coupled oligopeptide transporter using a dipeptide library. Nat. Commun. 2013, 4, 2502. [Google Scholar] [CrossRef] [PubMed]
- Biegel, A.; Knutter, I.; Hartrodt, B.; Gebauer, S.; Theis, S.; Luckner, P.; Kottra, G.; Rastetter, M.; Zebisch, K.; Thondorf, I.; et al. The renal type H+/peptide symporter PEPT2: Structure-affinity relationships. Amino Acids 2006, 31, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Léran, S.; Varala, K.; Boyer, J.-C.; Chiurazzi, M.; Crawford, N.; Daniel-Vedele, F.; David, L.; Dickstein, R.; Fernandez, E.; Forde, B.; et al. A unified nomenclature of nitrate transporter 1/peptide transporter family members in plants. Trends Plant Sci. 2014, 19, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Tsay, Y.-F.; Chiu, C.-C.; Tsai, C.-B.; Ho, C.-H.; Hsu, P.-K. Nitrate transporters and peptide transporters. FEBS Lett. 2007, 581, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Boursiac, Y.; Léran, S.; Corratgé-Faillie, C.; Gojon, A.; Krouk, G.; Lacombe, B. ABA transport and transporters. Trends Plant Sci. 2013, 18, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Nour-Eldin, H.H.; Andersen, T.G.; Burow, M.; Madsen, S.R. NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 2012, 488, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Newstead, S. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature 2014, 507, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Iwata, S.; Kaback, H.R. Lactose permease as a paradigm for membrane transport proteins (review). Mol. Membr. Biol. 2004, 21, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, O. Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970. [Google Scholar] [CrossRef] [PubMed]
- Doki, S.; Kato, H.E.; Solcan, N.; Iwaki, M.; Koyama, M.; Hattori, M.; Iwase, N.; Tsukazaki, T.; Sugita, Y.; Kandori, H.; et al. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter pot. Proc. Natl. Acad. Sci. USA 2013, 110, 11343–11348. [Google Scholar] [CrossRef] [PubMed]
- Solcan, N.; Kwok, J.; Fowler, P.W.; Cameron, A.D.; Drew, D.; Iwata, S.; Newstead, S. Alternating access mechanism in the pot family of oligopeptide transporters. EMBO J. 2012, 31, 3411–3421. [Google Scholar] [CrossRef] [PubMed]
- Newstead, S. Recent advances in understanding proton coupled peptide transport via the pot family. Curr. Opin. Struct. Biol. 2016, 45, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Forrest, L.R. Structural symmetry in membrane proteins. Annu. Rev. Biophys. 2015, 44, 311–337. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Shlykov, M.A.; Castillo, R.; Sun, E.I.; Saier, M.H. The major facilitator superfamily (MFS) revisited. FEBS J. 2012, 279, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.; Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 2016, 85, 543–572. [Google Scholar] [CrossRef] [PubMed]
- Law, C.J.; Maloney, P.C.; Wang, D.-N. Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Madej, M.G.; Dang, S.; Yan, N.; Kaback, H.R. Evolutionary mix-and-match with mfs transporters. Proc. Natl. Acad. Sci. USA 2013, 110, 5870–5874. [Google Scholar] [CrossRef] [PubMed]
- Madej, M.G.; Kaback, H.R. Evolutionary mix-and-match with MFS transporters II. Proc. Natl. Acad. Sci. USA 2013, 110, E4831–E4838. [Google Scholar] [CrossRef] [PubMed]
- Radestock, S.; Forrest, L.R. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J. Mol. Biol. 2011, 407, 698–715. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.W.; Orwick-Rydmark, M.; Radestock, S.; Solcan, N.; Dijkman, P.M.; Lyons, J.A.; Kwok, J.; Caffrey, M.; Watts, A.; Forrest, L.R.; et al. Gating topology of the proton-coupled oligopeptide symporters. Structure 2015, 23, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Forrest, L.R.; Krämer, R.; Ziegler, C. The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta 2011, 1807, 167–188. [Google Scholar] [CrossRef] [PubMed]
- Newstead, S. Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transporters. Biochim. Biophys. Acta 2015, 1850, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Newstead, S.; Drew, D.; Cameron, A.D.; Postis, V.L.G.; Xia, X.; Fowler, P.W.; Ingram, J.C.; Carpenter, E.P.; Sansom, M.S.P.; Mcpherson, M.J.; et al. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 2011, 30, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Madej, M.G.; Guan, L.; Nie, Y.; Kaback, H.R. An early event in the transport mechanism of LacY protein: INTERACTION BETWEEN HELICES V AND I. J. Biol. Chem. 2011, 286, 30415–30422. [Google Scholar] [CrossRef] [PubMed]
- Reginsson, G.W.; Schiemann, O. Pulsed electron-electron double resonance: Beyond nanometre distance measurements on biomacromolecules. Biochem. J. 2011, 434, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Guettou, F.; Quistgaard, E.M.; Trésaugues, L.; Moberg, P.; Jegerschöld, C.; Zhu, L.; Jong, A.J.O.; Nordlund, P.; Löw, C. Structural insights into substrate recognition in proton-dependent oligopeptide transporters. EMBO Rep. 2013, 14, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.A.; Parker, J.L.; Solcan, N.; Brinth, A.; Li, D.; Shah, S.T.; Caffrey, M.; Newstead, S. Structural basis for polyspecificity in the pot family of proton-coupled oligopeptide transporters. EMBO Rep. 2014, 15, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Guettou, F.; Quistgaard, E.M.; Raba, M.; Moberg, P.; Low, C.; Nordlund, P. Selectivity mechanism of a bacterial homolog of the human drug-peptide transporters PepT1 and PepT2. Nat. Struct. Mol. Biol. 2014, 21, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Samsudin, F.; Parker, J.L.; Sansom, M.S.; Newstead, S.; Fowler, P.W. Accurate prediction of ligand affinities for a proton-dependent oligopeptide transporter. Cell Chem. Biol. 2016, 23, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Newstead, S. Towards a structural understanding of drug and peptide transport within the proton-dependent oligopeptide transporter (POT) family. Biochem. Soc. Trans. 2011, 39, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.H.; Huang, C.Y.; Tsay, Y.F. Chl1 is a dual-affinity nitrate transporter of arabidopsis involved in multiple phases of nitrate uptake. Plant Cell 1999, 11, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-H.; Lin, S.-H.; Hu, H.-C.; Tsay, Y.-F. Chl1 functions as a nitrate sensor in plants. Cell 2009, 138, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Hsu, P.K.; Tsay, Y.F. Uptake, allocation and signaling of nitrate. Trends Plant Sci. 2012, 17, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Bankston, J.R.; Payandeh, J.; Hinds, T.R.; Zagotta, W.N.; Zheng, N. Crystal structure of the plant dual-affinity nitrate transporter NRT1.1. Nature 2014, 507, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Forrest, L.R. Structural biology. (Pseudo-)symmetrical transport. Science 2013, 339, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Smirnova, I.; Kasho, V.; Verner, G.; Kaback, H.R.; Iwata, S. Structure and mechanism of the lactose permease of escherichia coli. Science 2003, 301, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Serdiuk, T.; Madej, M.G.; Sugihara, J.; Kawamura, S.; Mari, S.A.; Kaback, H.R.; Muller, D.J. Substrate-induced changes in the structural properties of LacY. Proc. Natl. Acad. Sci. USA 2014, 111, E1571–E1580. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newstead, S. Symmetry and Structure in the POT Family of Proton Coupled Peptide Transporters. Symmetry 2017, 9, 85. https://doi.org/10.3390/sym9060085
Newstead S. Symmetry and Structure in the POT Family of Proton Coupled Peptide Transporters. Symmetry. 2017; 9(6):85. https://doi.org/10.3390/sym9060085
Chicago/Turabian StyleNewstead, Simon. 2017. "Symmetry and Structure in the POT Family of Proton Coupled Peptide Transporters" Symmetry 9, no. 6: 85. https://doi.org/10.3390/sym9060085
APA StyleNewstead, S. (2017). Symmetry and Structure in the POT Family of Proton Coupled Peptide Transporters. Symmetry, 9(6), 85. https://doi.org/10.3390/sym9060085