
Mirror Complementary Triplet Periodicity of Dispersed Repeats in Bacterial Genomes


Abstract
1. Introduction
2. Materials and Methods
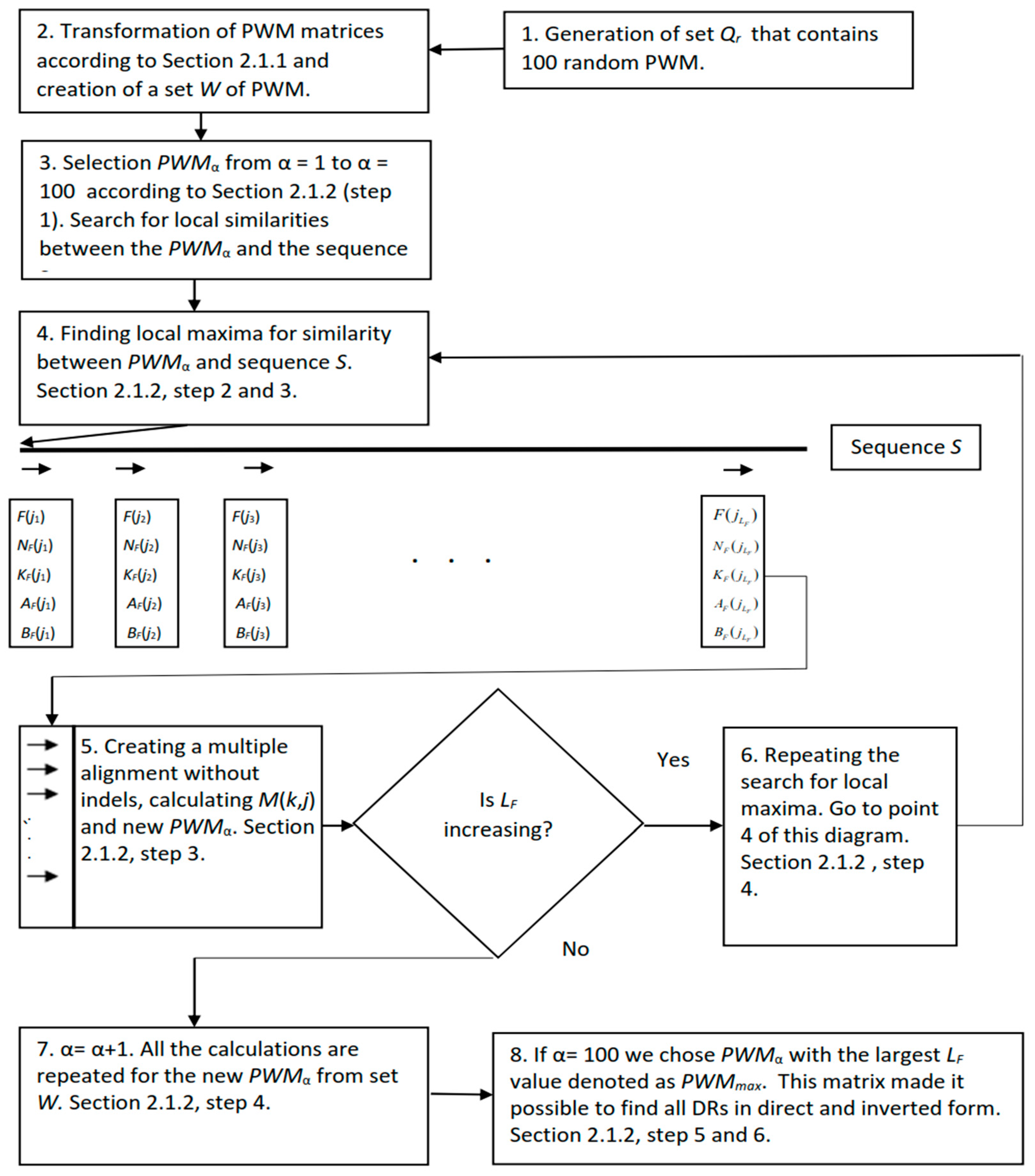
2.1. IP Method to Identify DRs in Bacterial Genomes
2.1.1. Generation of Random PWMs
2.1.2. Iterative Procedure to Search for DRs Using Matrices of Set W
2.1.3. Calculation of Statistical Significance
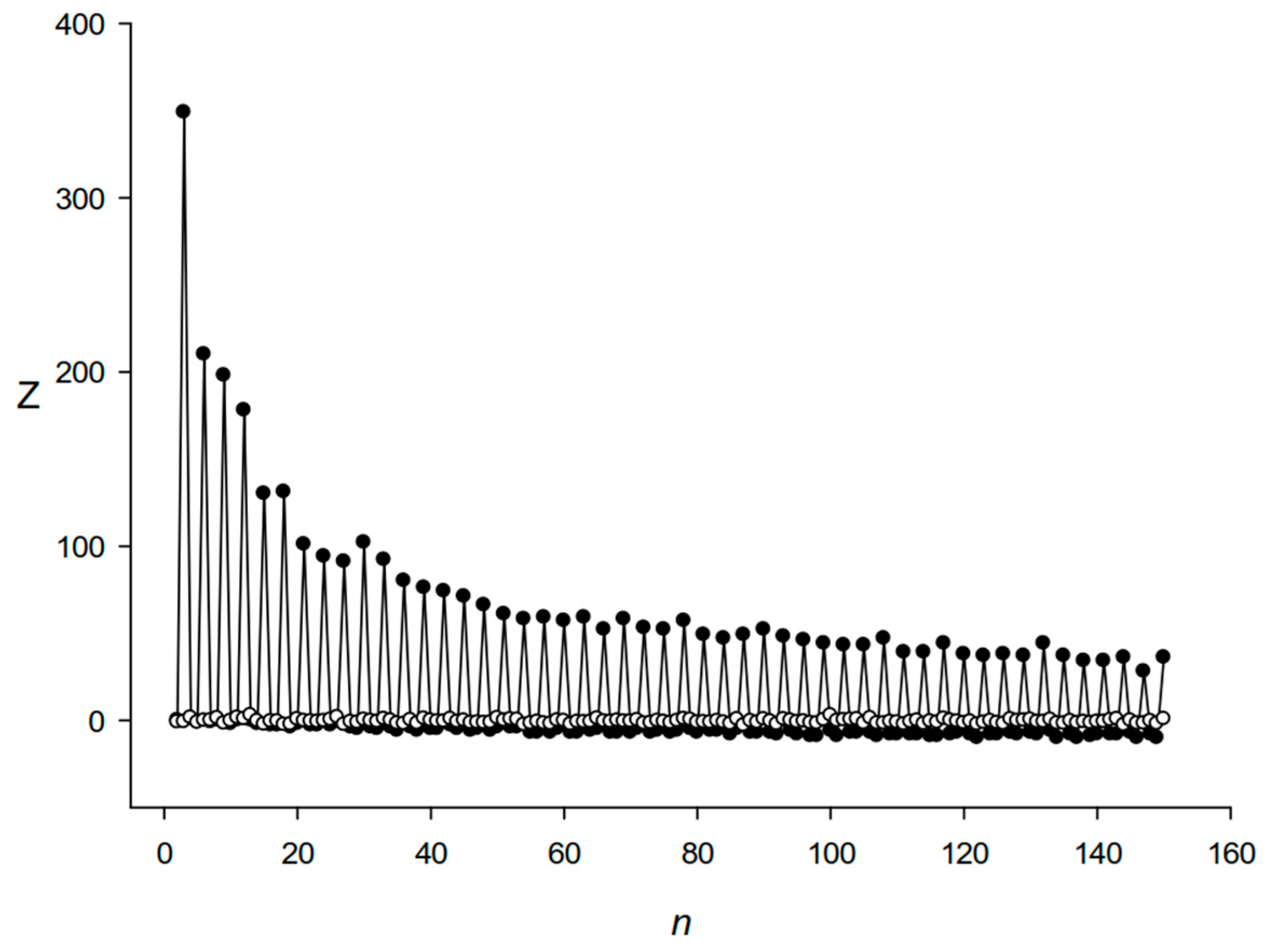
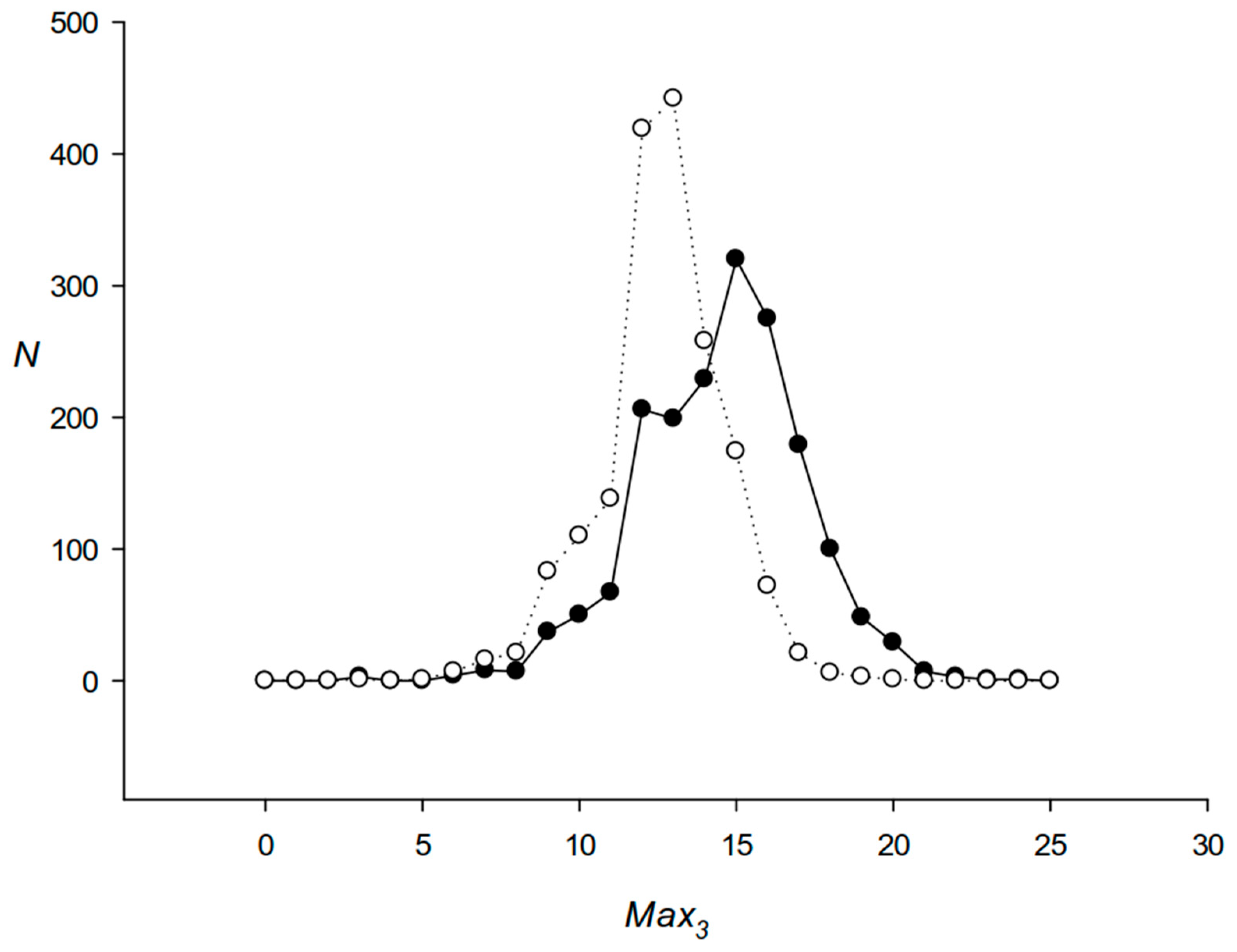
2.2. Algorithm to Search for the Length Periodicity in the Identified DRs
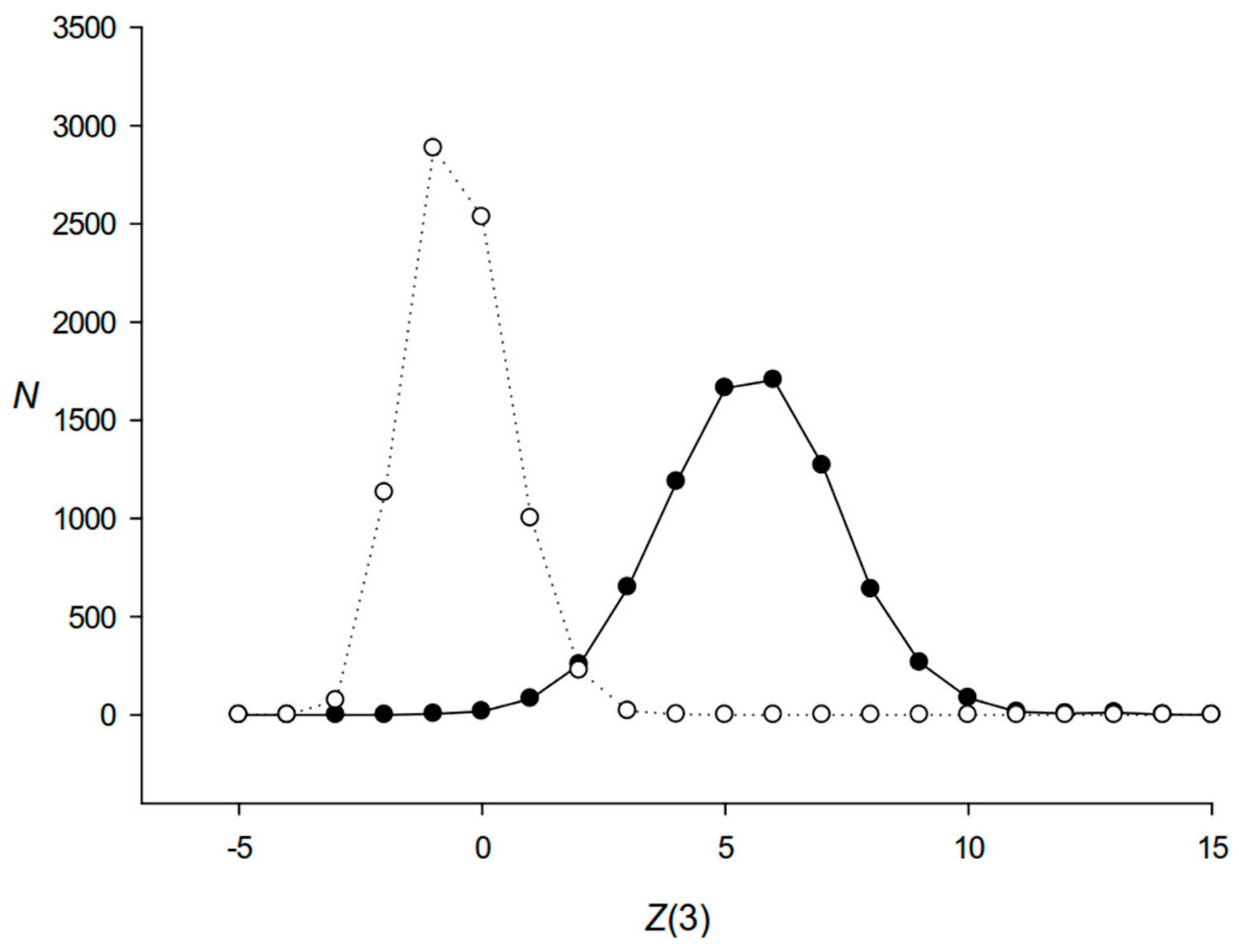
2.3. Calculation of Max3 for S12 Sequences
3. Results
3.1. Identification of DRs in the E. coli Genome
3.2. Intersection of DRs with the E. coli Genes
3.3. Analysis of Codon Frequencies in S12 and S34 Sequences
3.4. Search for DRs in the Genomes of Other Bacteria
4. Discussion
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Sherry, S.T.; Yankie, L.; Karsch-Mizrachi, I. GenBank 2024 Update. Nucleic Acids Res. 2024, 52, D134–D137. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, G.A.; Hunt, M.; Malone, K.M.; Lima, L.; Horesh, G.; Alako, B.T.F.; Thomson, N.R.; Iqbal, Z. Exploring bacterial diversity via a curated and searchable snapshot of archived DNA sequences. PLoS Biol. 2021, 19, e3001421. [Google Scholar] [CrossRef]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J. Clin. Med. 2020, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Liang, C. Generic repeat finder: A high-sensitivity tool for genome-wide de novo repeat detection. Plant Physiol. 2019, 180, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhu, W.; Zhou, J.; Li, H.; Xu, X.; Zhang, B.; Gao, X. Repetitive DNA sequence detection and its role in the human genome. Commun. Biol. 2023, 6, 954. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Kohany, O.; Jurka, M.V. Repetitive sequences in complex genomes: Structure and evolution. Annu. Rev. Genom. Hum. Genet. 2007, 8, 241–259. [Google Scholar] [CrossRef]
- Treangen, T.J.; Abraham, A.L.; Touchon, M.; Rocha, E.P.C. Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiol. Rev. 2009, 33, 539–571. [Google Scholar] [CrossRef]
- Versalovic, J.; Lupski, J.R. Bacterial Genomes. Physical Structure and Analysis; de Bruijn, F.J., Lupski, J.R., Weinstock, G.M., Eds.; Chapman & Hall: New York, NY, USA, 1998; pp. 38–48. [Google Scholar] [CrossRef]
- Storer, J.M.; Hubley, R.; Rosen, J.; Smit, A.F.A. Methodologies for the De novo Discovery of Transposable Element Families. Genes 2022, 13, 709. [Google Scholar] [CrossRef]
- Tempel, S. Mobile Genetic Elements. Protocols and Genomic Applications, 2nd ed.; Bigot, Y., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 29–51. [Google Scholar] [CrossRef]
- Jurka, J.; Klonowski, P.; Dagman, V.; Pelton, P. CENSOR—A program for identification and elimination of repetitive elements from DNA sequences. Comput. Chem. 1996, 20, 119–121. [Google Scholar]
- Bedell, J.A.; Korf, I.; Gish, W. MaskerAid: A performance enhancement to RepeatMasker. Bioinformatics 2000, 16, 1040–1041. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21, i152–i158. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef]
- Volfovsky, N.; Haas, B.J.; Salzberg, S.L. A clustering method for repeat analysis in DNA sequences. Genome Biol. 2001, 2, research0027.1. [Google Scholar] [CrossRef]
- Korotkov, E.; Suvorova, Y.; Kostenko, D.; Korotkova, M. Search for Dispersed Repeats in Bacterial Genomes Using an Iterative Procedure. Int. J. Mol. Sci. 2023, 24, 10964. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, E.; Korotkova, M. Detection of Dispersed Repeats in the Genomes of Bacteria from Different Phyla. IPSJ Trans. Bioinforma. 2024, 17, 55–63. [Google Scholar] [CrossRef]
- Suvorova, Y.M.; Korotkov, E.V. Study of triplet periodicity differences inside and between genomes. Stat. Appl. Genet. Mol. Biol. 2015, 14, 113–123. [Google Scholar] [CrossRef]
- Kullback, S. Statistics and Information Theory; J. Wiley and Sons: New York, NY, USA, 1959. [Google Scholar]
- Pugacheva, V.; Korotkov, A.; Korotkov, E. Search of latent periodicity in amino acid sequences by means of genetic algorithm and dynamic programming. Stat. Appl. Genet. Mol. Biol. 2016, 15, 381–400. [Google Scholar] [CrossRef]
- Mitchell, D.; Bridge, R. A test of Chargaff’s second rule. Biochem. Biophys. Res. Commun. 2006, 340, 90–94. [Google Scholar] [CrossRef]
- Shporer, S.; Chor, B.; Rosset, S.; Horn, D. Inversion symmetry of DNA k-mer counts: Validity and deviations. BMC Genom. 2016, 17, 696. [Google Scholar] [CrossRef]
- Matkarimov, B.T.; Saparbaev, M.K. Chargaff’s second parity rule lies at the origin of additive genetic interactions in quantitative traits to make omnigenic selection possible. PeerJ 2023, 11, e16671. [Google Scholar] [CrossRef]
- Hart, A.; Martínez, S.; Olmos, F. A Gibbs Approach to Chargaff’s Second Parity Rule. J. Stat. Phys. 2012, 146, 408–422. [Google Scholar] [CrossRef]
- Fariselli, P.; Taccioli, C.; Pagani, L.; Maritan, A. DNA sequence symmetries from randomness: The origin of the Chargaff’s second parity rule. Brief. Bioinform. 2021, 22, 2172–2181. [Google Scholar] [CrossRef]
- Albrecht-Buehler, G. Asymptotically increasing compliance of genomes with Chargaff’s second parity rules through inversions and inverted transpositions. Proc. Natl. Acad. Sci. USA 2006, 103, 17828–17833. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, T.; Marzi, S.; Romby, P. The role of mRNA structure in translational control in bacteria. RNA Biol. 2009, 6, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Forsdyke, D.R. Genomic compliance with Chargaff’s second parity rule may have originated non-adaptively, but stem-loops now function adaptively. J. Theor. Biol. 2024, 595, 111943. [Google Scholar] [CrossRef]
- Yevdokimov, Y.M.; Salyanov, V.I.; Nechipurenko, Y.D.; Skuridin, S.G.; Zakharov, M.A.; Spener, F.; Palumbo, M. Molecular Constructions (Superstructures) with Adjustable Properties Based on Double-Stranded Nucleic Acids. Mol. Biol. 2003, 37, 293–306. [Google Scholar] [CrossRef]
- Yevdokimov, Y.M.; Salyanov, V.I.; Skuridin, S.G. From liquid crystals to DNA nanoconstructions. Mol. Biol. 2009, 43, 284–300. [Google Scholar] [CrossRef]
- Skuridin, S.G.; Vereshchagin, F.V.; Salyanov, V.I.; Chulkov, D.P.; Kompanets, O.N.; Yevdokimov, Y.M. Ordering of double-stranded DNA molecules in a cholesteric liquid-crystalline phase and in dispersion particles of this phase. Mol. Biol. 2016, 50, 783–790. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Base | 1 | 2 | 3 |
|---|---|---|---|
| A | 186,352 | 218,972 | 113,679 |
| T | 111,922 | 217,757 | 183,613 |
| C | 192,006 | 140,503 | 229,042 |
| G | 227,398 | 140,446 | 191,344 |
| Base | 1 | 2 | 3 |
|---|---|---|---|
| A | 33.5 | 115.3 | −148.7 |
| T | −149.1 | 117.3 | 31.5 |
| C | 11.6 | −112.9 | 101.4 |
| G | 99.3 | −111.4 | 12.0 |
| Gene Location | Total Number of Genes | Number of Genes with at Least One + DR | Number of Genes with S12 or S34 Sequences |
|---|---|---|---|
| Plus strand | 2029 | 1448 | 1256 (1451 S12) |
| Minus strand | 2145 | 1479 | 1371 (1564 S34) |
| Base | 1 | 2 | 3 |
|---|---|---|---|
| A | 13.6 | 62.6 | −76.1 |
| T | −97.1 | 75.8 | 21.4 |
| C | −1.5 | −33.2 | 34.7 |
| G | 79.0 | −97.2 | 18.2 |
| Codon Number (i) | Codon | X(i) |
|---|---|---|
| 1 | GAA | 172.1028 |
| 2 | CTG | 159.4557 |
| 3 | AAA | 145.2018 |
| 4 | GCG | 95.5202 |
| 5 | GAT | 94.2846 |
| 6 | CAG | 88.2709 |
| 7 | GGC | 79.1911 |
| 8 | AAC | 63.4375 |
| 9 | ATG | 58.9810 |
| 10 | ATT | 58.2361 |
| 11 | ATC | 56.1757 |
| 12 | GCC | 55.1771 |
| 13 | ACC | 54.8639 |
| 14 | GTG | 46.0153 |
| 15 | GAC | 40.2726 |
| 16 | CGC | 36.9499 |
| 17 | CCG | 35.9462 |
| 18 | GAG | 32.4702 |
| 19 | GGT | 30.2339 |
| 20 | GCA | 27.5429 |
| 21 | AAT | 22.9861 |
| 22 | CAA | 20.2570 |
| 23 | AGC | 17.3211 |
| 24 | CGT | 10.5570 |
| 25 | ACG | 5.6548 |
| Number | Species | Number of Genes with | Number of | Genome Size (106 Bases) | ||
|---|---|---|---|---|---|---|
| At Least One S12 Sequence | At Least One S34 Sequence | S12 Sequences | S34 Sequences | |||
| 1 | Azotobacter vinelandi | 1034 | 1388 | 1199 | 1193 | 5.3 |
| 2 | Bacillis subtilis | 1033 | 1262 | 1176 | 1133 | 4.2 |
| 3 | Clostridium tetani | 792 | 891 | 913 | 1039 | 2.8 |
| 4 | Methylococcus capsula | 914 | 881 | 1143 | 1046 | 3.3 |
| 5 | Mycobacterium tuberculosis | 1370 | 1377 | 1160 | 1137 | 4.4 |
| 6 | Salinispora arenicola | 1737 | 1564 | 1460 | 1324 | 5.8 |
| 7 | Shigella sonnei | 1149 | 1202 | 1007 | 1065 | 4.9 |
| 8 | Thermosipho africanus | 632 | 718 | 559 | 624 | 2.0 |
| 9 | Treponema pallidum | 351 | 280 | 299 | 233 | 1.1 |
| 10 | Xanthomonas campestri | 1568 | 1583 | 1334 | 1326 | 5.2 |
| 11 | Yersinia pestis | 1446 | 1457 | 1268 | 1268 | 4.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korotkov, E.V. Mirror Complementary Triplet Periodicity of Dispersed Repeats in Bacterial Genomes. Symmetry 2025, 17, 549. https://doi.org/10.3390/sym17040549
Korotkov EV. Mirror Complementary Triplet Periodicity of Dispersed Repeats in Bacterial Genomes. Symmetry. 2025; 17(4):549. https://doi.org/10.3390/sym17040549
Chicago/Turabian StyleKorotkov, Eugene Vadimovitch. 2025. "Mirror Complementary Triplet Periodicity of Dispersed Repeats in Bacterial Genomes" Symmetry 17, no. 4: 549. https://doi.org/10.3390/sym17040549
APA StyleKorotkov, E. V. (2025). Mirror Complementary Triplet Periodicity of Dispersed Repeats in Bacterial Genomes. Symmetry, 17(4), 549. https://doi.org/10.3390/sym17040549