Chiroptical Properties and Conformation of Four Lasiocepsin-Related Antimicrobial Peptides: Structural Role of Disulfide Bridges

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
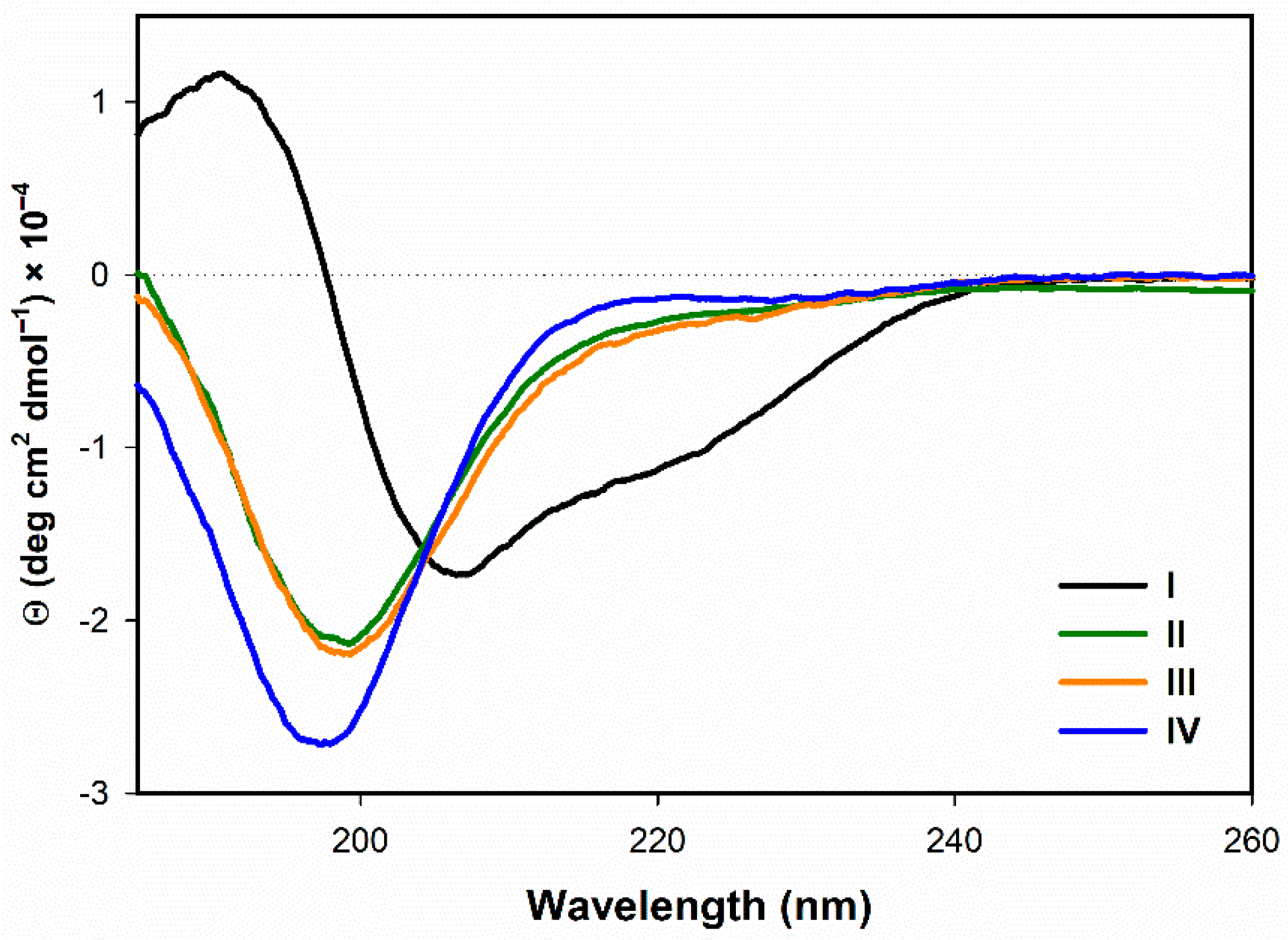
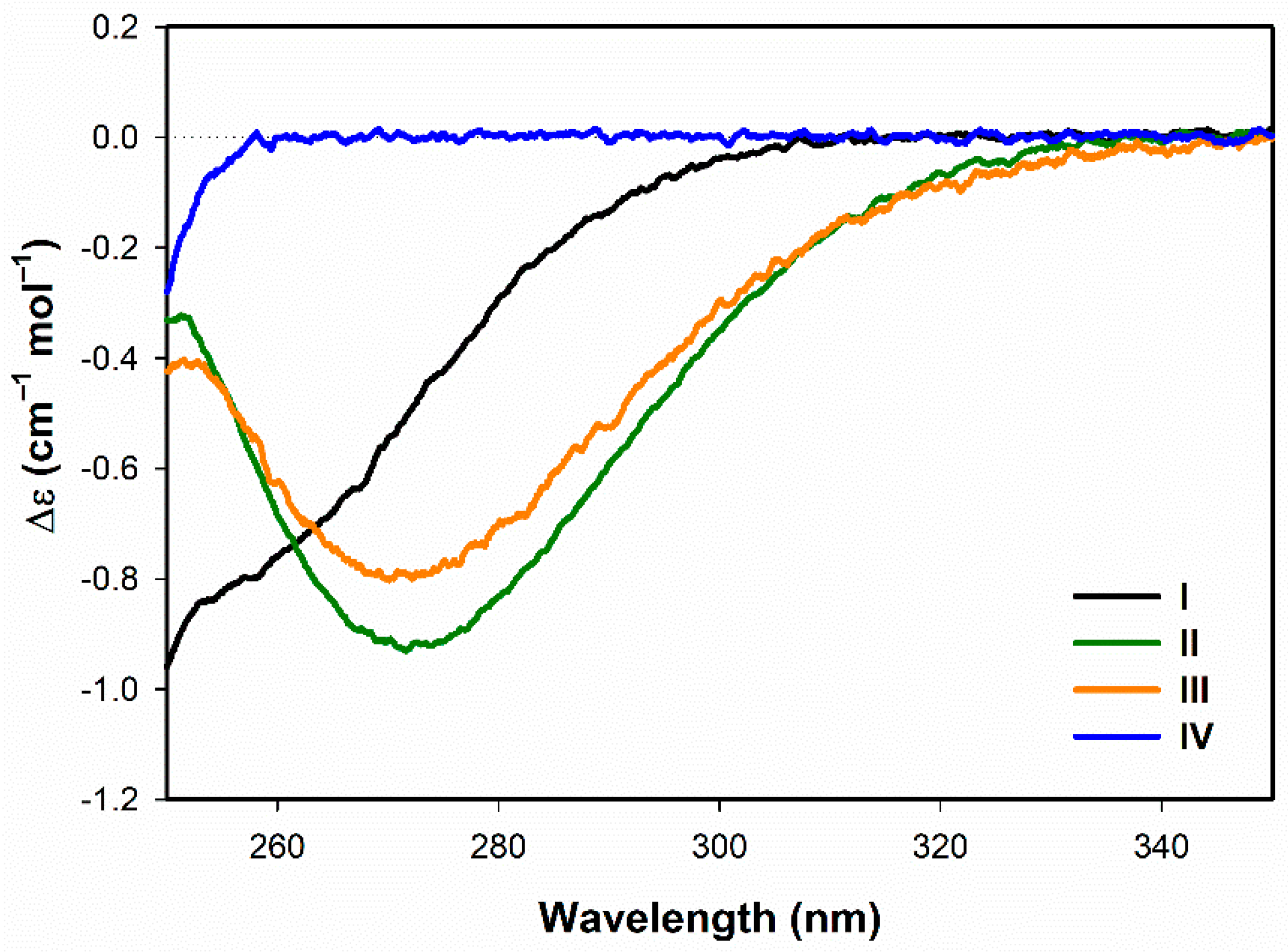
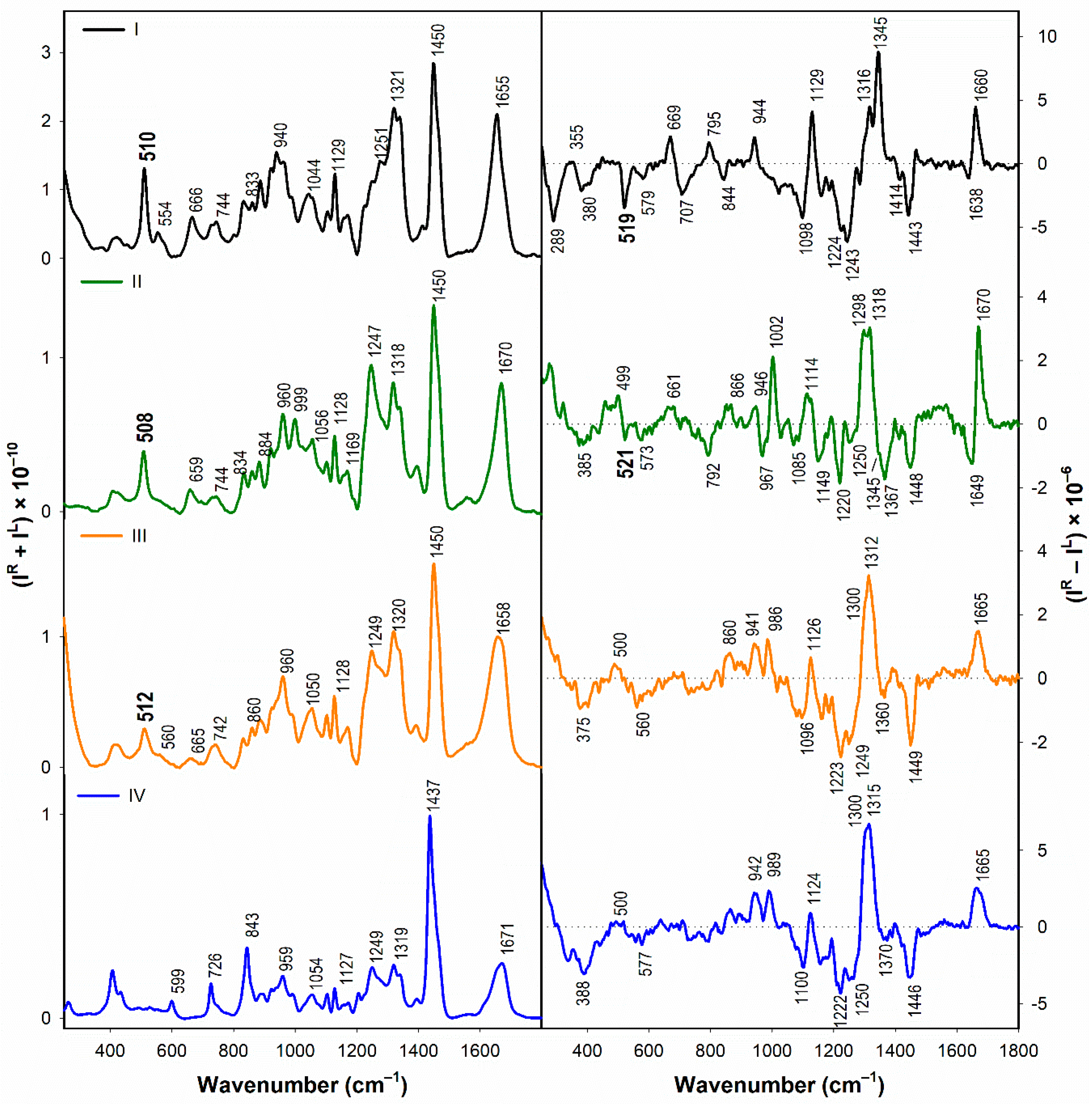
3.1. Secondary Structuture Assignement of I–IV in Water
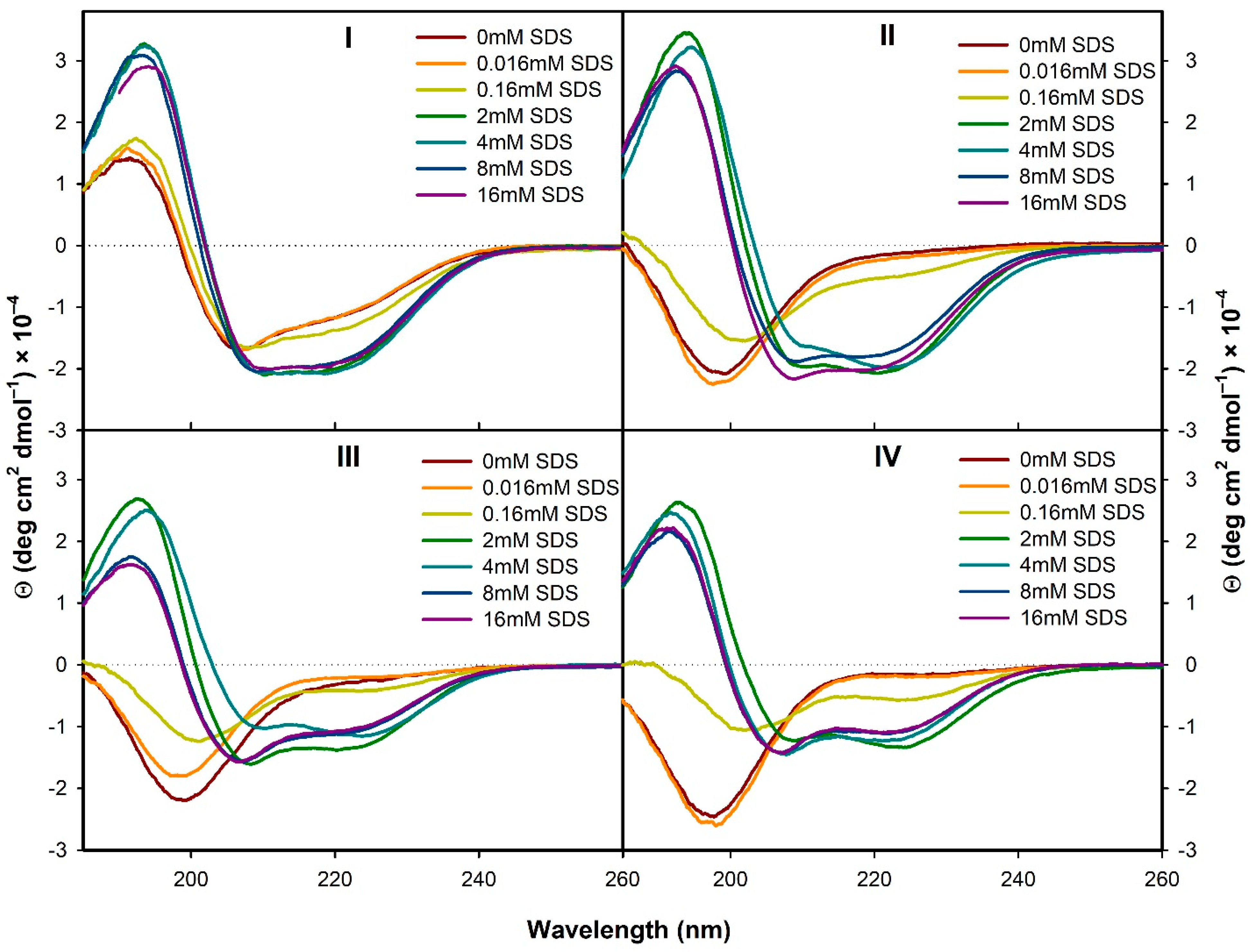
3.1.1. Electronic Circular Dichroism
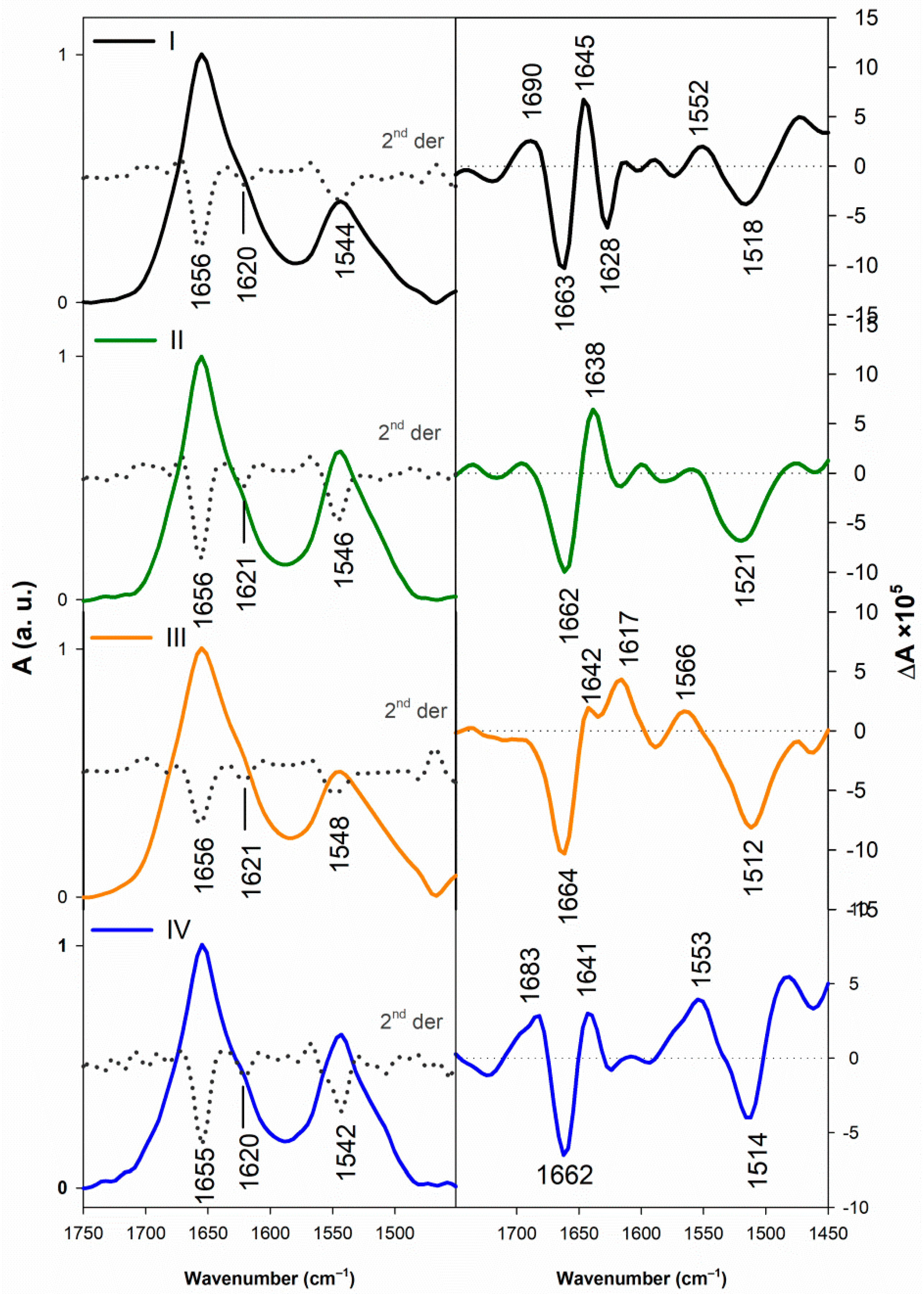
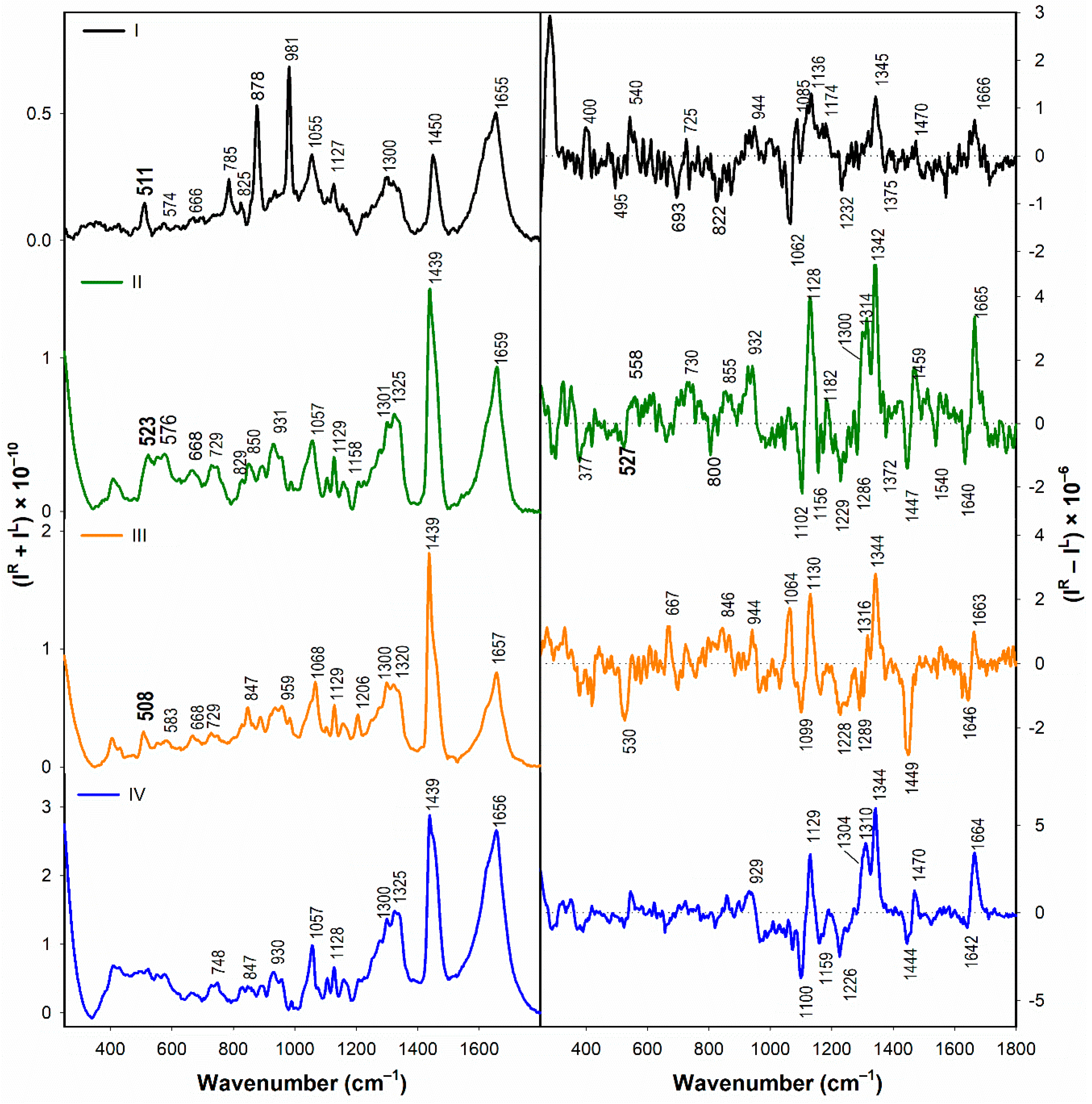
3.1.2. Vibrational Spectroscopy
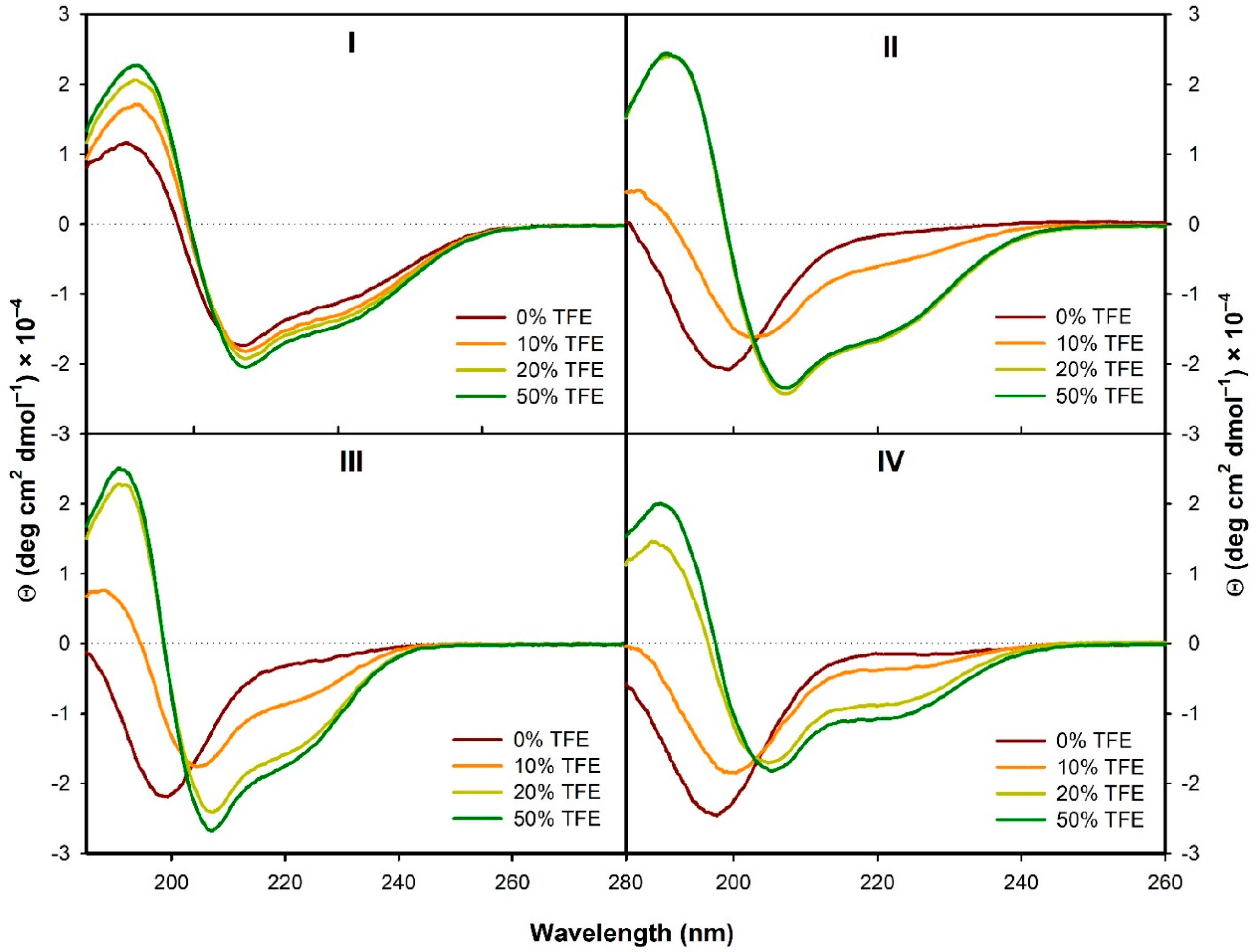
3.2. Structural Changes of I–IV due to the Presence of TFE and SDS
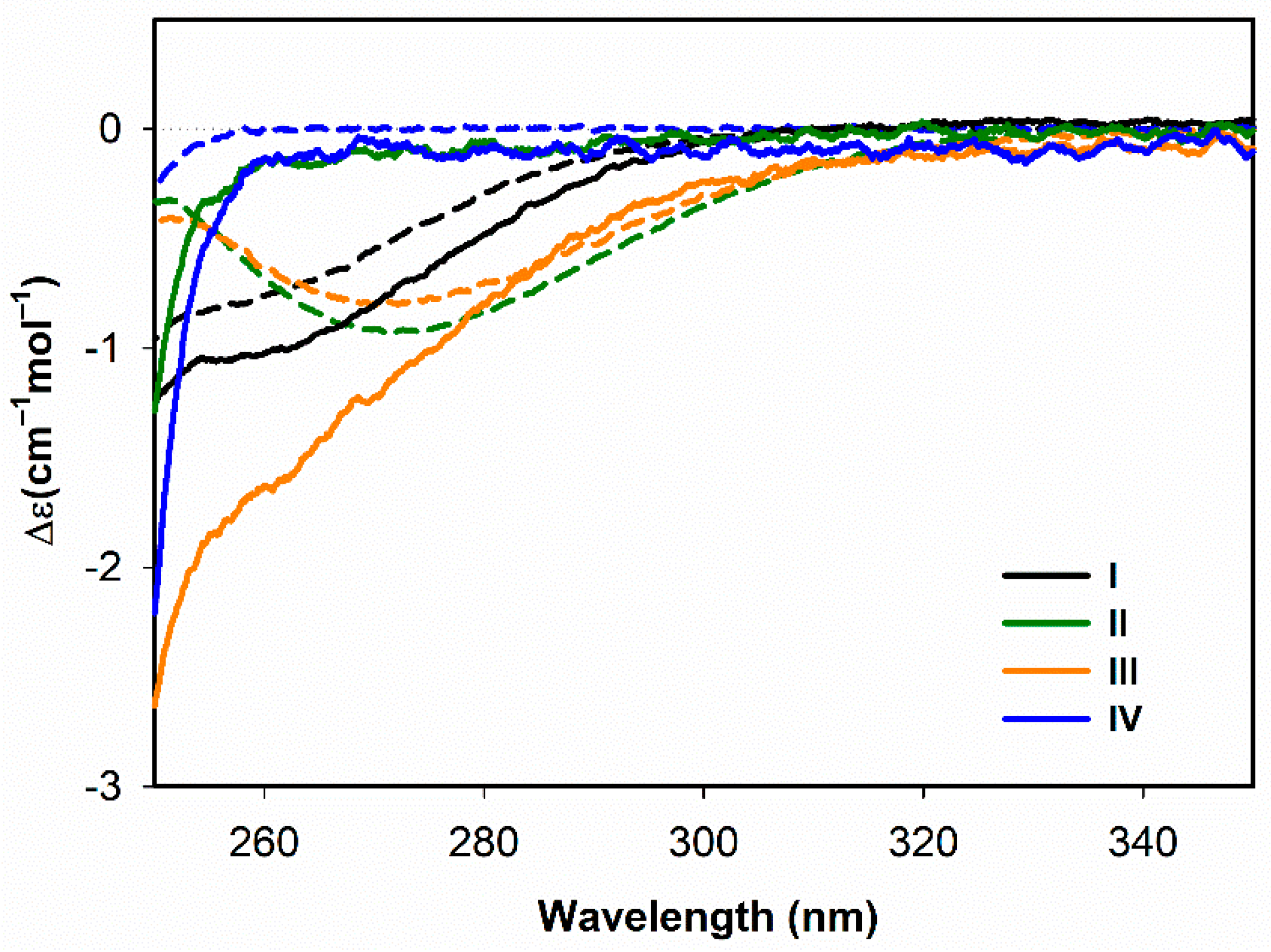
3.2.1. Structural Changes Followed by ECD
3.2.2. Structural Changes Followed by Vibrational Spectroscopy
3.3. Summarization of the Results
- Secondary structure of I in water, as derived from ECD, IR/VCD, and Raman/ROA data, corresponds well to the published NMR structure.
- The native peptide I manifests high structural and conformational rigidity, as evidenced by ROA data. While the analog II appears to be still conformationally rigid (although to somewhat lesser extent than I), the analogs III and IV seem to be rather flexible. The analog IV is the most flexible one.
- In the presence of TFE (>20%) or SDS (>2 mM), all the analogs form α-helical structure (at least to some extent) with some random coil/PPII structure participation. On the contrary, in H2O the α-helical structure is observed only for the natural peptide I.
- Based on the ROA spectra, hydrated form of α-helical structure seems to prevail in all the analogs I–IV in the presence of SDS. Analogs II and IV seem to also have a considerable portion of unhydrated α-helical structure.
- According to the Raman spectra, the disulfide groups of I–III in water adopt predominantly the gauche-gauche-gauche conformation. In the presence of SDS, gauche-gauche-gauche conformation remains dominant in I and III, while for II we observe a conformational change to gauche-gauche-trans and trans-gauche-trans conformation.
- Both ECD and ROA spectra indicate structural changes in absolute configuration of disulfide groups upon addition of SDS.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Monincová, L.; Slaninová, J.; Fučík, V.; Hovorka, O.; Voburka, Z.; Bednárová, L.; Maloň, P.; Štokrová, J.; Čeřovský, V. Lasiocepsin, a Novel Cyclic Antimicrobial Peptide from the Venom of Eusocial Bee Lasioglossum Laticeps (Hymenoptera: Halictidae). Amino Acids 2012, 43, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.M.; Nijnik, A.; Mayer, M.L.; Hancock, E.W. Potential of Immunomodulatory Host Defense Peptides as Novel Anti-Infectives. Trends Biotechnol. 2009, 27, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing Antimicrobial Peptides: Form Follows Function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Phoenix, D.A.; Dennison, S.R.; Harris, F. Antimicrobial Peptides; Wiley-VCH; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Scott, M.G. The Role of Antimicrobial Peptides in Animal Defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8856–8861. [Google Scholar] [CrossRef]
- Spohn, R.; Daruka, L.; Lázár, V.; Martins, A.; Vidovics, F.; Grézal, G.; Méhi, O.; Kintses, B.; Számel, M.; Jangir, P.K.; et al. Integrated Evolutionary Analysis Reveals Antimicrobial Peptides with Limited Resistance. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Epand, R.F.; Mor, A.; Epand, R.M. Lipid Complexes with Cationic Peptides and OAKs; Their Role in Antimicrobial Action and in the Delivery of Antimicrobial Agents. Cell. Mol. Life Sci. 2011, 68, 2177–2188. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F.; Arnusch, C.J.; Papahadjopoulos-Sternberg, B.; Wang, G.; Shai, Y. Lipid Clustering by Three Homologous Arginine-Rich Antimicrobial Peptides Is Insensitive to Amino Acid Arrangement and Induced Secondary Structure. Biochim. Biophys. Acta 2010, 1798, 1272–1280. [Google Scholar] [CrossRef]
- Shaw, J.E.; Epand, R.F.; Hsu, J.C.Y.; Mo, G.C.H.; Epand, R.M.; Yip, C.M. Cationic Peptide-Induced Remodelling of Model Membranes: Direct Visualization by in Situ Atomic Force Microscopy. J. Struct. Biol. 2008, 162, 121–138. [Google Scholar] [CrossRef]
- White, S.H.; Wimley, W.C. Hydrophobic Interactions of Peptides with Membrane Interfaces. Biochim. Biophys. Acta Rev. Biomembr. 1998, 1376, 339–352. [Google Scholar] [CrossRef]
- White, S.H.; Wimley, W.C. Membrane Protein Folding and Stability: Physical Principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [Google Scholar] [CrossRef]
- Lee, T.-H.; Hall, K.N.; Aguilar, M.-I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2015, 16, 25–39. [Google Scholar] [CrossRef]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E.W. Antimicrobial Peptides: Methods and Protocols; Hansen, P.R., Ed.; Springer Science+Business Media LLC: Berlin, Germany, 2017; Volume 1548. [Google Scholar]
- Taylor, K.; Barran, P.E.; Dorin, J.R. Structure-Activity Relationships in Β-Defensin Peptides. Pept. Sci. 2008, 90, 1–7. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. Can We Predict Biological Activity of Antimicrobial Peptides from Their Interactions with Model Phospholipid Membranes? Peptides 2003, 24, 1693–1703. [Google Scholar] [CrossRef]
- Wimley, W.C. Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef]
- Lavergne, V.; Taft, R.J.; Alewood, P.F. Cysteine-Rich Mini-Proteins in Human Biology. Curr. Top. Med. Chem. 2012, 12, 1514–1533. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, B. Evolutionary Origin of β-Defensins. Dev. Comp. Immunol. 2012, 39, 79–84. [Google Scholar] [CrossRef]
- Monincová, L.; Buděšínský, M.; Čujová, S.; Čeřovský, V.; Veverka, V. Structural Basis for Antimicrobial Activity of Lasiocepsin. ChemBioChem 2014, 15, 301–308. [Google Scholar] [CrossRef]
- Castaneda, O.; Sotolongo, V.; Amor, A.M.; Stocklin, R.; Anderson, A.J.; Harvey, A.L.; Engstrom, A.; Wernstedt, C.; Karlsson, E. Characterization of a Potassium Channel Toxin from the Caribbean Sea Anemone Stichodactyla Helianthus. Toxicon 1995, 33, 603–613. [Google Scholar] [CrossRef]
- Tudor, J.E.; Pallaghy, P.K.; Pennington, M.W.; Norton, R.S. Solution Structure of ShK Toxin, a Novel Potassium Channel Inhibitor from a Sea Anemone. Nat. Struct. Biol. 1996, 3, 317–320. [Google Scholar] [CrossRef]
- Zhang, L.; Chou, C.P.; Moo-Young, M. Disulfide Bond Formation and Its Impact on the Biological Activity and Stability of Recombinant Therapeutic Proteins Produced by Escherichia Coli Expression System. Biotechnol. Adv. 2011, 29, 923–929. [Google Scholar] [CrossRef]
- Berova, N.; Polavarapu, P.L.; Nakanishi, K.; Woody, R.W. (Eds.) Comprehensive Chiroptical Spectroscopy Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products and Biomolecules; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Nafie, L.A. Vibrational Optical Activity: Principles and Applications; John Wiley & Sons: Chichester, UK, 2011. [Google Scholar]
- Barron, L. The Development of Biomolecular Raman Optical Activity Spectroscopy. Biomed. Spectrosc. Imaging 2015, 4, 223–253. [Google Scholar] [CrossRef]
- Blondelle, S.E.; Lohner, K.; Aguilar, M.I. Lipid-Induced Conformation and Lipid-Binding Properties of Cytolytic and Antimicrobial Peptides: Determination and Biological Speci¢city. Biochim. Biophys. Acta 1999, 1462, 89–108. [Google Scholar] [CrossRef]
- Shanmugam, G.; Polavarapu, P.L.; Gopinath, D.; Jayakumar, R. The Structure of Antimicrobial Pexiganan Peptide in Solution Probed by Fourier Transform Infrared Absorption, Vibrational Circular Dichroism, and Electronic Circular Dichroism Spectroscopy. Biopolym. Pept. Sci. Sect. 2005, 80, 636–642. [Google Scholar] [CrossRef]
- Novotná, P.; Urbanová, M. Vibrational Circular Dichroism Study of Polypeptide Model-Membrane Systems. Anal. Biochem. 2012, 427, 211–218. [Google Scholar] [CrossRef]
- Kocourková, L.; Novotná, P.; Čujová, S.; Čeřovský, V.; Urbanová, M.; Setnička, V. Conformational Study of Melectin and Antapin Antimicrobial Peptides in Model Membrane Environments. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 170, 247–255. [Google Scholar] [CrossRef]
- Mijiddorj, B.; Kaneda, S.; Sato, H.; Kitahashi, Y.; Javkhlantugs, N.; Naito, A.; Ueda, K.; Kawamura, I. The Role of D-Allo-Isoleucine in the Deposition of the Anti-Leishmania Peptide Bombinin H4 as Revealed by 31P Solid-State NMR, VCD Spectroscopy, and MD Simulation. Biochim. Biophys. Acta Proteins Proteomics 2018, 1866, 789–798. [Google Scholar] [CrossRef]
- Pazderková, M.; Maloň, P.; Zíma, V.; Hofbauerová, K.; Kopecký, V.J.; Kočišová, E.; Pazderka, T.; Čeřovský, V.; Bednárová, L. Interaction of Halictine-Related Antimicrobial Peptides with Membrane Models. Int. J. Mol. Sci. 2019, 20, 631. [Google Scholar] [CrossRef]
- Keiderling, T.A. Structure of Condensed Phase Peptides: Insights from Vibrational Circular Dichroism and Raman Optical Activity Techniques. Chem. Rev. 2020, 120, 3381–3419. [Google Scholar] [CrossRef]
- Barth, A.; Zscherp, C. What Vibrations Tell Us about Proteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef]
- Keiderling, T.A. Protein and Peptide Secondary Structure and Conformational Determination with Vibrational Circular Dichroism. Curr. Opin. Chem. Biol. 2002, 6, 682–688. [Google Scholar] [CrossRef]
- Ma, S.; Freedman, T.B.; Dukor, R.K.; Nafie, L.A. Near-Infrared and Mid-Infrared Fourier Transform Vibrational Circular Dichroism of Proteins in Aqueous Solution. Appl. Spectrosc. 2010, 64, 615–626. [Google Scholar] [CrossRef]
- Dukor, R.K.; Keiderling, T.A. Reassessment of the Random Coil Conformation: Vibrational CD Study of Proline Oligopeptides and Related Polypeptides. Biopolymers 1991, 31, 1747–1761. [Google Scholar] [CrossRef]
- Bochicchio, B.; Tamburro, A.M. Polyproline II Structure in Proteins: Identification by Chiroptical Spectroscopies, Stability, and Functions. Chirality 2002, 14, 782–792. [Google Scholar] [CrossRef]
- Havel, H.A. Spectroscopic Methods for Determining Protein Structure in Solution; VCH Publishers, Inc.: New York, NY, USA, 1996. [Google Scholar]
- Zhu, F.J.; Isaacs, N.W.; Hecht, L.; Barron, L.D. Raman Optical Activity: A Tool for Protein Structure Analysis. Structure 2005, 13, 1409–1419. [Google Scholar] [CrossRef]
- Kapitán, J.; Baumruk, V.; Kopecký, V.J.; Bouř, P. Conformational Flexibility of L-Alanine Zwitterion Determines Shapes of Raman and Raman Optical Activity Spectral Bands. J. Phys. Chem. A 2006, 110, 4689–4696. [Google Scholar] [CrossRef]
- Bednárová, L.; Bouř, P.; Maloň, P. Vibrational and Electronic Optical Activity of the Chiral Disulphide Group: Implications for Disulphide Bridge Conformation. Chirality 2010, 22, 514–526. [Google Scholar] [CrossRef]
- Maloň, P.; Bednárová, L.; Straka, M.; Krejčí, L.; Kumprecht, L.; Kraus, T.; Kubáňová, M.; Baumruk, V. Disulfide Chromophore and Its Optical Activity. Chirality 2010, 22, E47–E55. [Google Scholar] [CrossRef]
- Pazderková, M.; Bednárová, L.; Dlouhá, H.; Flegel, M.; Lebl, M.; Hlaváček, J.; Setnička, V.; Urbanová, M.; Hynie, S.; Klenerová, V.; et al. Electronic and Vibrational Optical Activity of Several Peptides Related to Neurohypophyseal Hormones: Disulfide Group Conformation. Biopolymers 2012, 97, 923–932. [Google Scholar] [CrossRef]
- Tanford, C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes; Wiley-Interscience: New York, NY, USA, 1980. [Google Scholar]
- Andrushchenko, V.V.; Vogel, H.J.; Prenner, E.J. Optimization of the Hydrochloric Acid Concentration Used for Trifluoroacetate Removal from Synthetic Peptides. J. Pept. Sci. 2007, 13, 37–43. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Provencher, S.W.; Glöckner, J. Estimation of Globular Protein Secondary Structure from Circular Dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W. A Constrained Regularization Method for Inverting Data Represented by Linear Algebraic or Integral Equations. Comp. Phys. Com. 1982, 27, 213–227. [Google Scholar] [CrossRef]
- Greenfield, N.J. Methods to Estimate the Conformation of Proteins and Polypeptides from Circular Dichroism Data. Anal. Biochem. 1996, 235, 1–10. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an Online Server for Protein Secondary Structure Analyses from Circular Dichroism Spectroscopic Data. Nucleic Acids Res. 2004, 32, 668–673. [Google Scholar] [CrossRef]
- Nafie, L.A.; Buijs, H.; Rilling, A.; Cao, X.; Dukor, R.K. Dual Source Fourier Transform Polarization Modulation Spectroscopy: An Improved Method for the Measurement of Circular and Linear Dichroism. Appl. Spectrosc. 2004, 58, 647–654. [Google Scholar] [CrossRef]
- Nafie, L.A. Dual Polarization Modulation: Real-Time, Spectral Multiplex Separation of Circular Dichroism from Linear Birefringence Spectral Intensities. Appl. Spectrosc. 2000, 54, 1634–1645. [Google Scholar] [CrossRef]
- Drake, A.F.; Siligardi, G.; Gibbons, W.A. Reassessment of the Electronic Circular Dichroism Criteria for Random Coil Conformations of Poly(L-Lysine) and the Implications for Protein Folding and Denaturation Studies. Biophys. Chem. 1988, 31, 143–146. [Google Scholar] [CrossRef]
- Shi, Z.; Olson, C.A.; Rose, G.D.; Baldwin, R.L.; Kallenbach, N.R. Polyproline II Structure in a Sequence of Seven Alanine Residues. Proc. Natl. Acad. Sci. USA 2002, 99, 9190–9195. [Google Scholar] [CrossRef]
- Bergson, G. Molecular Orbital Treatment of the 3para-Alpha-Interaction in Five-Membered Cyclic Disulphides. Ark. Kemi 1958, 12, 233–237. [Google Scholar]
- Bergson, G. A Semi-Empirical Study of Interaction between Lone-Pair Electrons with Special Reference to Problem of Hybridization and Theory of Restricted Rotation About Single Bonds—Applications to Structure and Reactivity of Sulphur Chains. Ark. Kemi 1962, 18, 409–434. [Google Scholar]
- Linderberg, J.; Michl, J. On Inherent Optical Activity of Organic Disulfides. J. Am. Chem. Soc. 1970, 92, 2619–2625. [Google Scholar] [CrossRef]
- Barth, A. Infrared Spectroscopy of Proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar] [CrossRef]
- Barth, A.; Haris, P.I. Biological and Biomedical Infrared Spectroscopy; Haris, P.I., Ed.; IOS Press: Amsterdam, The Netherlands, 2009; Volume 2. [Google Scholar]
- Ma, S.; Cao, X.; Mak, M.; Sadik, A.; Walkner, C.; Freedman, T.B.; Lednev, I.K.; Dukor, R.K.; Nafie, L.A. Vibrational Circular Dichroism Shows Unusual Sensitivity to Protein Fibril Formation and Development in Solution. J. Am. Chem. Soc. 2007, 129, 12364–12365. [Google Scholar] [CrossRef]
- Kurouski, D.; Dukor, R.K.; Lu, X.; Nafie, L.A.; Lednev, I.K. Normal and Reversed Supramolecular Chirality of Insulin Fibrils Probed by Vibrational Circular Dichroism at the Protofilament Level of Fibril Structure. Biophys. J. 2012, 103, 522–531. [Google Scholar] [CrossRef]
- Kurouski, D.; Lu, X.; Popova, L.; Wan, W.; Shanmugasundaram, M.; Stubbs, G.; Dukor, R.K.; Lednev, I.K.; Nafie, L.A. Is Supramolecular Filament Chirality the Underlying Cause of Major Morphology Differences in Amyloid Fibrils? J. Am. Chem. Soc. 2014, 136, 2302–2312. [Google Scholar] [CrossRef]
- McColl, I.H.; Blanch, E.W.; Gill, A.C.; Rhie, A.G.; Ritchie, M.A.; Hecht, L.; Nielsen, K.; Barron, L.D. A New Perspective on Beta-Sheet Structures Using Vibrational Raman Optical Activity: From Poly(L-Lysine) to the Prion Protein. J. Am. Chem. Soc. 2003, 125, 10019–10026. [Google Scholar] [CrossRef]
- Barron, L.D.; Hecht, L.; Blanch, E.W.; Bell, A.F. Solution Structure and Dynamics of Biomolecules from Raman Optical Activity. Prog. Biophys. Mol. Biol. 2000, 73, 1–49. [Google Scholar] [CrossRef]
- Zhu, F.; Tranter, G.E.; Isaacs, N.W.; Hecht, L.; Barron, L.D. Delineation of Protein Structure Classes from Multivariate Analysis of Protein Raman Optical Activity Data. J. Mol. Biol. 2006, 363, 19–26. [Google Scholar] [CrossRef]
- Qian, W.; Zhao, W.; Krimm, S. Vibrational Studies of the Disulfied Group in Proteins Part IV. SS and CS Stretch Frequencies of Known Peptide and Protein Disulfide Bridges. J. Mol. Struct. 1991, 250, 89–102. [Google Scholar] [CrossRef]
- Kapitán, J.; Baumruk, V.; Hulačová, H.; Maloň, P. Raman Optical Activity of the Hinge Peptide. Vib. Spectrosc. 2006, 42, 88–92. [Google Scholar] [CrossRef]
- Buck, M. Trifluoroethanol and Colleagues: Cosolvents Come of Age. Recent Studies with Peptides and Proteins. Q. Rev. Biophys. 1998, 31, 297–355. [Google Scholar] [CrossRef]
- McColl, I.H.; Blanch, E.W.; Hecht, L.; Barron, L.D. A Study of Alpha-Helix Hydration in Polypeptides, Proteins, and Viruses Using Vibrational Raman Optical Activity. J. Am. Chem. Soc. 2004, 126, 8181–8188. [Google Scholar] [CrossRef]
- Schweitzer-Stenner, R. Conformational Propensities and Residual Structures in Unfolded Peptides and Proteins. Mol. Biosyst. 2012, 8, 122–133. [Google Scholar] [CrossRef]
- Woody, R.W. Application of the Bergson Model to the Optical Properties of Chiral Disulfides. Tetrahedron 1973, 29, 1273–1283. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ILasiocepsin Analogs | Antimicrobial Activity MIC (μM) | |||||
|---|---|---|---|---|---|---|
| (mg/mL) | ||||||
| B.s. | S.a. | E.c. | P.a. | C.a. | ||
| I | GLPRKILCAIAKKKGKCKGPLKLVCKC | 0.4 | 93 | 8.6 | 15 | 3.6 |
| 0.001 | 0.269 | 0.025 | 0.04 | 0.01 | ||
| II | GLPRKILAAIAKKKGKCKGPLKLVAKC | 4.2 | >100 | >100 | >100 | 50 |
| 0.012 | >0.283 | >0.283 | >0.283 | 0.0141 | ||
| III | GLPRKILCAIAKKKGKAKGPLKLVCKA | 0.5 | >100 | 55 | 65 | 70 |
| 0.001 | >0.283 | 0.156 | 0.184 | 0.0198 | ||
| IV | GLPRKILAAIAKKKGKAKGPLKLVAKA | 12 | >100 | >100 | >100 | >100 |
| 0.033 | >0.277 | >0.277 | >0.277 | >0.277 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pazderková, M.; Profant, V.; Maloň, P.; Dukor, R.K.; Čeřovský, V.; Baumruk, V.; Bednárová, L. Chiroptical Properties and Conformation of Four Lasiocepsin-Related Antimicrobial Peptides: Structural Role of Disulfide Bridges. Symmetry 2020, 12, 812. https://doi.org/10.3390/sym12050812
Pazderková M, Profant V, Maloň P, Dukor RK, Čeřovský V, Baumruk V, Bednárová L. Chiroptical Properties and Conformation of Four Lasiocepsin-Related Antimicrobial Peptides: Structural Role of Disulfide Bridges. Symmetry. 2020; 12(5):812. https://doi.org/10.3390/sym12050812
Chicago/Turabian StylePazderková, Markéta, Václav Profant, Petr Maloň, Rina K. Dukor, Václav Čeřovský, Vladimír Baumruk, and Lucie Bednárová. 2020. "Chiroptical Properties and Conformation of Four Lasiocepsin-Related Antimicrobial Peptides: Structural Role of Disulfide Bridges" Symmetry 12, no. 5: 812. https://doi.org/10.3390/sym12050812
APA StylePazderková, M., Profant, V., Maloň, P., Dukor, R. K., Čeřovský, V., Baumruk, V., & Bednárová, L. (2020). Chiroptical Properties and Conformation of Four Lasiocepsin-Related Antimicrobial Peptides: Structural Role of Disulfide Bridges. Symmetry, 12(5), 812. https://doi.org/10.3390/sym12050812