

Thioureas Derived from (S)-1-(2-pyridyl)ethylamine Enantiomer: Synthesis and Selected Applications as an Organocatalyst
 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
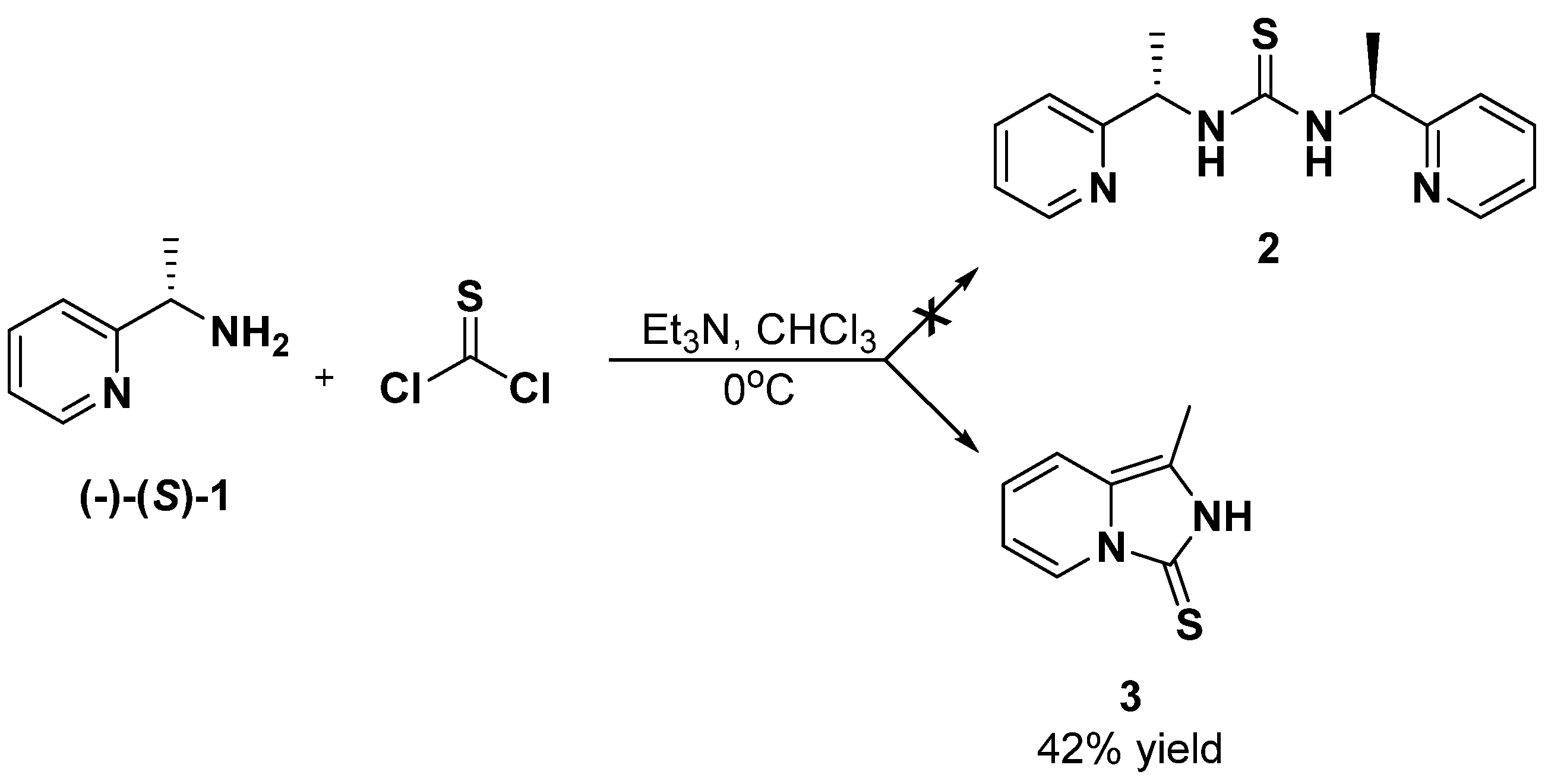
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. Materials and Methods
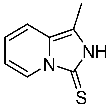
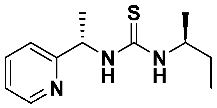
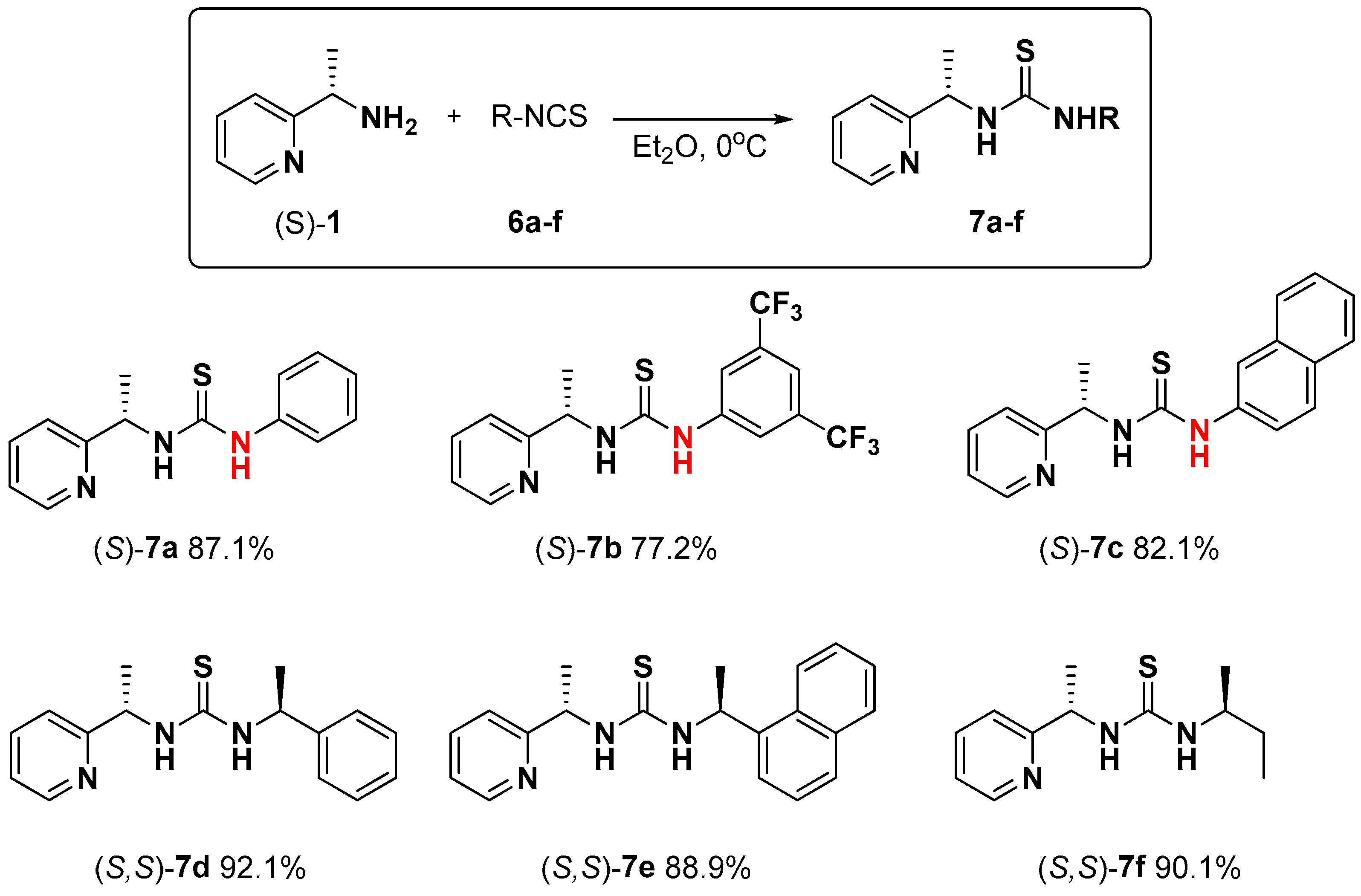
4.2. General Procedure for the Synthesis of Asymmetric Thioureas 7a–f
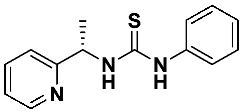
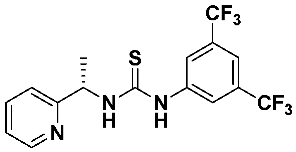


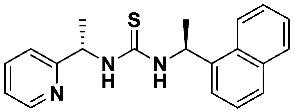
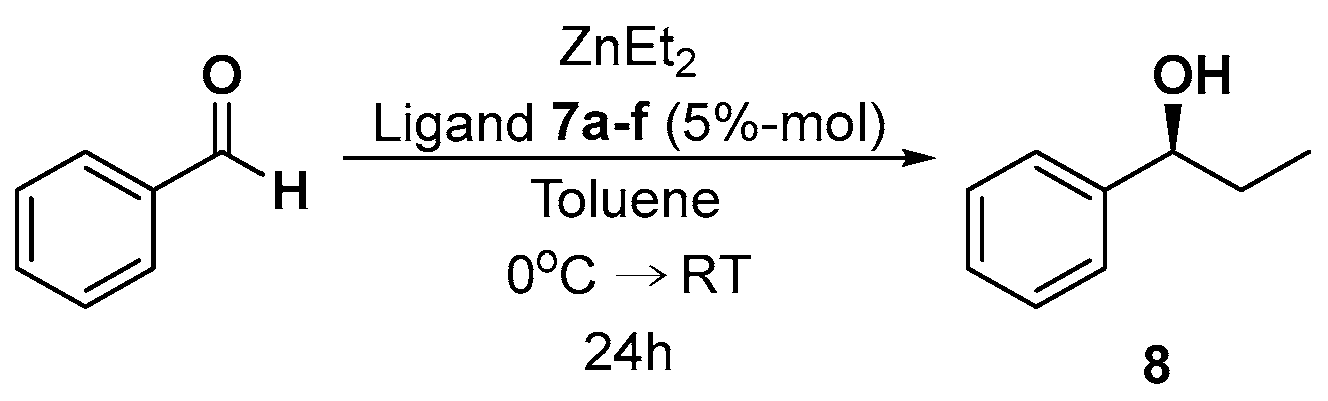
4.3. Asymmetric Addition of Diethylzinc to Benzaldehyde
4.4. Asymmetric Aldol Condensation
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels–Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Reetz, M.T.; List, B.; Jaroch, S.; Weinmann, H. (Eds.) Organocatalysis; Ernst Schering Foundation Symposium Proceedings; Springer: Berlin/Heidelberg, Germany, 2008; Volume 2007/2. [Google Scholar] [CrossRef]
- List, B. (Ed.) Asymmetric Organocatalysis; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2009; Volume 291. [Google Scholar] [CrossRef]
- Dalko, P.I. (Ed.) Comprehensive Enantioselective Organocatalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar] [CrossRef]
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis; Wiley: New York, NY, USA, 2005. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric Organocatalytic Tandem/Domino Reactions to Access Bioactive Products. Curr. Org. Chem. 2021, 25, 1457–1471. [Google Scholar] [CrossRef]
- Carlone, A.; Bernardi, L. Enantioselective Organocatalytic Approaches to Active Pharmaceutical Ingredients—Selected Industrial Examples. Phys. Sci. Rev. 2019, 4, 401–434. [Google Scholar] [CrossRef]
- Hughes, D.L. Asymmetric Organocatalysis in Drug Development—Highlights of Recent Patent Literature. Org. Process Res. Dev. 2018, 22, 574–584. [Google Scholar] [CrossRef]
- Ardevines, S.; Marqués-López, E.; Herrera, R.P. Horizons in Asymmetric Organocatalysis: En Route to the Sustainability and New Applications. Catalysts 2022, 12, 101. [Google Scholar] [CrossRef]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic Cascade Reactions as a New Tool in Total Synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef]
- Howell, G.P. Asymmetric and Diastereoselective Conjugate Addition Reactions: C–C Bond Formation at Large Scale. Org. Process Res. Dev. 2012, 16, 1258–1272. [Google Scholar] [CrossRef]
- Simmons, E.M.; Mudryk, B.; Lee, A.G.; Qiu, Y.; Razler, T.M.; Hsiao, Y. Development of a Kilogram-Scale Process for the Enantioselective Synthesis of 3-Isopropenyl-Cyclohexan-1-One via Rh/DTBM-SEGPHOS-Catalyzed Asymmetric Hayashi Addition Enabled by 1,3-Diol Additives. Org. Process Res. Dev. 2017, 21, 1659–1667. [Google Scholar] [CrossRef]
- Dalko, P.I.; Moisan, L. Enantioselective Organocatalysis. Angew. Chem. Int. Ed. 2001, 40, 3726–3748. [Google Scholar] [CrossRef]
- Dalko, P.I.; Moisan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef] [PubMed]
- Houk, K.N.; List, B. Asymmetric Organocatalysis. Acc. Chem. Res. 2004, 37, 487. [Google Scholar] [CrossRef]
- List, B.; Yang, J.W. The Organic Approach to Asymmetric Catalysis. Science 2006, 313, 1584–1586. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric Organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- Dondoni, A.; Massi, A. Asymmetric Organocatalysis: From Infancy to Adolescence. Angew. Chem. Int. Ed. 2008, 47, 4638–4660. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric Aminocatalysis-Gold Rush in Organic Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef] [PubMed]
- Sigman, M.S.; Jacobsen, E.N. Schiff Base Catalysts for the Asymmetric Strecker Reaction Identified and Optimized from Parallel Synthetic Libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Schreiner, P.R.; Wittkopp, A. H-Bonding Additives Act Like Lewis Acid Catalysts. Org. Lett. 2002, 4, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Highly Enantioselective Catalytic Acyl-Pictet–Spengler Reactions. J. Am. Chem. Soc. 2004, 126, 10558–10559. [Google Scholar] [CrossRef]
- Abermil, N.; Masson, G.; Zhu, J. Highly Enantioselective Aza Morita–Baylis–Hillman Reaction Catalyzed by Bifunctional β-Isocupreidine Derivatives. J. Am. Chem. Soc. 2008, 130, 12596–12597. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Shih, H.-W.; Deng, L. Asymmetric Mannich Reactions with in Situ Generation of Carbamate-Protected Imines by an Organic Catalyst. Org. Lett. 2007, 9, 603–606. [Google Scholar] [CrossRef]
- Vera, S.; Garcia-Urricelquui, A.; Mielgo, A. Oriabide Progress in (Thio)urea- and Squaramide-Based Brønsted Base Catalysts with Multiple H-Bond Donors. Eur. J. Org. Chem. 2023, 26, e202201254. [Google Scholar] [CrossRef]
- Steppeler, F.; Iwan, D.; Wojaczynska, E.; Wojaczynski, J. Chiral Thioureas-Preparation and Significance in Asymmetric Synthesis. Molecules 2020, 25, 401. [Google Scholar] [CrossRef]
- Gandhi, S.; Sivadas, V.; Baire, B. Thio-urea-Amine Promoted Cascade Catalysis: A Tool for Complexity generation. Eur. J. Org. Chem. 2021, 2021, 220–234. [Google Scholar] [CrossRef]
- Drabowicz, J.; Kłos, M.; Chrzanowski, J. Sposób Rozdzielania Racemicznych 1-(2 lub 3)-pirydyloetyloamin na Enancjomery. Polish Patent P-411027, 13 May 2021. [Google Scholar]
- Brunner, H.; Fisch, H. Asymmetrische Katalysen, 39. Mitt.: Monohydrosilane in der enantioselektiven katalytischen Hydrosilylierung prochiraler Ketone. Monatshefte Chem./Chem. Mon. 1988, 119, 525–532. [Google Scholar] [CrossRef]
- Brunner, H.; Fisch, H. Asymmetrische katalysen XXXVI. Neue mehrzähnige liganden mit dem (S)-(α)-(2-Pyridyl)ethylrest; Rh-komplexe und enantioselektive hydrosilylierungen. J. Organomet. Chem. 1987, 335, 1–14. [Google Scholar] [CrossRef]
- Ma, Z.; Kuloor, C.; Kreyenschulte, C.; Bartling, S.; Malina, O.; Haumann, M.; Menezes, P.W.; Zboril, R.; Beller, M.; Jagadeesh, R.V. Development of iron- based single atom materials for general a nd efficient synthesis of amines. Angew. Chem. Int. Ed. 2024, 63, e20240785. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Wang, C.; Miao, H.; Qin, Z.; Huang, M.; Xu, Y.; Xue, W.; Yang, S.; Liu, C.; Bai, C.; et al. Novel bidentate N-coordinated alkylaluminum complexes: Synthesis, characterization, andefficient catalysis for hydrophosphonylation. Dalton Trans. 2024, 53, 4186–4193. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.-R.; Qi, T.-T.; Liu, S.-J.; Liu, C.-M.; Tang, Y.-Z.; Chen, J.-L. Syntheses and structures of chiral tri- and tetranuclear Cd(II) clusters with luminescent and ferroelectric properties. Polyhedron 2015, 85, 894–899. [Google Scholar] [CrossRef]
- Marinescu, M.; Popa, C.-V. Pyridine compounds with antimicrobial and antivarius activity. Int. J. Mol. Sci. 2022, 23, 5650. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.J.; Jecs, E.; Miller, E.J.; Nguyen, H.H.; Tahirovic, Y.A.; Truax, V.M.; Kim, M.B.; Kuo, K.M.; Wang, T.; Sum, C.S.; et al. Synthesis and SAR of 1,2,3,4-Tetrahydroisoquinoline-Based CXCR4 Antagonists. ACS Med. Chem. Lett. 2018, 9, 17–22. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Chen, C.-R.; Chen, C.Y.; Liang, P.-H. Synthesis of Quillaic Acid through Sustainable C–H Bond Activations. J. Org. Chem. 2024, 89, 5491–5497. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Lee, G.H.; No, Z.S. Direct Access to Various 1-Substituted-Imidazo[1,5-a]Pyridine-3(2H)-Thione Derivatives. Bull. Korean Chem. Soc. 2007, 28, 685–688. [Google Scholar] [CrossRef]
- Ferreira, R.B.; Tormena, C.F.; Almeida, W.P. Synthesis and Spectroscopic Analysis of Substituted 2-Aminothiazolines. J. Mol. Struct. 2013, 1037, 186–190. [Google Scholar] [CrossRef]
- Leśniak, S.; Rachwalski, M.; Sznajder, E.; Kiełbasiński, P. New highly efficient aziridine-functionalized tridentate sulfinyl catalysts for enantioselective diethylzinc addition to carbonyl compounds. Tetrahedron Asymmetry 2009, 20, 2311–2314. [Google Scholar] [CrossRef]
- Muramatsu, Y.; Harada, T. Catalytic Asymmetric Alkylation of Aldehydes with Grignard Reagents. Angew. Chem. Int. Ed. 2008, 47, 1088. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Tomihama, M.; Yamamoto, K.; Matsubara, N.; Harada, T.J. Method for Catalytic Enantioselective Alkylation of Aldehydes Using Grignard Reagents as Alkyl Sources. J. Org. Chem. 2018, 83, 6127. [Google Scholar] [CrossRef] [PubMed]
- Rachwalski, M.; Leśniak, S.; Kiełbasiński, P. Highly enantioselective asymmetric direct aldol reaction catalyzed by amine-functionalized tridentate sulfinyl ligands. Tetrahedron Asymmetry 2011, 22, 1325–1327. [Google Scholar] [CrossRef]
- Fotaras, S.; Kokotos, C.G.; Kokotos, G. A Tripeptide-like Prolinamide-Thiourea as an Aldol Reaction Catalyst. Org. Biomol. Chem. 2012, 10, 5613. [Google Scholar] [CrossRef]
- Mei, K.; Zhang, S.; He, S.; Li, P.; Jin, M.; Xue, F.; Luo, G.; Zhang, H.; Song, L.; Duan, W.; et al. (S)-Pyrrolidine sulfonamide catalyzed asymmetric direct aldol reactions of aryl methyl ketones with aryl aldehydes. Tetrahedron Lett. 2008, 49, 2681–2685. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Product 8 | |||
|---|---|---|---|---|---|
| Yield [%] | [α]D b | ee [%] a | Abs. Conf. [41] | ||
| 1 | 7a | 6 | −3.08 | 7 | S |
| 2 | 7b | 20 | −3.96 | 9 | S |
| 3 | 7c | 5 | −2.20 | 5 | S |
| 4 | 7d | 12 | −8.80 | 20 | S |
| 5 | 7e | 10 | −9.68 | 22 | S |
| 6 | 7f | 5 | 6.60 | 15 | S |
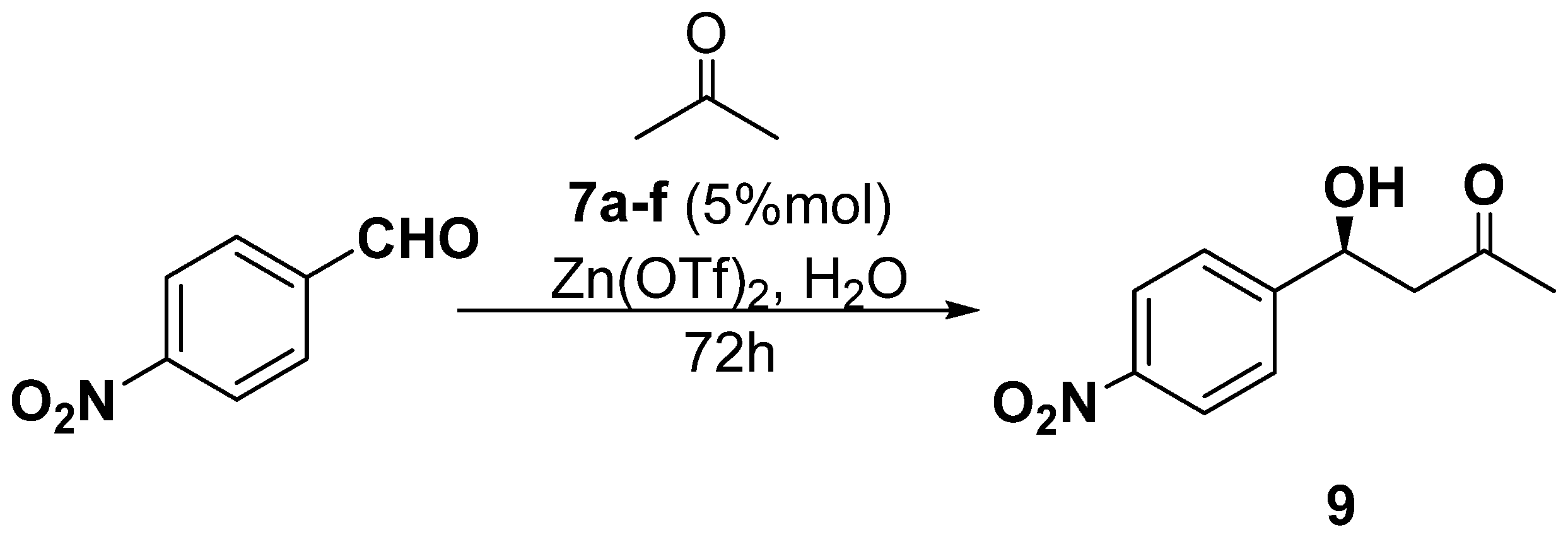
| Entry | Catalyst | Product 9 | |||
|---|---|---|---|---|---|
| Yield [%] | [α]D b | ee [%] a | Abs. Conf. [44] | ||
| 1 | 7a | 10 | −9.08 | 15 | S |
| 2 | 7b | 40 | −19.99 | 33 | S |
| 3 | 7c | 11 | −8.48 | 14 | S |
| 4 | 7d | 35 | −12.72 | 21 | S |
| 5 | 7e | 42 | −21.20 | 35 | S |
| 6 | 7f | 12 | −11.51 | 19 | S |
| 7 | 7e b | 81 | 0.00 | 0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrzanowski, J.; Sancineto, L.; Deska, M.; Rachwalski, M.; Drabowicz, J. Thioureas Derived from (S)-1-(2-pyridyl)ethylamine Enantiomer: Synthesis and Selected Applications as an Organocatalyst. Symmetry 2025, 17, 216. https://doi.org/10.3390/sym17020216
Chrzanowski J, Sancineto L, Deska M, Rachwalski M, Drabowicz J. Thioureas Derived from (S)-1-(2-pyridyl)ethylamine Enantiomer: Synthesis and Selected Applications as an Organocatalyst. Symmetry. 2025; 17(2):216. https://doi.org/10.3390/sym17020216
Chicago/Turabian StyleChrzanowski, Jacek, Luca Sancineto, Malgorzata Deska, Michal Rachwalski, and Jozef Drabowicz. 2025. "Thioureas Derived from (S)-1-(2-pyridyl)ethylamine Enantiomer: Synthesis and Selected Applications as an Organocatalyst" Symmetry 17, no. 2: 216. https://doi.org/10.3390/sym17020216
APA StyleChrzanowski, J., Sancineto, L., Deska, M., Rachwalski, M., & Drabowicz, J. (2025). Thioureas Derived from (S)-1-(2-pyridyl)ethylamine Enantiomer: Synthesis and Selected Applications as an Organocatalyst. Symmetry, 17(2), 216. https://doi.org/10.3390/sym17020216