
Photophysical Properties and Electronic Structure of Symmetrical Curcumin Analogues and Their BF2 Complexes, Including a Phenothiazine Substituted Derivative


Abstract
:
1. Introduction
2. Materials and Methods
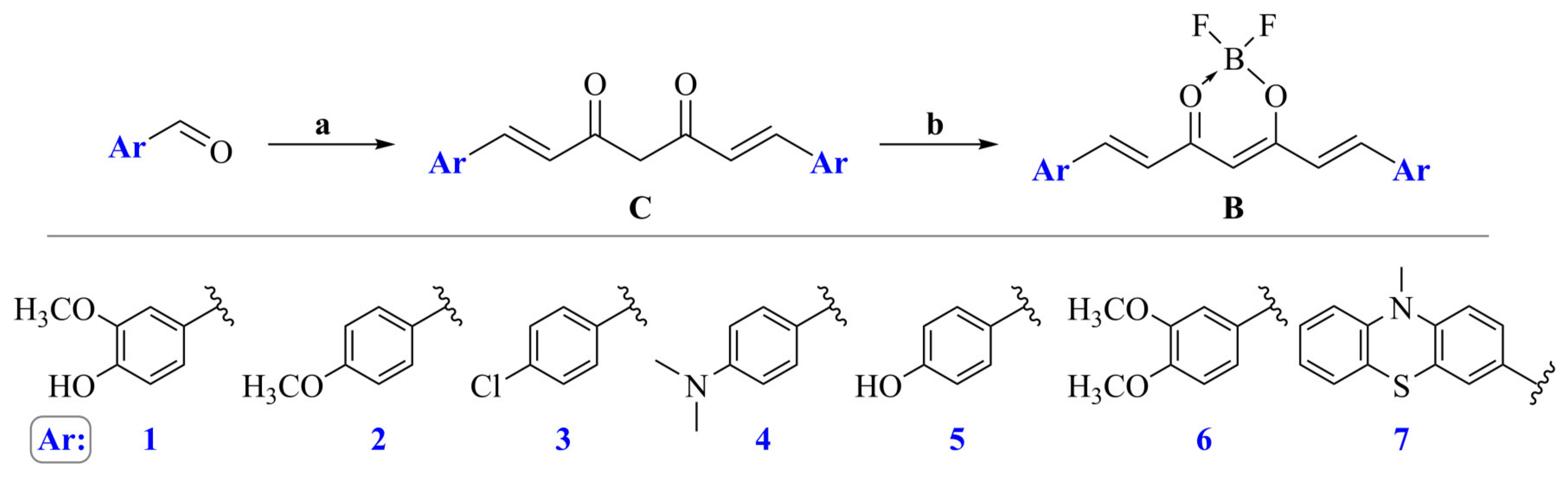
2.1. General Procedure for the Synthesis of Curcumin Derivatives (C1–C7)
2.2. General Procedure for the Synthesis of Curcumin Derivatives (B1–B7)
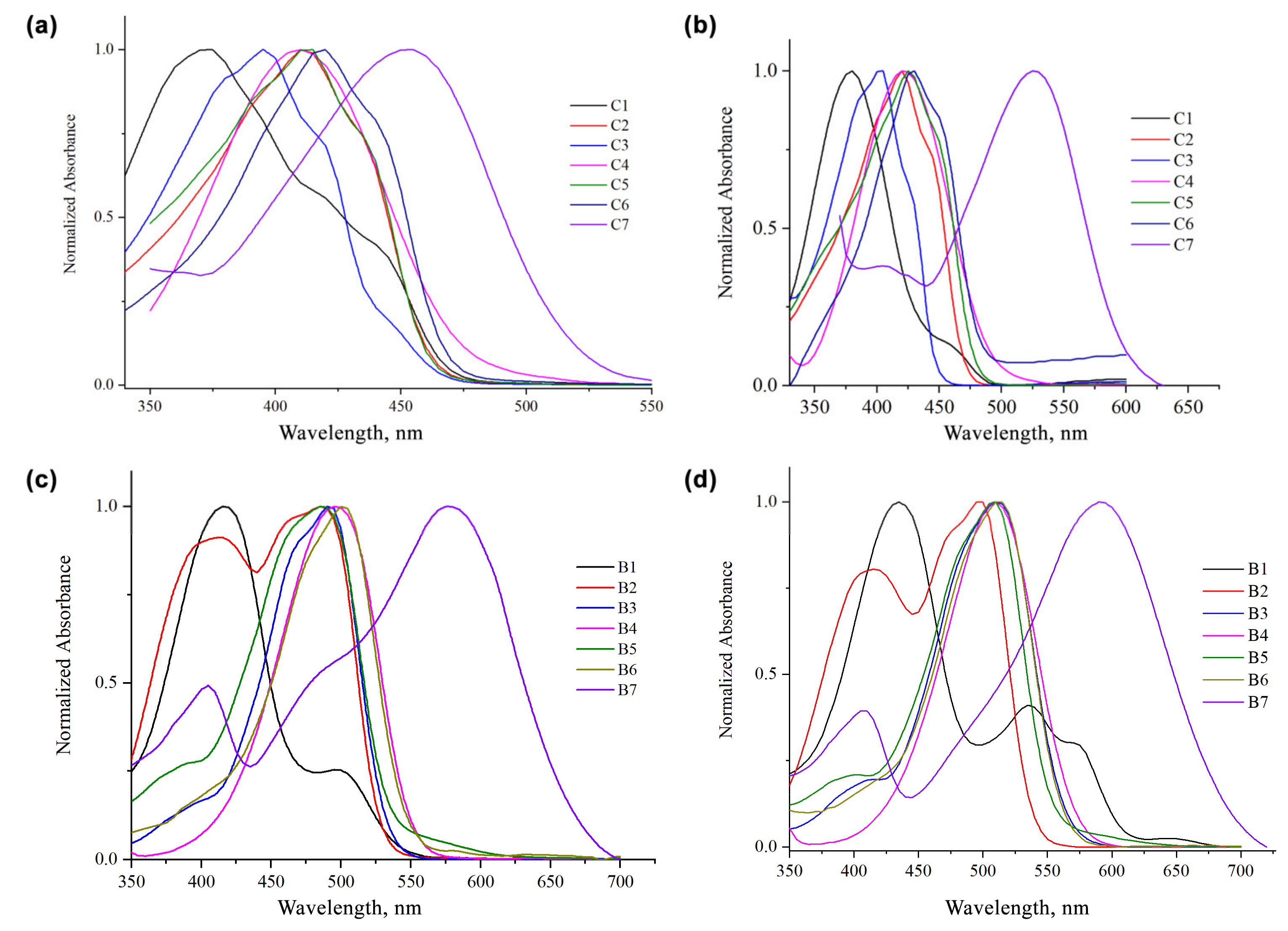
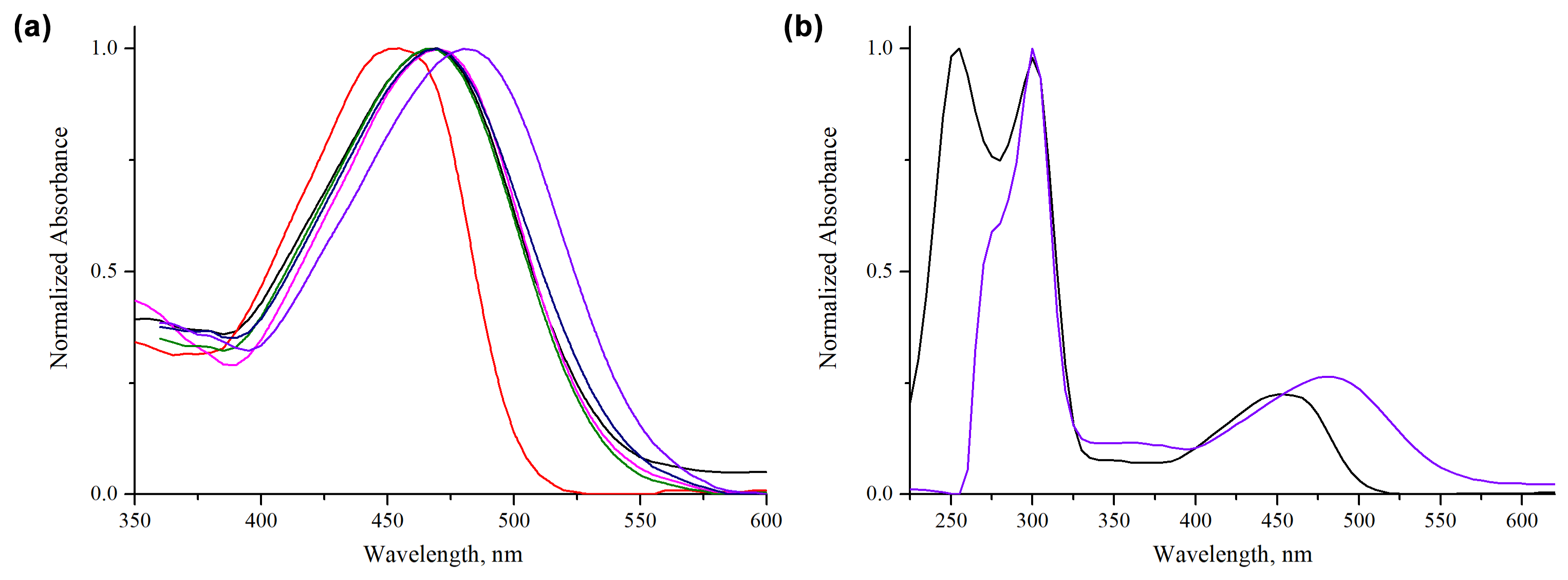
3. Results
4. Theoretical Study
4.1. Computational Details
4.2. Computational Results
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Canard, G.; Ponce-Vargas, M.; Jacquemin, D.; Le Guennic, B.; Felouat, A.; Rivoal, M.; Zaborova, E.; D’Aléo, A.; Fages, F. Influence of the electron donor groups on the optical and electrochemical properties of borondifluoride complexes of curcuminoid derivatives: A joint theoretical and experimental study. RSC Adv. 2017, 7, 10132–10142. [Google Scholar] [CrossRef] [Green Version]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.A.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin-from molecule to biological function. Angew. Chem. Int. Ed. 2012, 51, 5308–5332. [Google Scholar] [CrossRef]
- Sui, Z.; Salto, R.; Li, J.; Craik, C.; Ortiz de Montellano, P.R. Inhibition of the HIV-1 and HIV-2 proteases by curcumin and curcumin boron complexes. Bioorg. Med. Chem. 1993, 1, 415–422. [Google Scholar] [CrossRef]
- Bai, G.; Yu, C.; Cheng, C.; Hao, E.; Wei, Y.; Mu, X.; Jiao, L. Syntheses and photophysical properties of BF2 complexes of curcumin analogues. Org. Biomol. Chem. 2014, 12, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Wang, D.; Cai, L.; Wang, D.J.; Li, X.M. Synthesis, photophysical and solvatochromic properties of diacetoxyboron complexes with curcumin derivatives. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 220, 117126. [Google Scholar] [CrossRef] [PubMed]
- Margar, S.N.; Rhyman, L.; Ramasami, P.; Sekar, N. Fluorescent difluoroboron-curcumin analogs: An investigation of the electronic structures and photophysical properties. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2016, 152, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, L.; Kanaan, H.; Camerel, F.; Jolinat, P.; Retailleau, P.; Ziessel, R. Fine-tuning of yellow or red photo- and electroluminescence of functional difluoro-boradiazaindacene films. Adv. Funct. Mater. 2008, 18, 401–413. [Google Scholar] [CrossRef]
- Chen, P.Z.; Niu, L.Y.; Chen, Y.Z.; Yang, Q.Z. Difluoroboron β-diketonate dyes: Spectroscopic properties and applications. Coord. Chem. Rev. 2017, 350, 196–216. [Google Scholar] [CrossRef]
- Kazantzis, K.T.; Koutsonikoli, K.; Mavroidi, B.; Zachariadis, M.; Alexiou, P.; Pelecanou, M.; Politopoulos, K.; Alexandratou, E.; Sagnou, M. Curcumin derivatives as photosensitizers in photodynamic therapy: Photophysical properties and: In vitro studies with prostate cancer cells. Photochem. Photobiol. Sci. 2020, 19, 193–206. [Google Scholar] [CrossRef]
- Ding, L.; Ma, S.; Lou, H.; Sun, L.; Ji, M. Synthesis and biological evaluation of curcumin derivatives with water-soluble groups as potential antitumor agents: An in vitro investigation using tumor cell lines. Molecules 2015, 20, 21501–21514. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Chang, S.; He, J.; Zhang, C.; Zhao, J.; Chen, T.; Wong, W.Y.; Wong, W.K.; Zhu, X. Molecular engineering of simple phenothiazine-based dyes to modulate dye aggregation, charge recombination, and dye regeneration in highly efficient dye-sensitized solar cells. Chem. Eur. J. 2014, 20, 6300–6308. [Google Scholar] [CrossRef] [PubMed]
- Al-Busaidi, I.J.; Haque, A.; Al Rasbi, N.K.; Khan, M.S. Phenothiazine-based derivatives for optoelectronic applications: A review. Synth. Met. 2019, 257, 116189. [Google Scholar] [CrossRef]
- Luo, J.S.; Wan, Z.Q.; Jia, C.Y. Recent advances in phenothiazine-based dyes for dye-sensitized solar cells. Chin. Chem. Lett. 2016, 27, 1304–1318. [Google Scholar] [CrossRef]
- Lampe, V.; Milobedzka, J. Studien über Curcumin. Ber. Dtsch. Chem. Ges. 1913, 46, 2235–2240. [Google Scholar] [CrossRef]
- Pavolini, T.; Gambarin, F.; Grinzato, A.M. Curcumina e curcuminoidi. Ann. Chim. 1950, 40, 280–291. [Google Scholar]
- Pabon, H.J.J. A synthesis of curcumin and related compounds. Recl. Trav. Chim. Pays-Bas 1964, 83, 379–386. [Google Scholar] [CrossRef]
- Weiss, H.; Reichel, J.; Görls, H.; Schneider, K.R.A.; Micheel, M.; Pröhl, M.; Gottschaldt, M.; Dietzek, B.; Weigand, W. Curcuminoid-BF2 complexes: Synthesis, fluorescence and optimization of BF2 group cleavage. Beilstein J. Org. Chem. 2017, 13, 2264–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkata Rao, E.; Sudheer, P. Revisiting curcumin chemistry part I: A new strategy for the synthesis of curcuminoids. Indian J. Pharm. Sci. 2011, 73, 262–270. [Google Scholar] [CrossRef]
- Rao, E.V.; Prasad, Y.R.; Sudheer, P. Revisiting Curcumin Chemistry-Part II: Synthesis of Monomethylcurcumin and Isomeric Demethoxycurcumins and their Characterization. Indian J. Pharm. Sci. 2017, 79, 820–828. [Google Scholar] [CrossRef]
- Deck, L.M.; Hunsaker, L.A.; Vander Jagt, T.A.; Whalen, L.J.; Royer, R.E.; Vander Jagt, D.L. Activation of anti-oxidant Nrf2 signaling by enone analogues of curcumin. Eur. J. Med. Chem. 2018, 143, 854–865. [Google Scholar] [CrossRef]
- Weber, W.M.; Hunsaker, L.A.; Abcouwer, S.F.; Deck, L.M.; Vander Jagt, D.L. Anti-oxidant activities of curcumin and related enones. Bioorg. Med. Chem. 2005, 13, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Sherin, D.R.; Thomas, S.G.; Rajasekharan, K.N. Mechanochemical synthesis of 2,2-difluoro-4, 6-bis(β-styryl)-1,3,2-dioxaborines and their use in cyanide ion sensing. Heterocycl. Commun. 2015, 21, 381–385. [Google Scholar] [CrossRef]
- Felouat, A.; D’Aléo, A.; Fages, F. Synthesis and photophysical properties of difluoroboron complexes of curcuminoid derivatives bearing different terminal aromatic units and a meso-aryl ring. J. Org. Chem. 2013, 78, 4446–4455. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Chen, J.; Chojnacki, J.; Zhang, S. BF3·OEt2-promoted concise synthesis of difluoroboron-derivatized curcumins from aldehydes and 2,4-pentanedione. Tetrahedron Lett. 2013, 54, 2070–2073. [Google Scholar] [CrossRef] [Green Version]
- Laali, K.K.; Rathman, B.M.; Bunge, S.D.; Qi, X.; Borosky, G.L. Fluoro-curcuminoids and curcuminoid-BF2adducts: Synthesis, X-ray structures, bioassay, and computational/docking study. J. Fluor. Chem. 2016, 191, 29–41. [Google Scholar] [CrossRef]
- Khopde, S.M.; Priyadarsini, K.I.; Palit, D.K.; Mukherjee, T. Effect of solvent on the excited-state photophysical properties of curcumin. Photochem. Photobiol. 2000, 72, 625–631. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Mary, C.P.V.; Vijayakumar, S.; Shankar, R. Metal chelating ability and antioxidant properties of Curcumin-metal complexes—A DFT approach. J. Mol. Graph. Modell. 2018, 79, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Parimita, S.P.; Ramshankar, Y.V.; Suresh, S.; Row, T.N.G. Redetermination of curcumin: (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxy-phenyl)hepta-1,4,6-trien-3-one. Acta Crystallogr. Sect. Sect. E Struct. Rep. Online 2007, 63, o860–o862. [Google Scholar] [CrossRef]
- Satalkar, V.; Rusmore, T.A.; Phillips, E.; Pan, X.; Benassi, E.; Wu, Q.; Ran, C.; Shao, Y. Computational modeling of curcumin-based fluorescent probe molecules. Theor. Chem. Acc. 2019, 138, 29. [Google Scholar] [CrossRef]
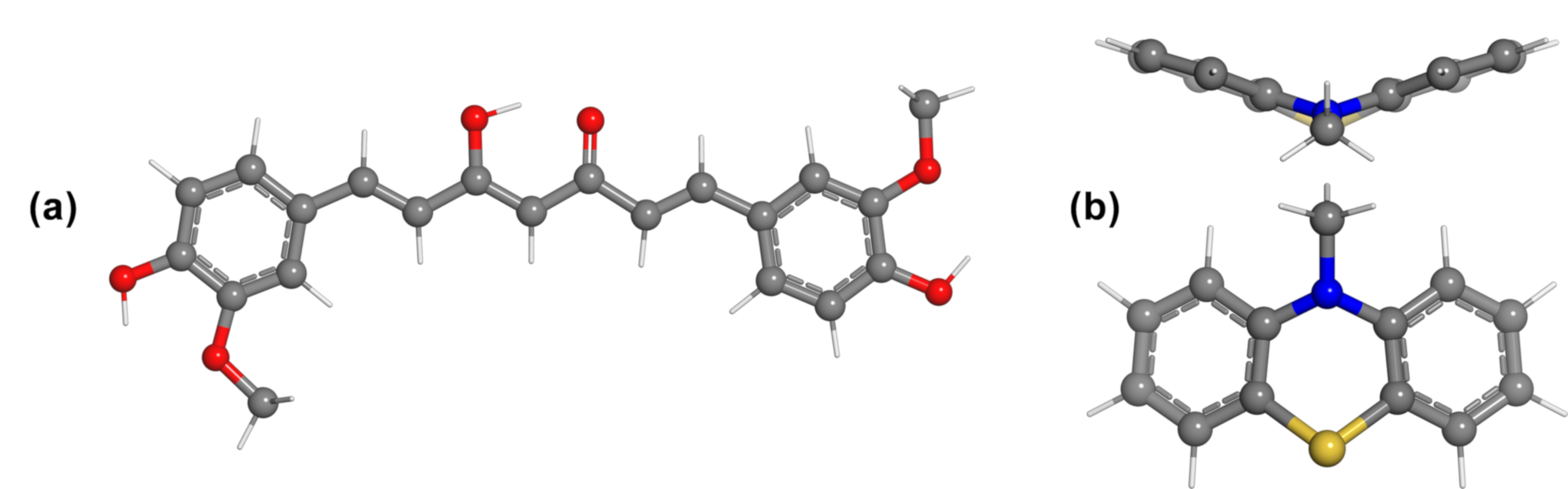







{kind=link}
{kind=link}
{kind=link}
{kind=link}
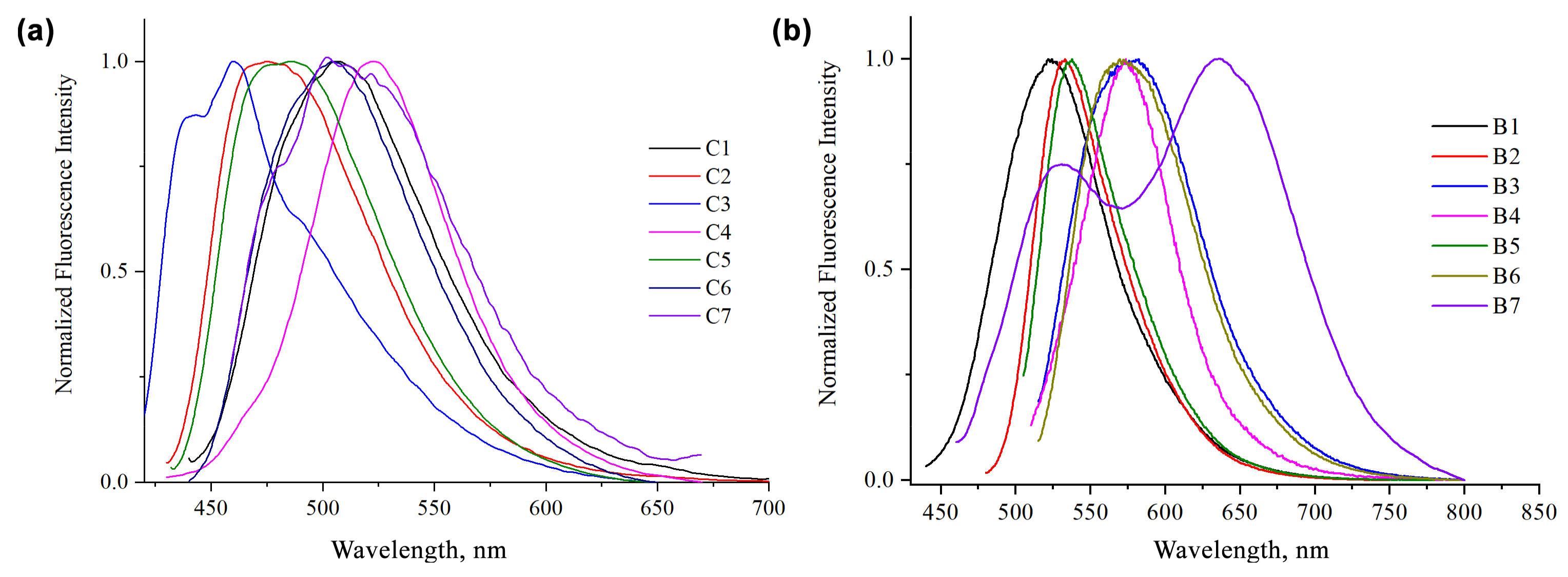{kind=link}
{kind=link}
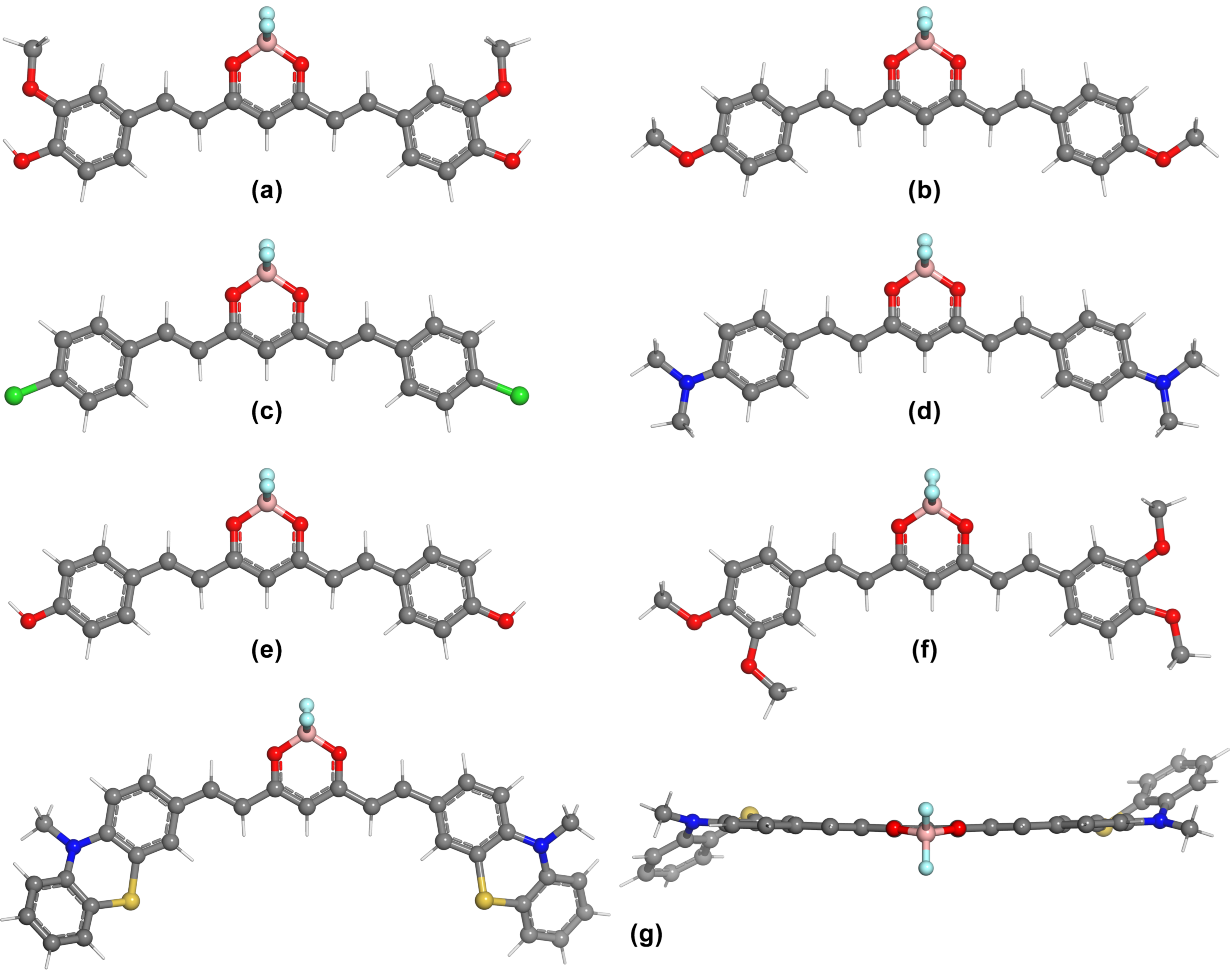{kind=link}
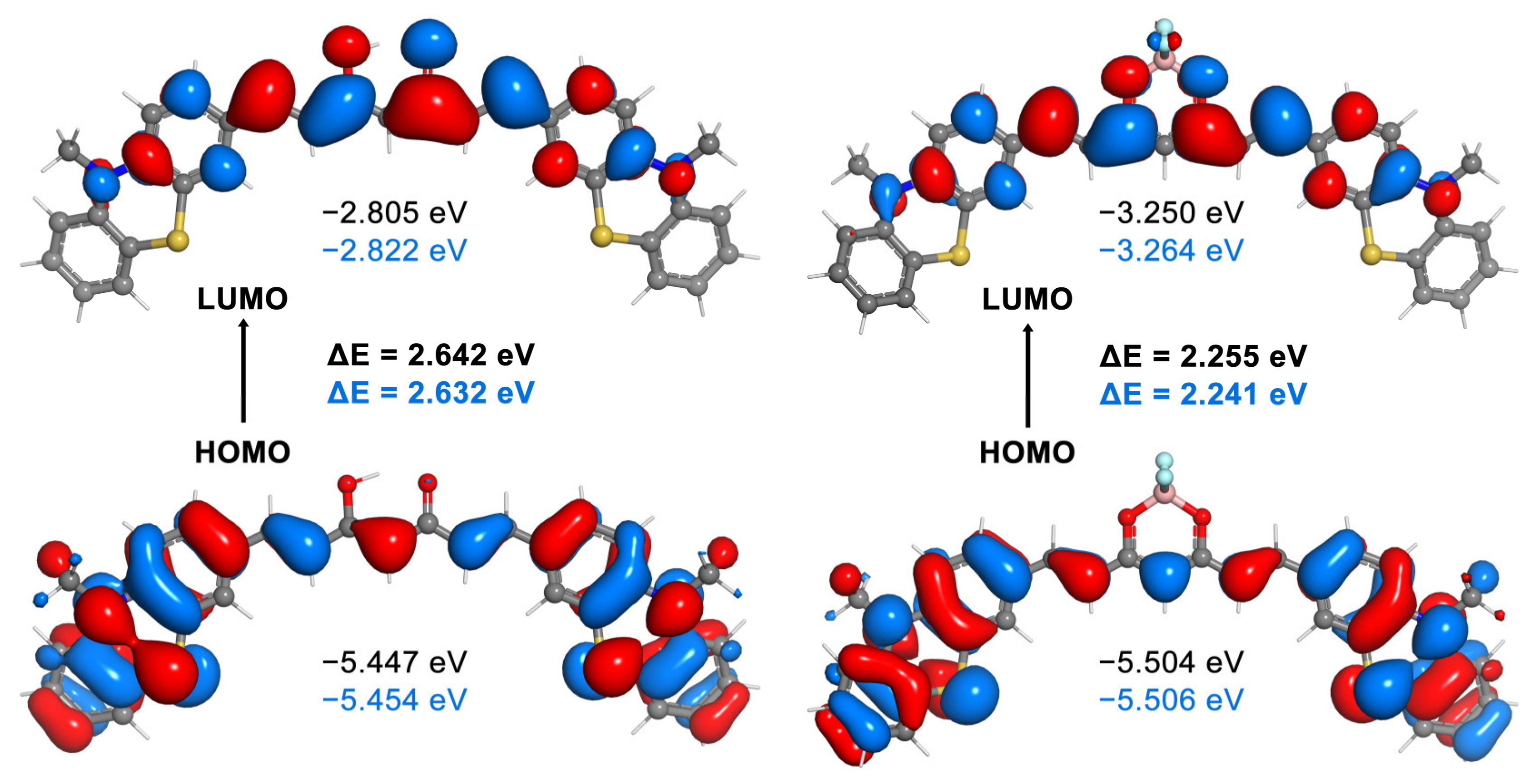{kind=link}
{kind=link}
| Comp. | Acetone | DMSO | ||||
|---|---|---|---|---|---|---|
| λabs (nm) | λem (nm) | Stokes Shift (cm−1) | ε | λabs (nm) | ε | |
| C1 | 374, 422 | 525 | 7690 | 64,900 | 396, 423 | 17,689 |
| C2 | 411 | 475 | 3278 | 52,140 | 420 | 26,592 |
| C3 | 396, 420 | 461 | 3561 | 30,980 | 403 | 18,107 |
| C4 | 409 | 522 | 5293 | 20,730 | 422 | 19,281 |
| C5 | 413 | 485 | 3595 | 48,020 | 425 | 35,730 |
| C6 | 418 | 504 | 4082 | 47,340 | 430 | 11,903 |
| C7 | 350, 468 | 522 | 2210 | 89,712 | 404, 526 | 45,035 |
| B1 | 418, 502 | 524 | 4839 | 69,500 | 435, 536, 572 | 134,245 |
| B2 | 405, 462, 482 | 532 | 5894 | 68,620 | 414, 498 | 87,471 |
| B3 | 500 | 534 | 1273 | 45,324 | 514 | 66,212 |
| B4 | 343, 490 | 575 | 3017 | 47,790 | 509 | 67,862 |
| B5 | 465, 486 | 538 | 2918 | 32,081 | 510 | 69,607 |
| B6 | 474, 498 | 570 | 3553 | 54,810 | 515 | 45,778 |
| B7 | 399, 565 | 528, 634 | 9289 | 123,700 | 408, 592 | 150,760 |
| Solvent | Curcumin Analogs | Difluoroboron-Curcumin Analogs | ||||||
|---|---|---|---|---|---|---|---|---|
| λabs (nm) | Eve (eV) | f | λabs (nm) | Eve (eV) | f | |||
| Gas phase | C1 | 426 | 2.909 | 1.525 | B1 | 460 | 2.693 | 1.618 |
| Acetone | 461 | 2.685 | 1.677 | 516 | 2.399 | 1.783 | ||
| DMSO | 464 | 2.668 | 1.693 | 521 | 2.379 | 1.801 | ||
| Gas phase | C2 | 414 | 2.995 | 1.741 | B2 | 441 | 2.808 | 1.892 |
| Acetone | 450 | 2.758 | 1.928 | 495 | 2.501 | 2.128 | ||
| DMSO | 453 | 2.739 | 1.943 | 500 | 2.478 | 2.146 | ||
| Gas phase | C3 | 403 | 3.074 | 1.637 | B3 | 425 | 2.915 | 1.803 |
| Acetone | 426 | 2.909 | 1.823 | 462 | 2.682 | 2.018 | ||
| DMSO | 428 | 2.894 | 1.837 | 465 | 2.663 | 2.035 | ||
| Gas phase | C4 | 455 | 2.724 | 1.813 | B4 | 491 | 2.521 | 1.986 |
| Acetone | 522 | 2.374 | 2.051 | 580 | 2.134 | 2.334 | ||
| DMSO | 527 | 2.350 | 2.072 | 588 | 2.107 | 2.361 | ||
| Gas phase | C5 | 409 | 3.034 | 1.610 | B5 | 433 | 2.858 | 1.761 |
| Acetone | 443 | 2.802 | 1.809 | 485 | 2.552 | 2.002 | ||
| DMSO | 446 | 2.782 | 1.825 | 490 | 2.528 | 2.021 | ||
| Gas phase | C6 | 435 | 2.850 | 1.496 | B6 | 471 | 2.635 | 1.564 |
| Acetone | 471 | 2.628 | 1.680 | 528 | 2.349 | 1.807 | ||
| DMSO | 474 | 2.612 | 1.698 | 532 | 2.330 | 1.830 | ||
| Gas phase | C7 | 495 | 2.501 | 1.107 | B7 | 556 | 2.228 | 1.130 |
| Acetone | 547 | 2.266 | 1.314 | 642 | 1.930 | 1.431 | ||
| DMSO | 550 | 2.252 | 1.335 | 648 | 1.912 | 1.459 | ||
| Acetonitrile | 548 | 2.261 | 1.311 | |||||
| Dichloromethane | 543 | 2.279 | 1.329 | |||||
| Ethanol | 547 | 2.263 | 1.316 | |||||
| n-Hexane | 520 | 2.38 | 1.297 | |||||
| Tetrahydrofuran | 542 | 2.287 | 1.321 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gál, E.; Nagy, L.C. Photophysical Properties and Electronic Structure of Symmetrical Curcumin Analogues and Their BF2 Complexes, Including a Phenothiazine Substituted Derivative. Symmetry 2021, 13, 2299. https://doi.org/10.3390/sym13122299
Gál E, Nagy LC. Photophysical Properties and Electronic Structure of Symmetrical Curcumin Analogues and Their BF2 Complexes, Including a Phenothiazine Substituted Derivative. Symmetry. 2021; 13(12):2299. https://doi.org/10.3390/sym13122299
Chicago/Turabian StyleGál, Emese, and Levente Csaba Nagy. 2021. "Photophysical Properties and Electronic Structure of Symmetrical Curcumin Analogues and Their BF2 Complexes, Including a Phenothiazine Substituted Derivative" Symmetry 13, no. 12: 2299. https://doi.org/10.3390/sym13122299
APA StyleGál, E., & Nagy, L. C. (2021). Photophysical Properties and Electronic Structure of Symmetrical Curcumin Analogues and Their BF2 Complexes, Including a Phenothiazine Substituted Derivative. Symmetry, 13(12), 2299. https://doi.org/10.3390/sym13122299