Stereoselective Synthesis of Chiral α-SCF3-β-Ketoesters Featuring a Quaternary Stereocenter


 and
and {kind=link}
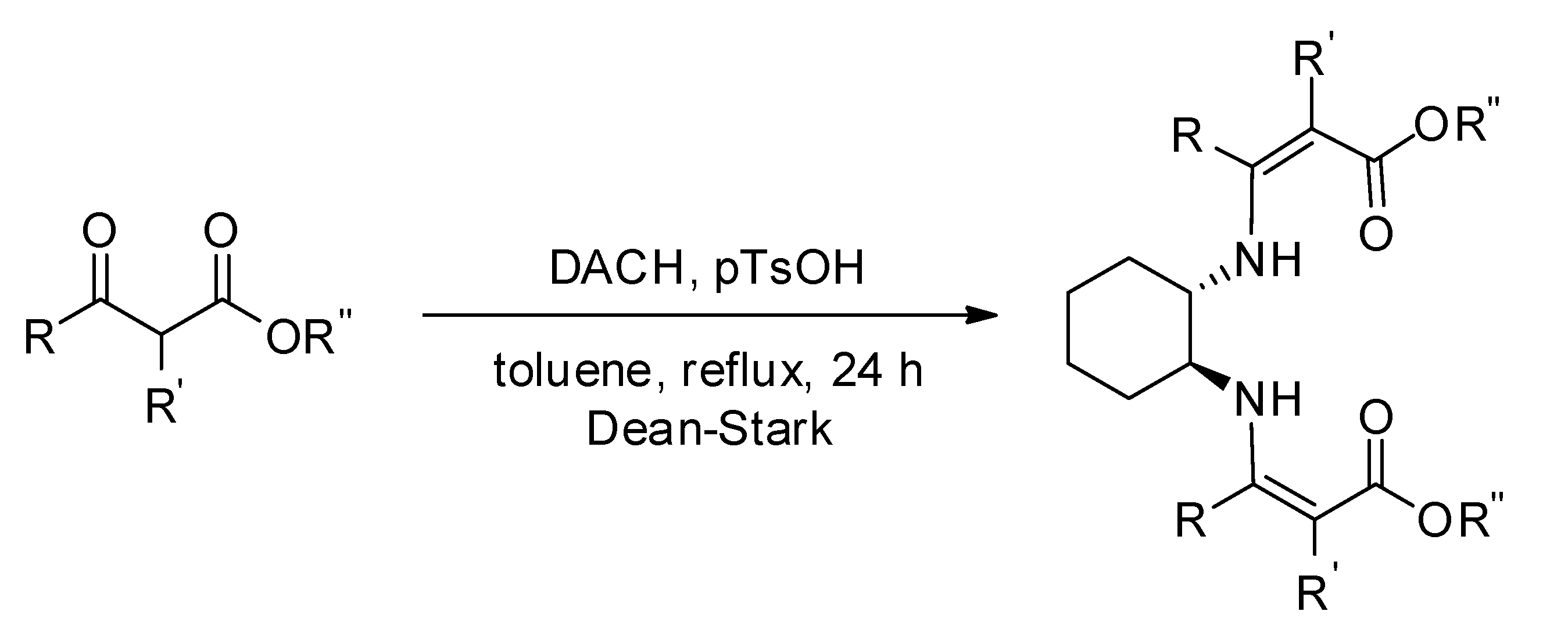{kind=link}
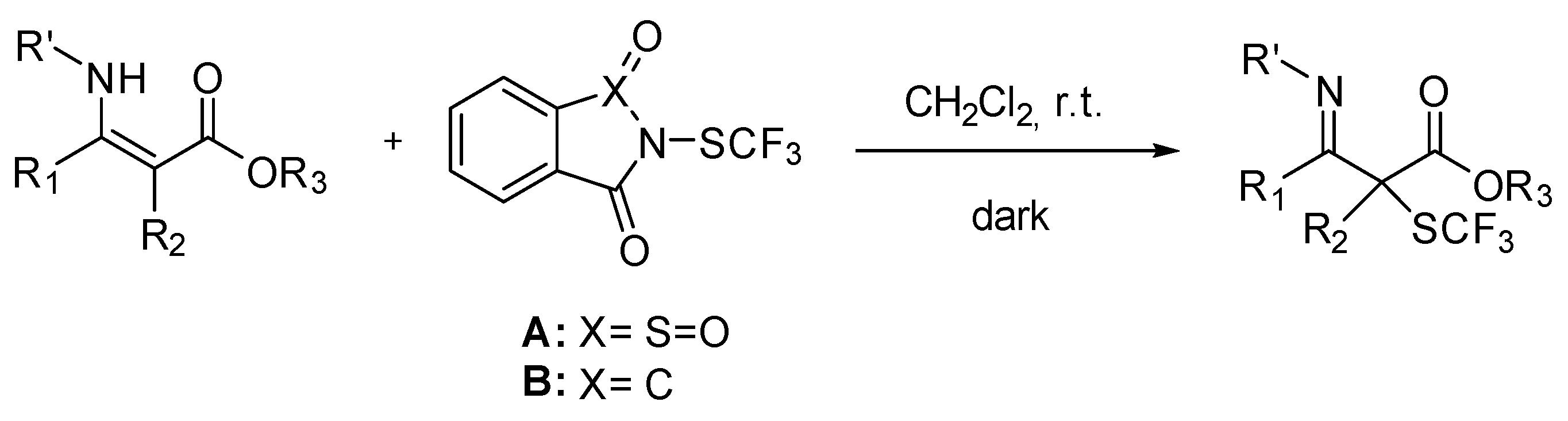{kind=link}
{kind=link}
{kind=link}
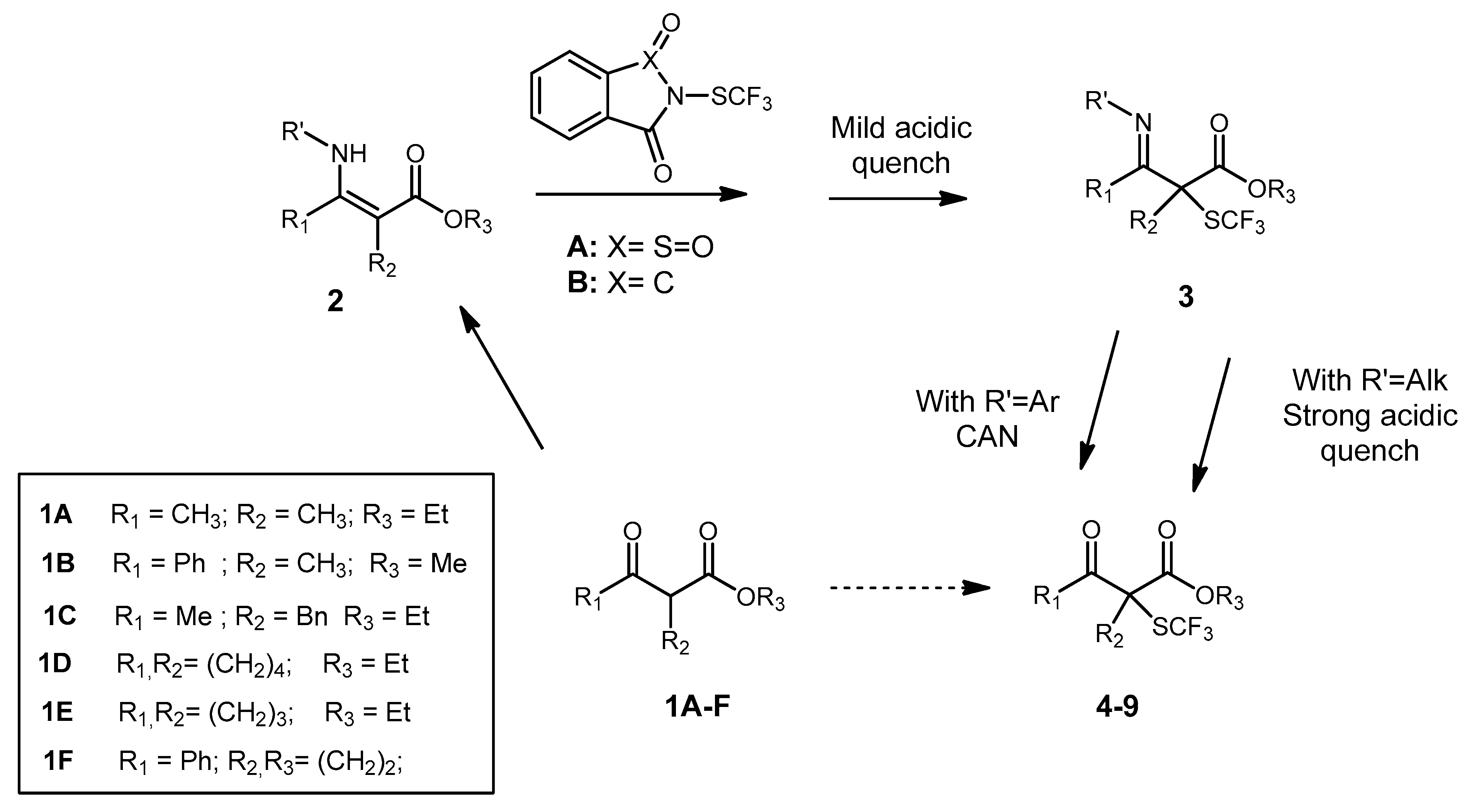{kind=link}
{kind=link}
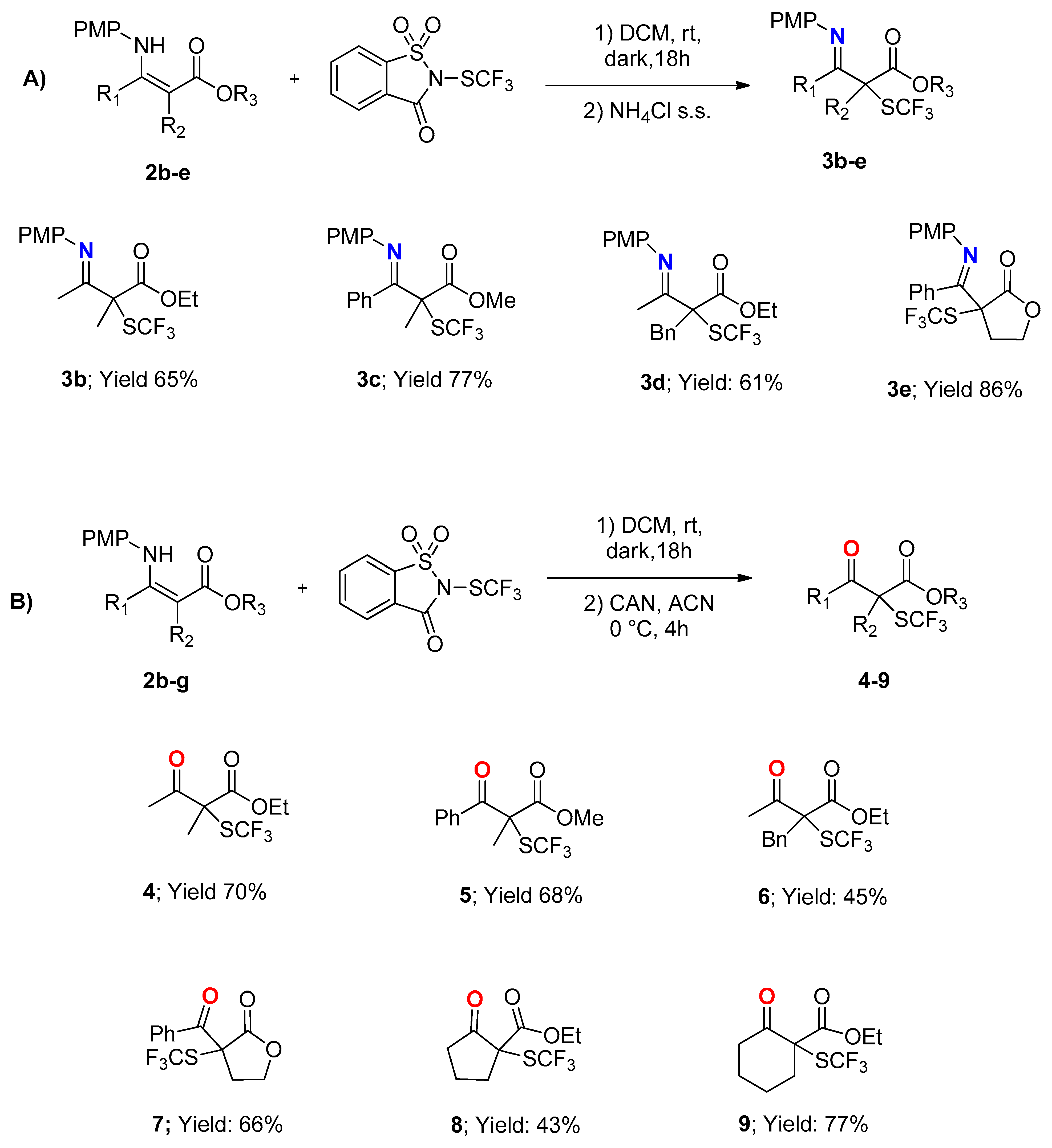{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
3. Experimental
3.1. Materials
3.2. Preparation of Chiral Enamines
3.2.1. Diethyl 3,3′-((1S,2S)-Cyclohexane-1,2-diylbis(azanediyl))bis(2-methylbut-2-enoate) 13

3.2.2. Diethyl 3,3′-((1S,2S)-Cyclohexane-1,2-diylbis(azanediyl))bis(2-benzylbut-2-enoate) 16

3.2.3. (3Z,3′Z)-3,3′-(((1S,2S)-Cyclohexane-1,2-diylbis(azanediyl))bis(phenylmethanylylidene))bis(dihydrofuran-2(3H)-one) 17

3.2.4. Diethyl 2,2′-((1S,2S)-Cyclohexane-1,2-diylbis(azanediyl))bis(cyclopent-1-enecarboxylate) 18

3.2.5. Diethyl 2,2′-(((1S,2S)-Cyclohexane-1,2-diyl)bis(azanediyl))bis(cyclohex-1-ene-1-carboxylate) 19

3.3. General Procedure for of α-SCF3 Substituted β-Imino Esters
3.3.1. Ethyl 3-((4-Methoxyphenyl)imino)-2-methyl-2-((trifluoromethyl)thio)butanoate 3b

3.3.2. Methyl 3-((4-Methoxyphenyl)imino)-2-methyl-3-phenyl-2-((trifluoromethyl)thio)propanoate 3c

3.3.3. Ethyl 2-Benzyl-3-((4-methoxyphenyl)imino)-2-((trifluoromethyl)thio)butanoate 3d

3.3.4. (E)-3-(((4-Methoxyphenyl)imino)(phenyl)methyl)-3-((trifluoromethyl)thio)dihydrofuran-2(3H)-one 3e

3.3.5. Ethyl 3-(Hexylimino)-2-methyl-2-((trifluoromethyl)thio)butanoate 3a

3.4. General Non Enantioselective Procedure for α-SCF3 SUBSTITUTED β-Ketoesters
3.5. General Enantioselective Procedure for Chiral α-SCF3 Substituted β-Ketoesters
3.5.1. Ethyl 2-Methyl-3-oxo-2-((trifluoromethyl)thio)butanoate 4

3.5.2. Methyl 2-Methyl-3-oxo-3-phenyl-2-((trifluoromethyl)thio)propanoate 5

3.5.3. Ethyl 2-Benzyl-3-oxo-2-((trifluoromethyl)thio)butanoate 6

3.5.4. 3-Benzoyl-3-((trifluoromethyl)thio)dihydrofuran-2(3H)-one 7

3.5.5. Ethyl 2-Oxo-1-((trifluoromethyl)thio)cyclopentanecarboxylate 8

3.5.6. Ethyl 2-Oxo-1-((trifluoromethyl)thio)cyclohexanecarboxylate 9

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III, Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Cahard, D.; Bizet, V. The influence of fluorine in asymmetric catalysis. Chem. Soc. Rev. 2014, 43, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Moschner, J.; Stulberg, V.; Fernandes, R.; Huhmann, S.; Leppkes, J.; Koksch, B. Approaches to Obtaining Fluorinated α-Amino Acids. Chem. Rev. 2019, 119, 10718–10801. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P. The Unique Role of Halogen Substituents in the Design of Modern Agrochemicals. Pest Manag. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Leroux, F.; Jeschke, P.; Schlosser, M. α-Fluorinated Ethers, Thioethers, and Amines: Anomerically Biased Species. Chem. Rev. 2005, 105, 827–856. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Wang, J.; Sanchez-Rosello, M.; Acena, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Hardy, M.A.; Chachignon, H.; Cahard, D. Advances in Asymmetric Di-and Trifluoromethylthiolation, and Di- and Trifluoromethoxylation Reactions. Asian J. Org. Chem. 2019, 8, 591–609. [Google Scholar] [CrossRef]
- Sicignano, M.; Rodríguez, R.I.; Capaccio, V.; Borello, F.; Cano, R.; De Riccardis, F.; Bernardi, L.; Díaz-Tendero, S.; Della Sala, G.; Alemán, J. Asymmetric trifluoromethylthiolation of azlactones under chiral phase transfer catalysis. Org. Biomol. Chem. 2020, 18, 2914–2920. [Google Scholar] [CrossRef]
- Capaccio, V.; Sicignano, M.; Rodríguez, R.I.; Della Sala, G.; Alemán, J. Asymmetric Synthesis of α-Trifluoromethylthio-β-Amino Acids under Phase Transfer Catalysis. Org. Lett. 2020, 22, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ji, J.; Zhang, X.; Jiang, Q.; Luo, J.; Zhao, X. Enantioselective Construction of Axially Chiral Amino Sulfide Vinyl Arenes by Chiral Sulfide Catalyzed Electrophilic Carbothiolation of Alkynes. Angew. Chem. Int. Ed. 2020, 59, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Hung, C.-I.J.; Mata, G.; Liu, Y.; Lu, Y.; Gnanamani, E. Direct Enantio- and Diastereoselective Zn-ProPhenol-Catalyzed Mannich Reactions of CF3- and SCF3-Substituted Ketones. Org. Lett. 2020, 22, 2437–2441. [Google Scholar] [CrossRef] [PubMed]
- Eitzinger, A.; Brière, J.F.; Cahard, D.; Waser, M. Enantioselective catalytic synthesis of α-aryl-α-SCF3-β2,2-amino acids. Org. Biomol. Chem. 2020, 18, 405–408. [Google Scholar] [CrossRef]
- Rossi, S.; Puglisi, A.; Raimondi, L.; Benaglia, M. Synthesis of alpha-trifluoromethylthio carbonyl compounds: A survey of the methods for the direct introduction of the SCF3 group on to organic molecules. ChemCatChem 2018, 10, 2717–2733. [Google Scholar] [CrossRef]
- Wang, X.; Yang, T.; Cheng, X.; Shen, Q. Enantioselective electrophilic trifluoromethylthiolation of β-ketoesters: A case of reactivity and selectivity bias for organocatalysis. Angew. Chem. Int. Ed. 2013, 52, 12860–12864. [Google Scholar] [CrossRef]
- Bootwicha, T.; Liu, X.; Pluta, R.; Atodiresei, I.; Rueping, M. N-trifluoromethylthiophthalimide: A stable electrophilic SCF3-reagent and its application in the catalytic asymmetric. Angew. Chem. Int. Ed. 2013, 52, 12856–12859. [Google Scholar] [CrossRef]
- Zhang, H.; Leng, X.; Wan, X.; Shen, Q. (1S)-(−)-N-Trifluoromethylthio-2,10-camphorsultam and its derivatives: Easily available, optically pure reagents for asymmetric trifluoromethylthiolation. Org. Chem. Front. 2017, 4, 1051–1057. [Google Scholar] [CrossRef]
- Hu, L.Q.; Wu, M.H.; Wan, H.X.; Wang, J.; Wang, G.Q.; Guo, H.B.; Sun, S.F. Efficient catalytic α-trifluoromethylthiolation of aldehydes. New J. Chem. 2016, 40, 6550–6553. [Google Scholar] [CrossRef]
- Jin, M.Y.; Li, Y.; Huang, R.; Zhou, Y.; Chung, L.W.; Wan, J. Catalytic asymmetric trifluoro-methylthiolation of carbonyl compounds via a diastereo and enantioselective Cu-catalyzed tandem reaction. Chem. Commun. 2018, 54, 4581–4584. [Google Scholar] [CrossRef]
- Chachignon, H.; Kondrashov, E.V.; Cahard, D. Diastereoselective Electrophilic Trifluoro-methylthiolation of Chiral Oxazolidinones: Access to Enantiopure α-SCF3 Alcohols. Adv. Synth. Catal. 2018, 360, 965–971. [Google Scholar] [CrossRef]
- Abubakar, S.S.; Benaglia, M.; Rossi, S.; Annunziata, R. Organocatalytic trifluoro-methylthiolation of silylenol ethers: Batch vs. continuous flow reactions. Catal. Today 2018, 308, 94–101. [Google Scholar] [CrossRef]
- Yang, X.; Zheng, K.; Zhang, C. Electrophilic Hypervalent Trifluoromethylthio-Iodine(III) Reagent. Org. Lett. 2020, 22, 2026–2031. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boselli, M.F.; Faverio, C.; Massolo, E.; Raimondi, L.; Puglisi, A.; Benaglia, M. Stereoselective Synthesis of Chiral α-SCF3-β-Ketoesters Featuring a Quaternary Stereocenter. Symmetry 2021, 13, 92. https://doi.org/10.3390/sym13010092
Boselli MF, Faverio C, Massolo E, Raimondi L, Puglisi A, Benaglia M. Stereoselective Synthesis of Chiral α-SCF3-β-Ketoesters Featuring a Quaternary Stereocenter. Symmetry. 2021; 13(1):92. https://doi.org/10.3390/sym13010092
Chicago/Turabian StyleBoselli, Monica Fiorenza, Chiara Faverio, Elisabetta Massolo, Laura Raimondi, Alessandra Puglisi, and Maurizio Benaglia. 2021. "Stereoselective Synthesis of Chiral α-SCF3-β-Ketoesters Featuring a Quaternary Stereocenter" Symmetry 13, no. 1: 92. https://doi.org/10.3390/sym13010092
APA StyleBoselli, M. F., Faverio, C., Massolo, E., Raimondi, L., Puglisi, A., & Benaglia, M. (2021). Stereoselective Synthesis of Chiral α-SCF3-β-Ketoesters Featuring a Quaternary Stereocenter. Symmetry, 13(1), 92. https://doi.org/10.3390/sym13010092