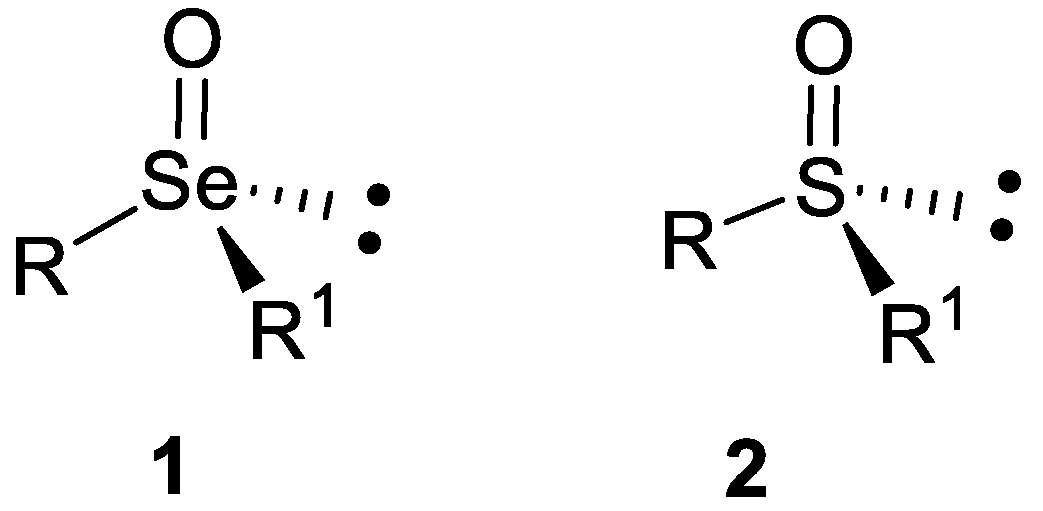
Optically Active Selenoxides: Structural and Synthetic Aspects




{kind=link}
{kind=link}
{kind=link}
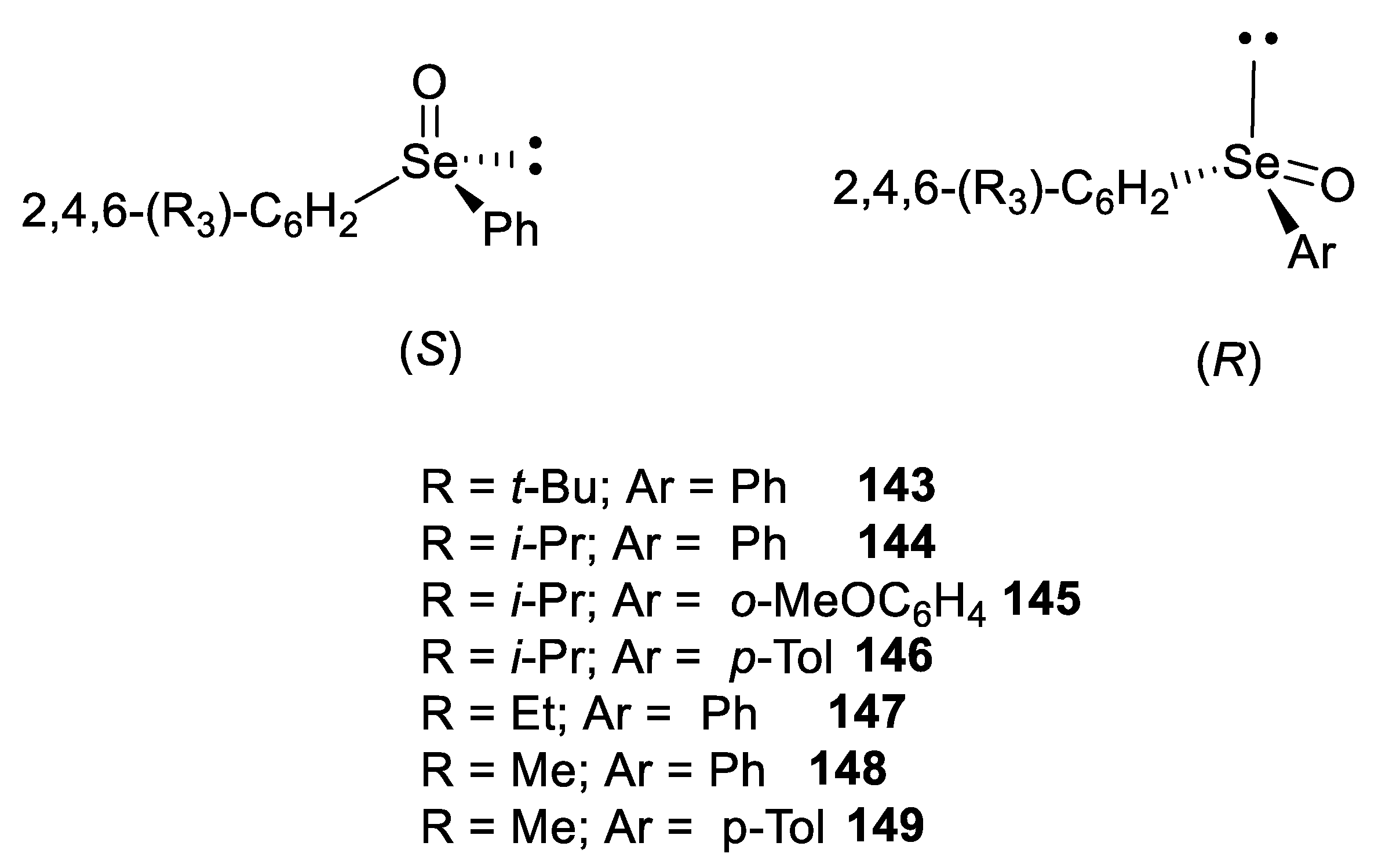{kind=link}
{kind=link}
{kind=link}
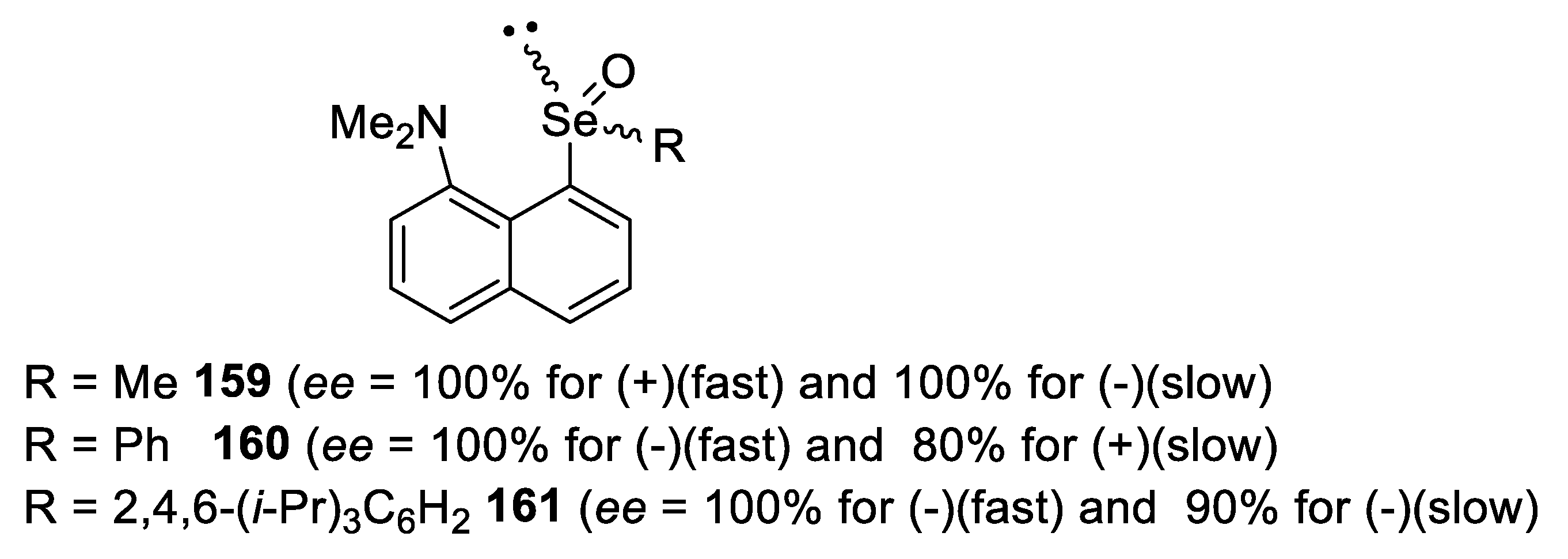{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
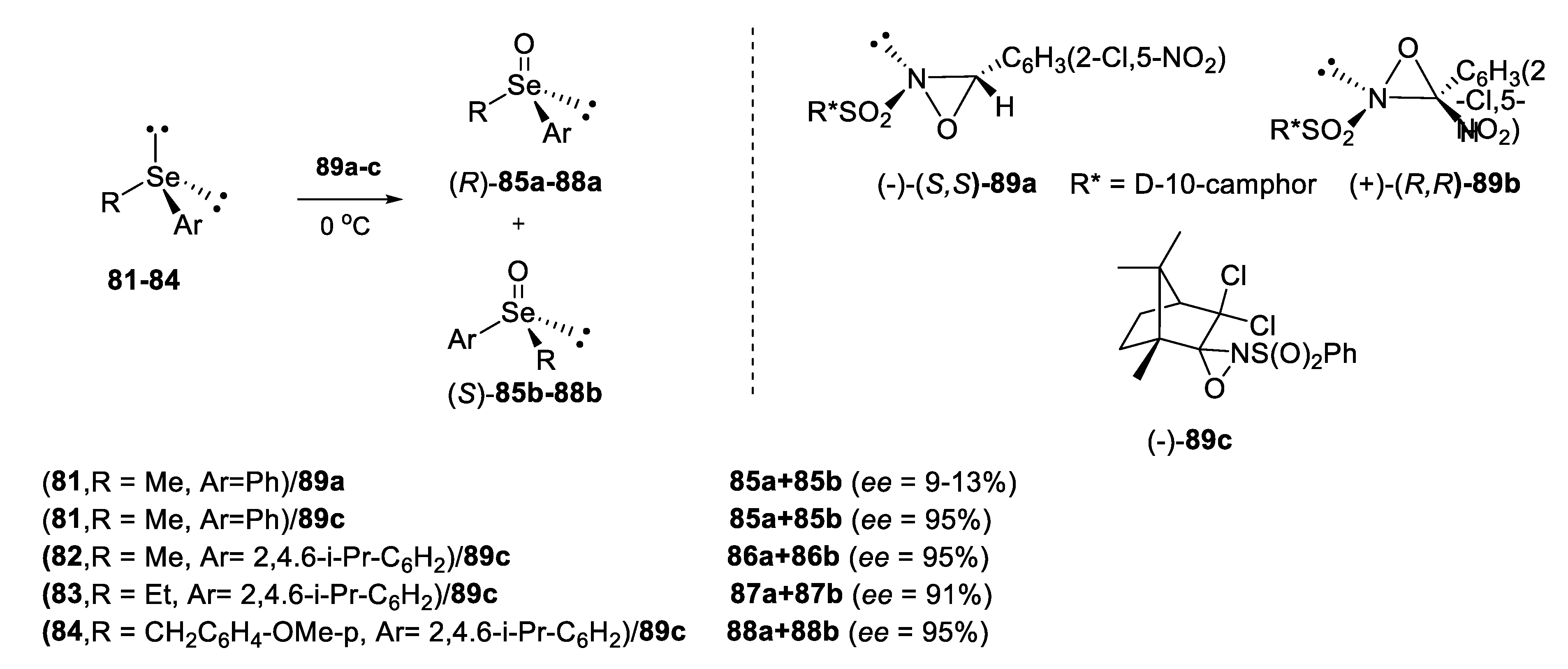{kind=link}
{kind=link}
{kind=link}
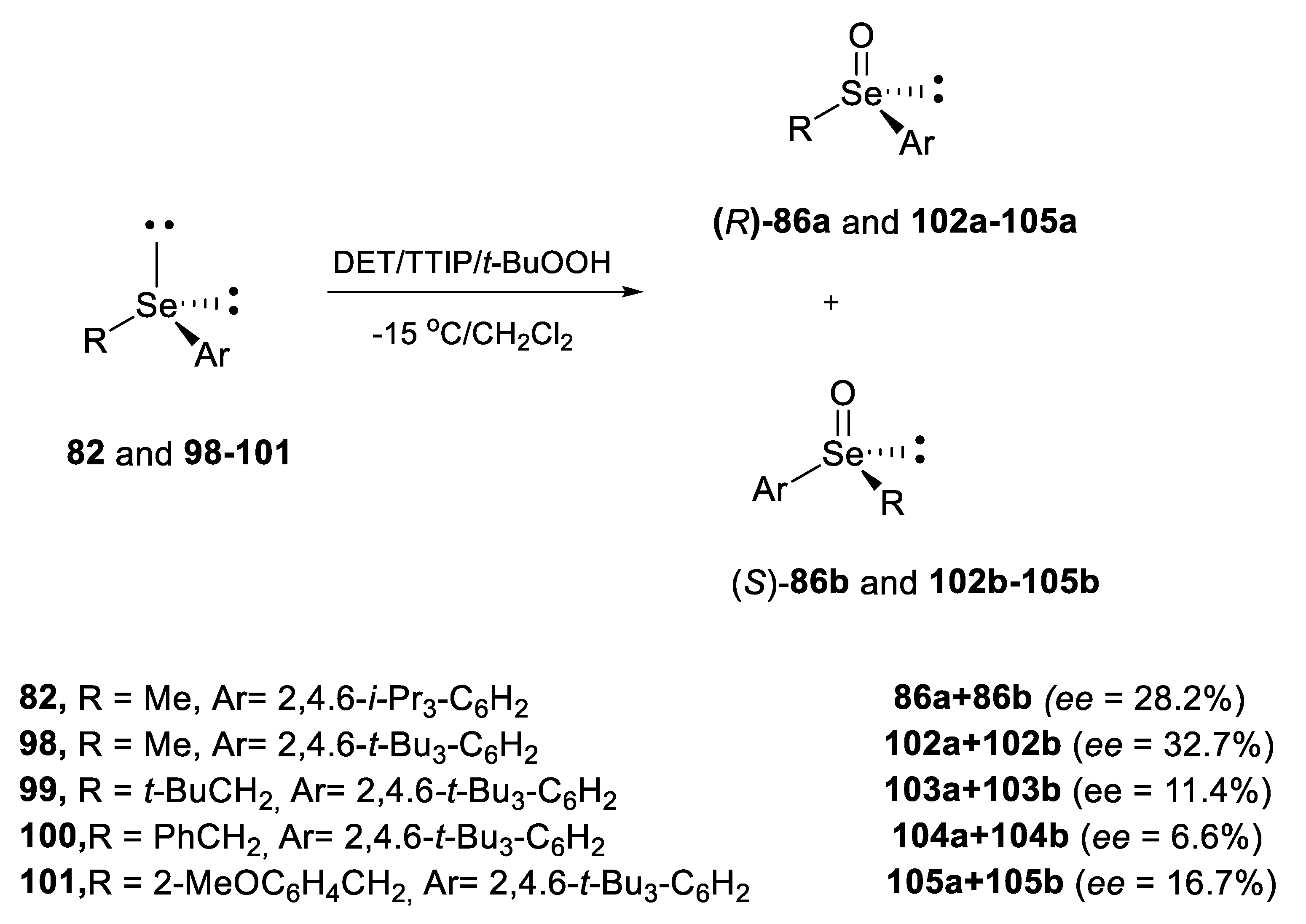{kind=link}
{kind=link}
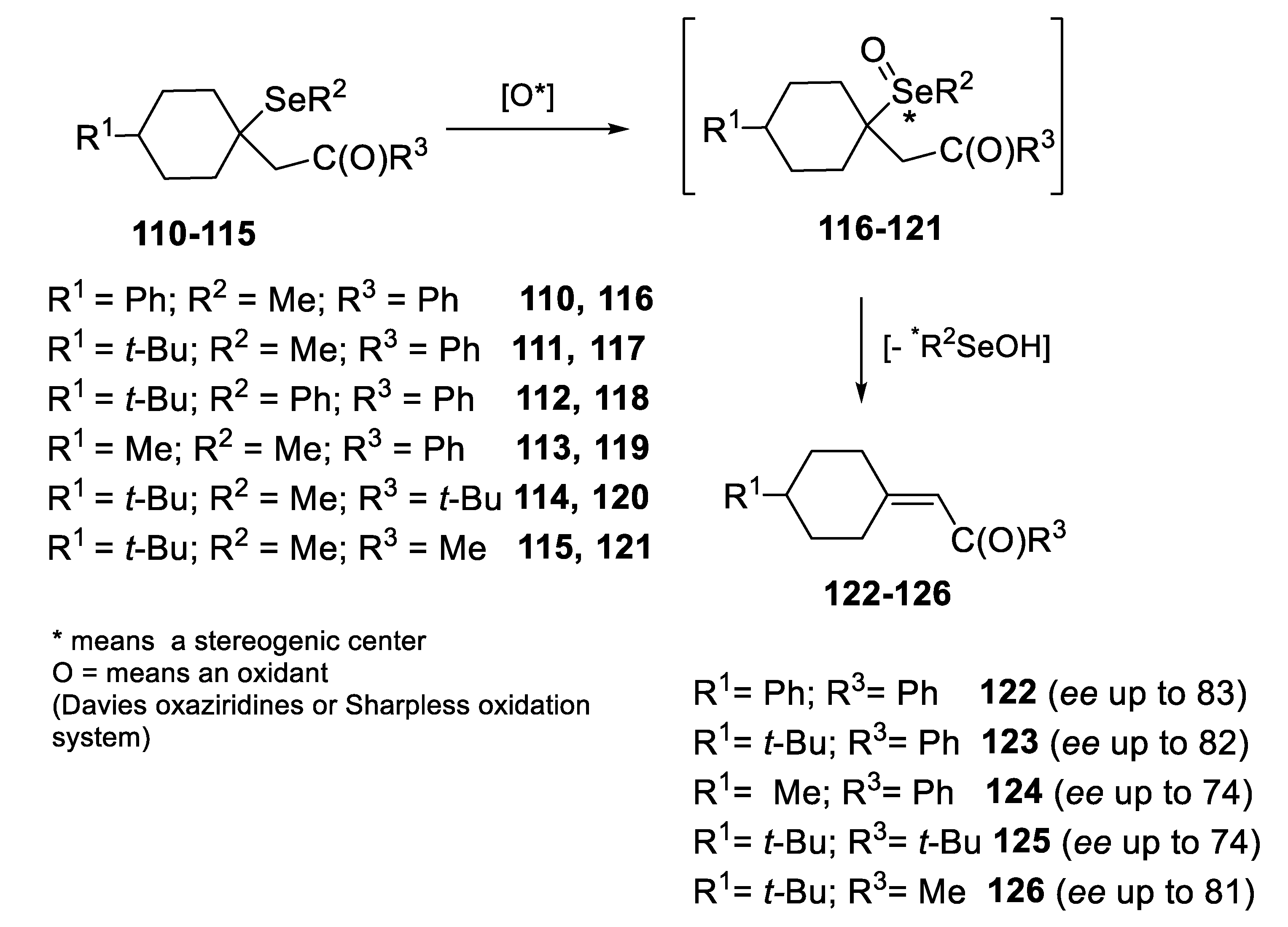{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- a)
- b)
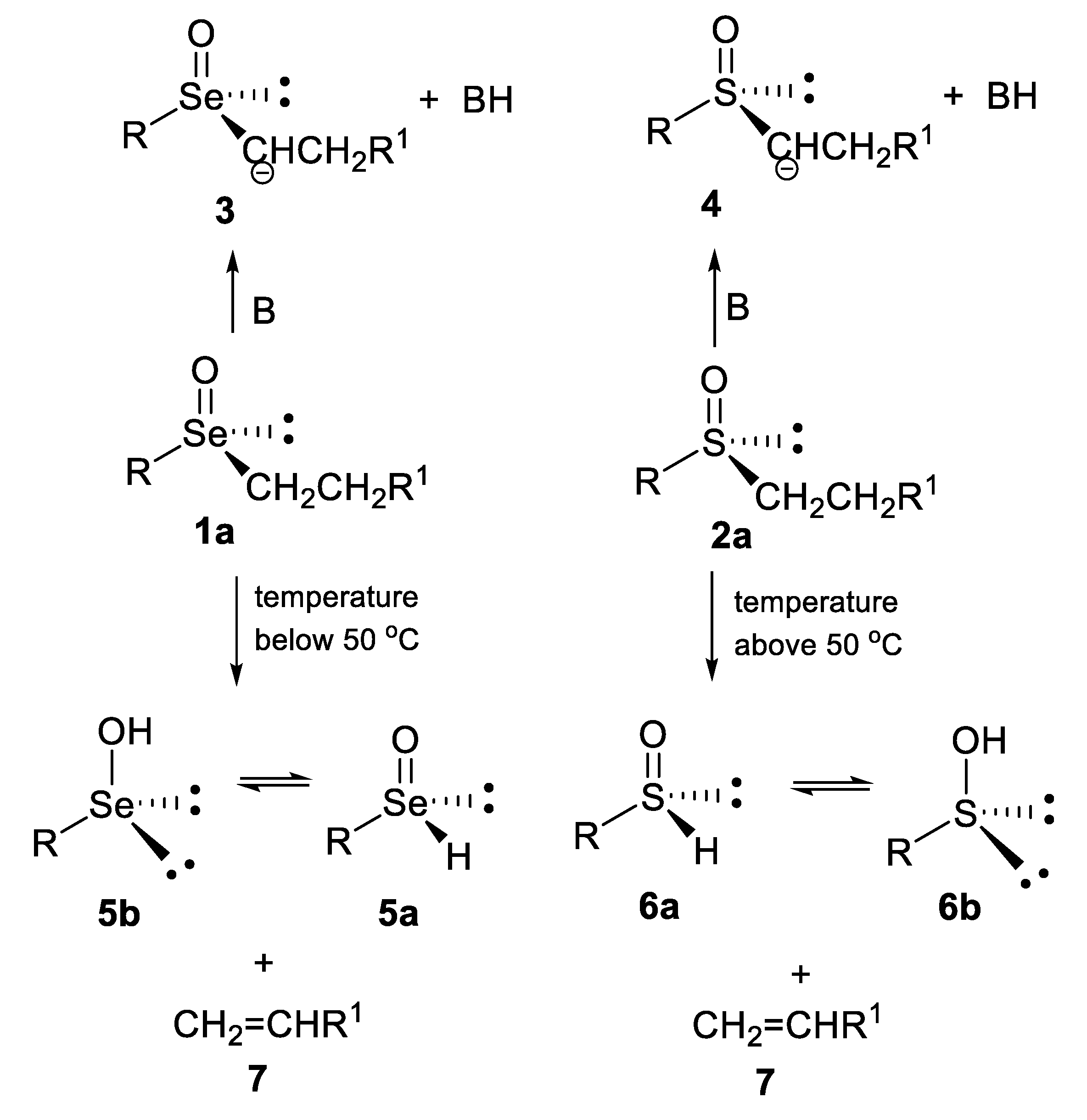
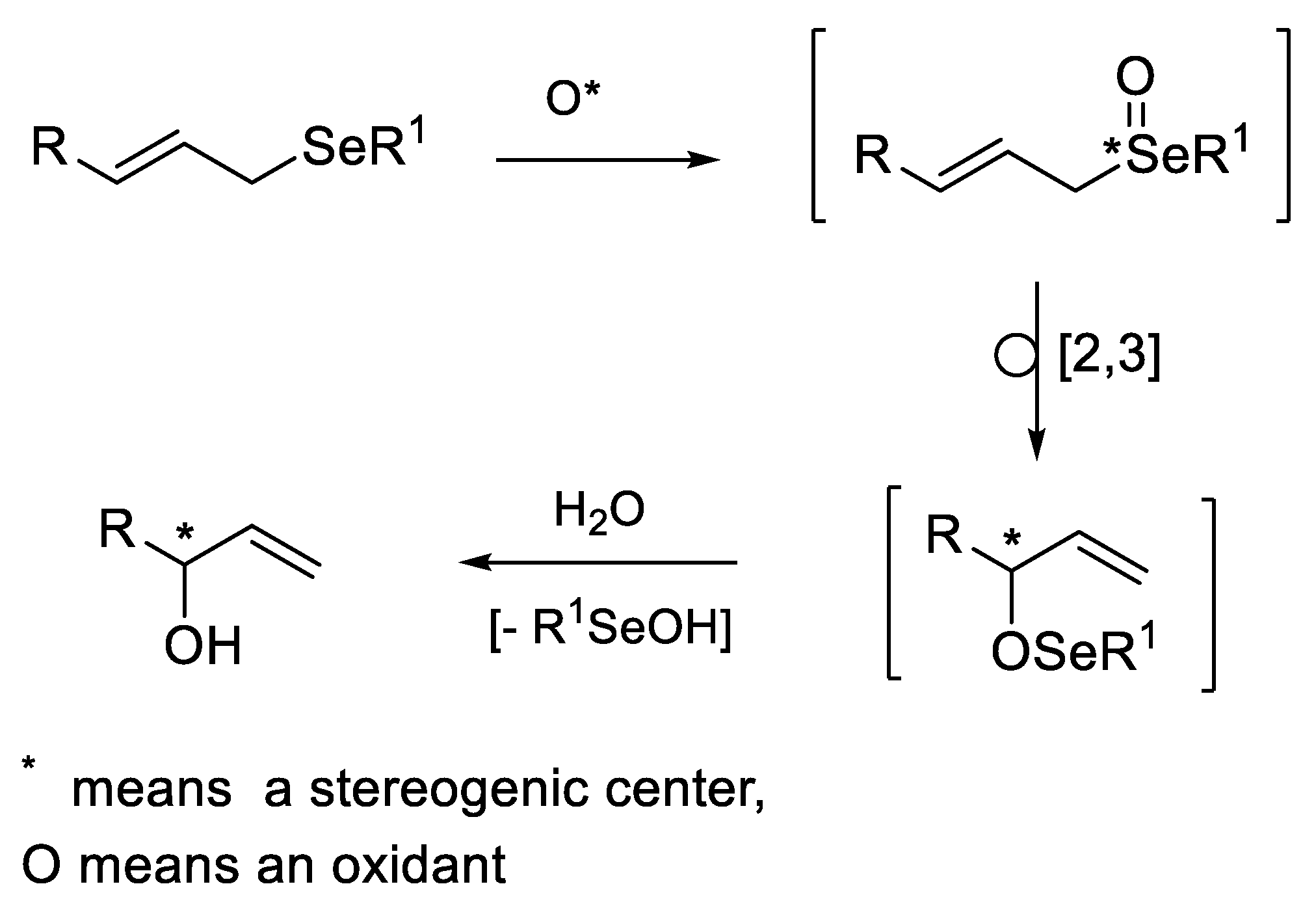
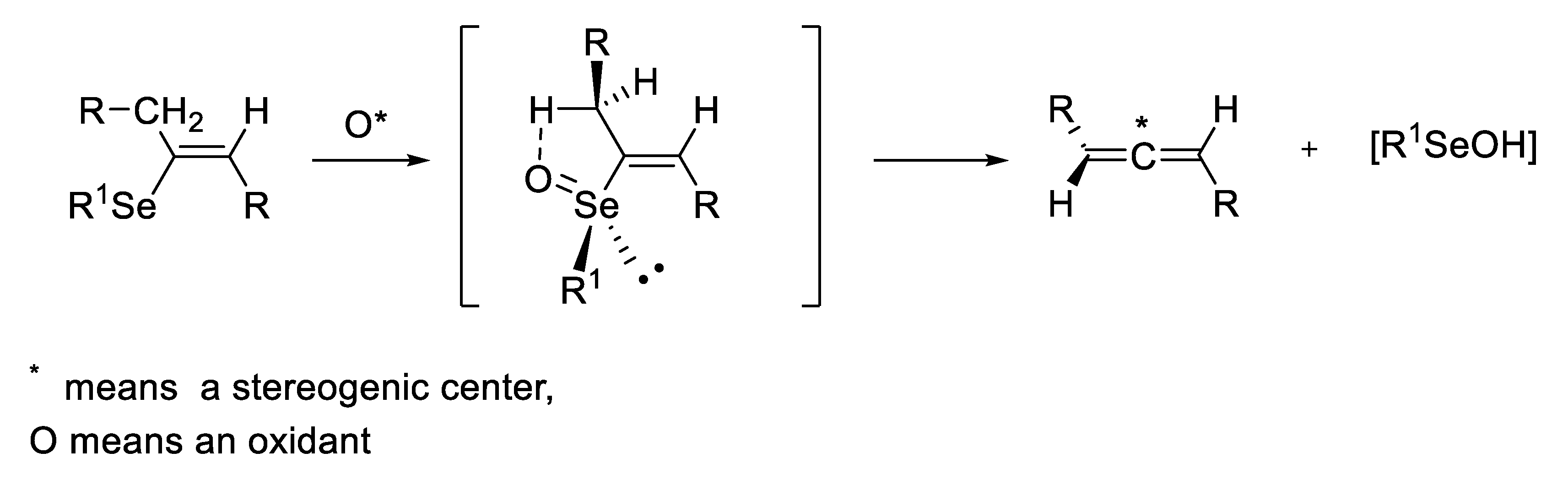
- undergo an internal type elimination of the E2 type for compounds containing β-hydrogen atoms, which leads to the formation of the corresponding, generally very unstable, seleninic 5 or sulfenic 6 acids and unsaturated carbon derivatives 7 (Scheme 1) [5,6]. It should be noted here that both acids can exist as chiral tetravalent (5a or 6a) or achiral divalent (5b [7,8,9] or 6b [10,11,12]) tautomers.
- a)
- reaction of diastereoisomerically pure precursors;
- b)
- asymmetric oxidation of prochiral selenides;
- c)
- chromatographic and nonclassical resolution of racemates by forming complexes with an optically active hydrogen bond donor;
- d)
- kinetic resolution of racemates;
- e)
- reaction of enantiopure, cyclic seleninic esters with organometallic reagents.
2. Synthesis of Optically Active Selenoxides
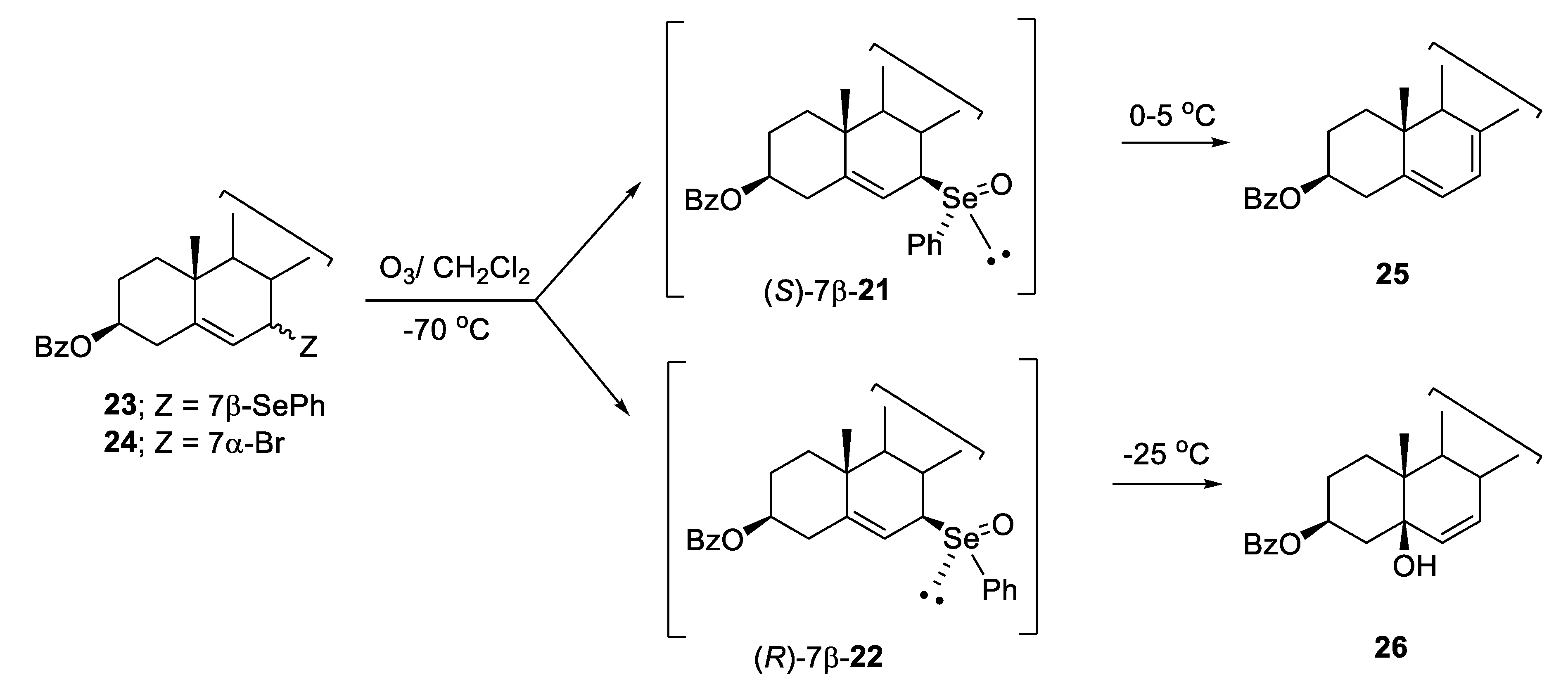
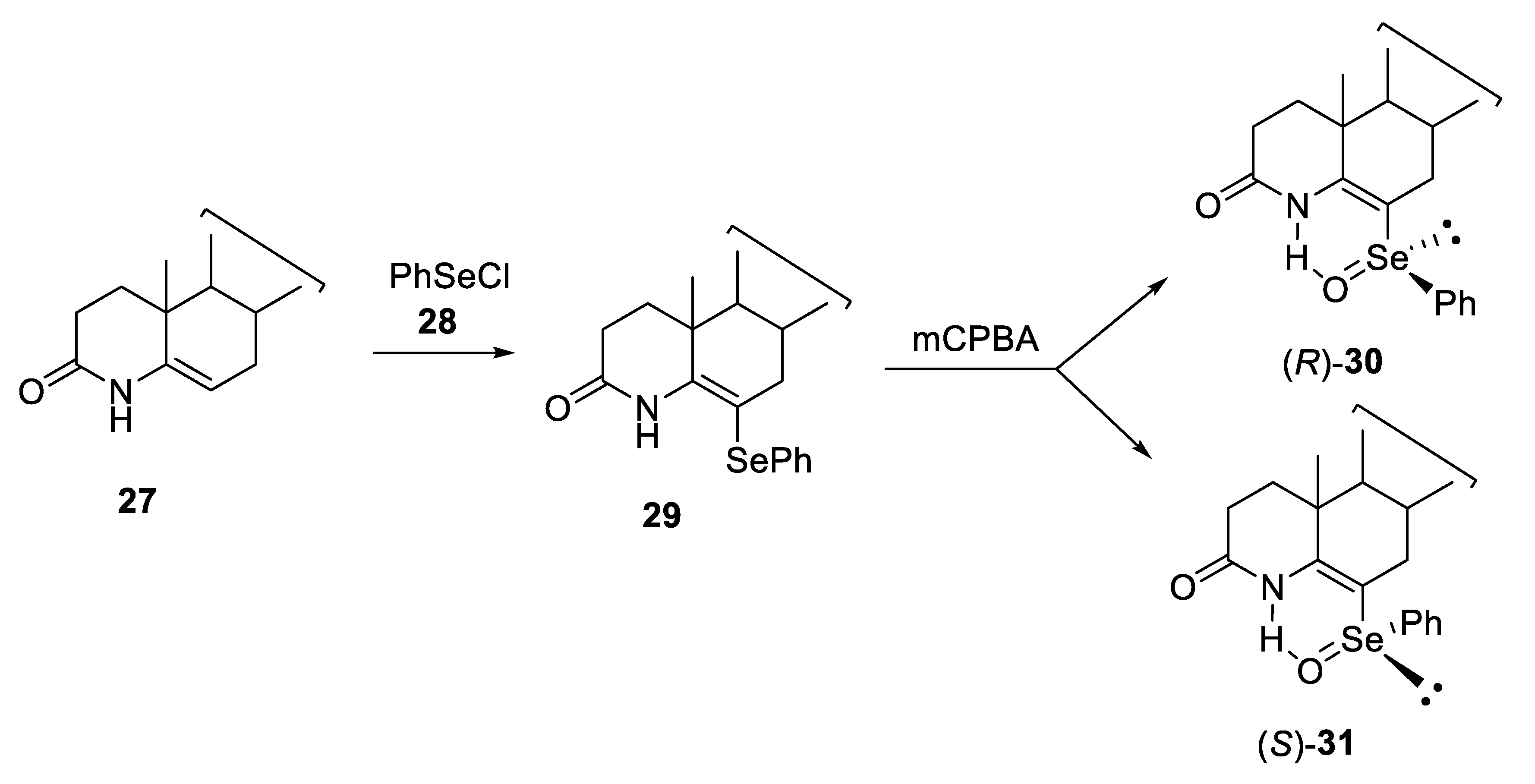
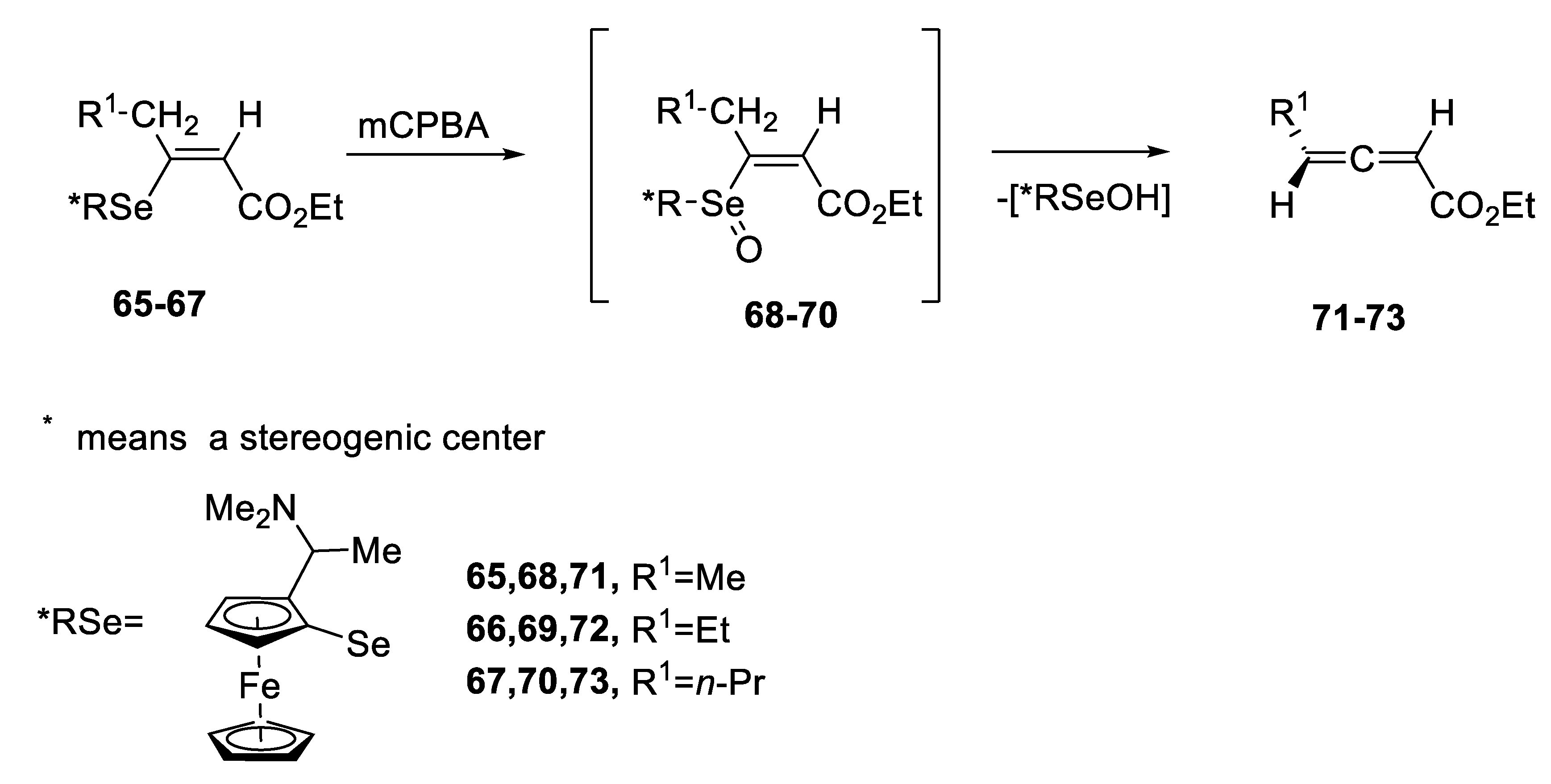
2.1. Diastereoisomeric Selenoxides
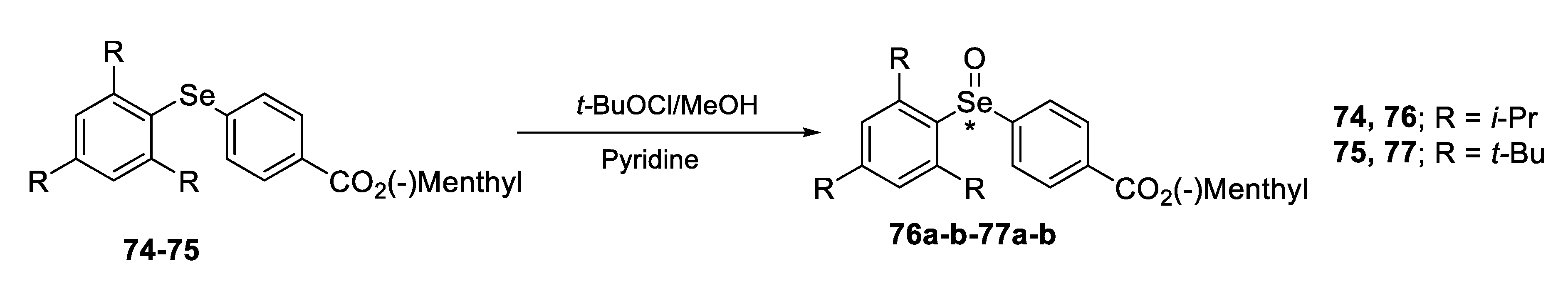
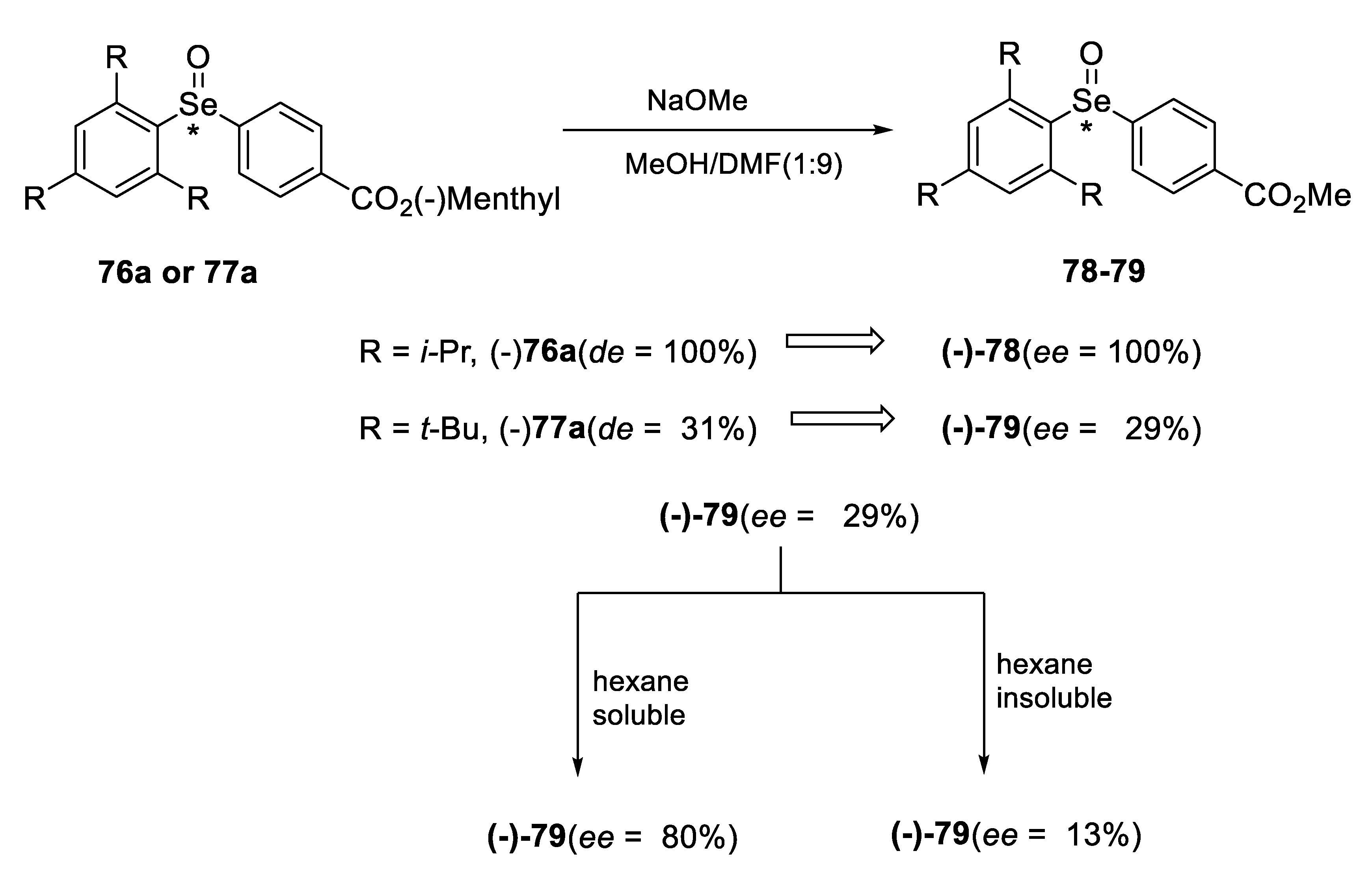
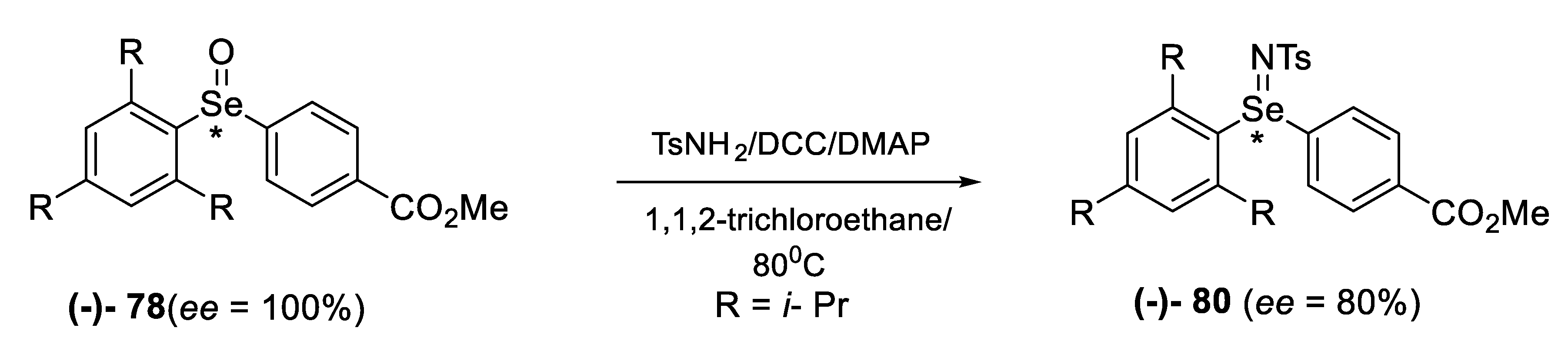
2.2. Enantiomeric Selenoxides
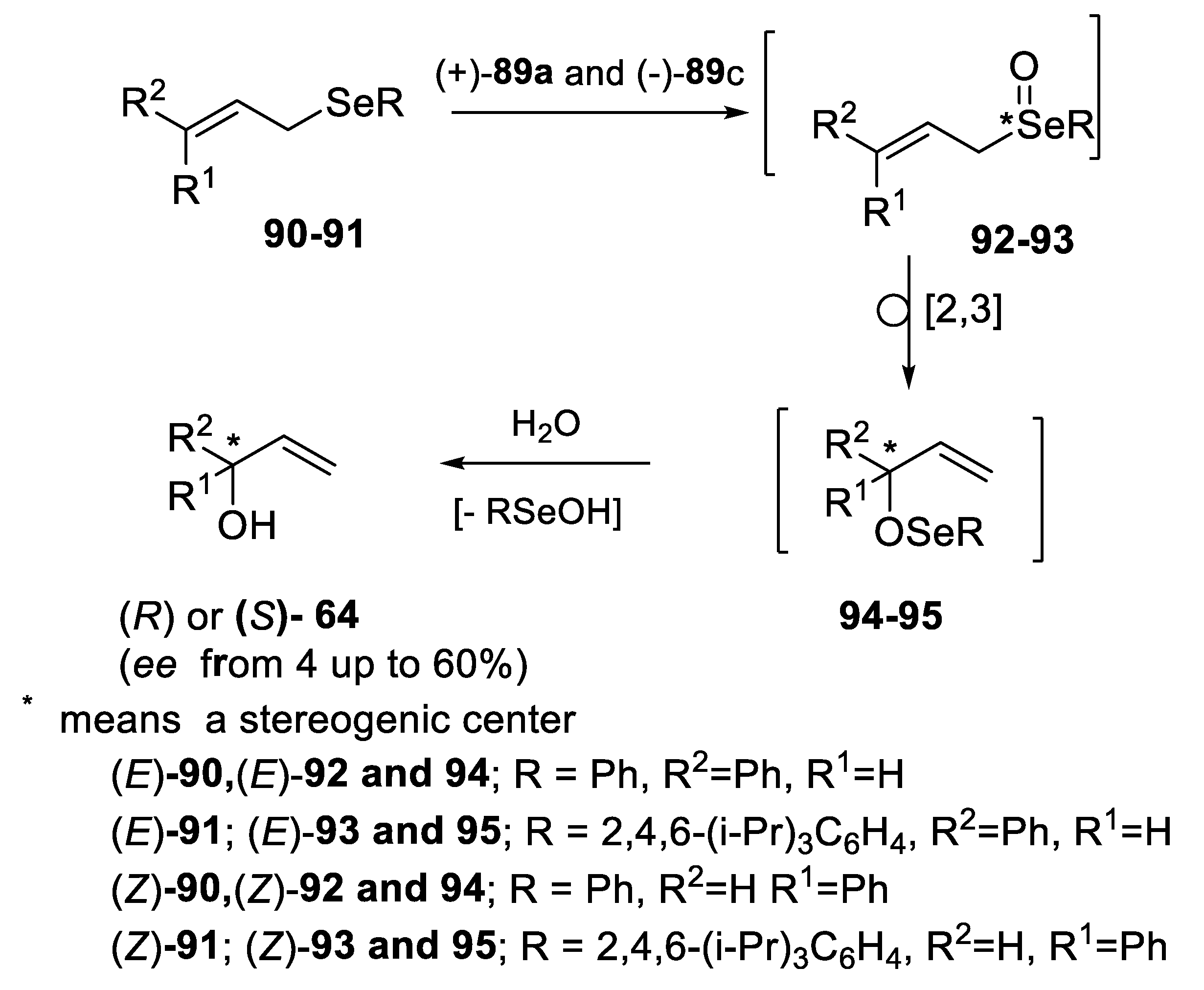
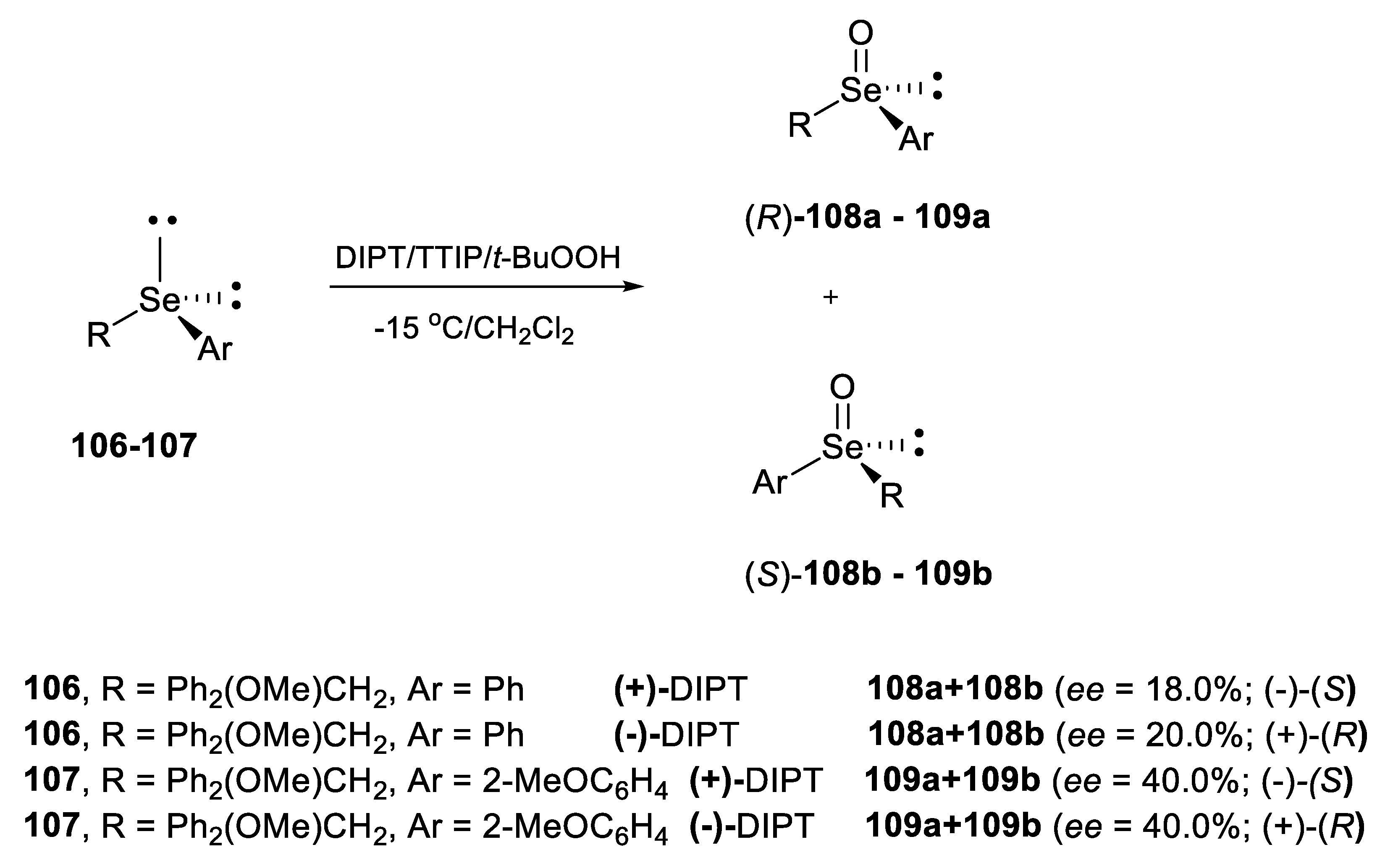
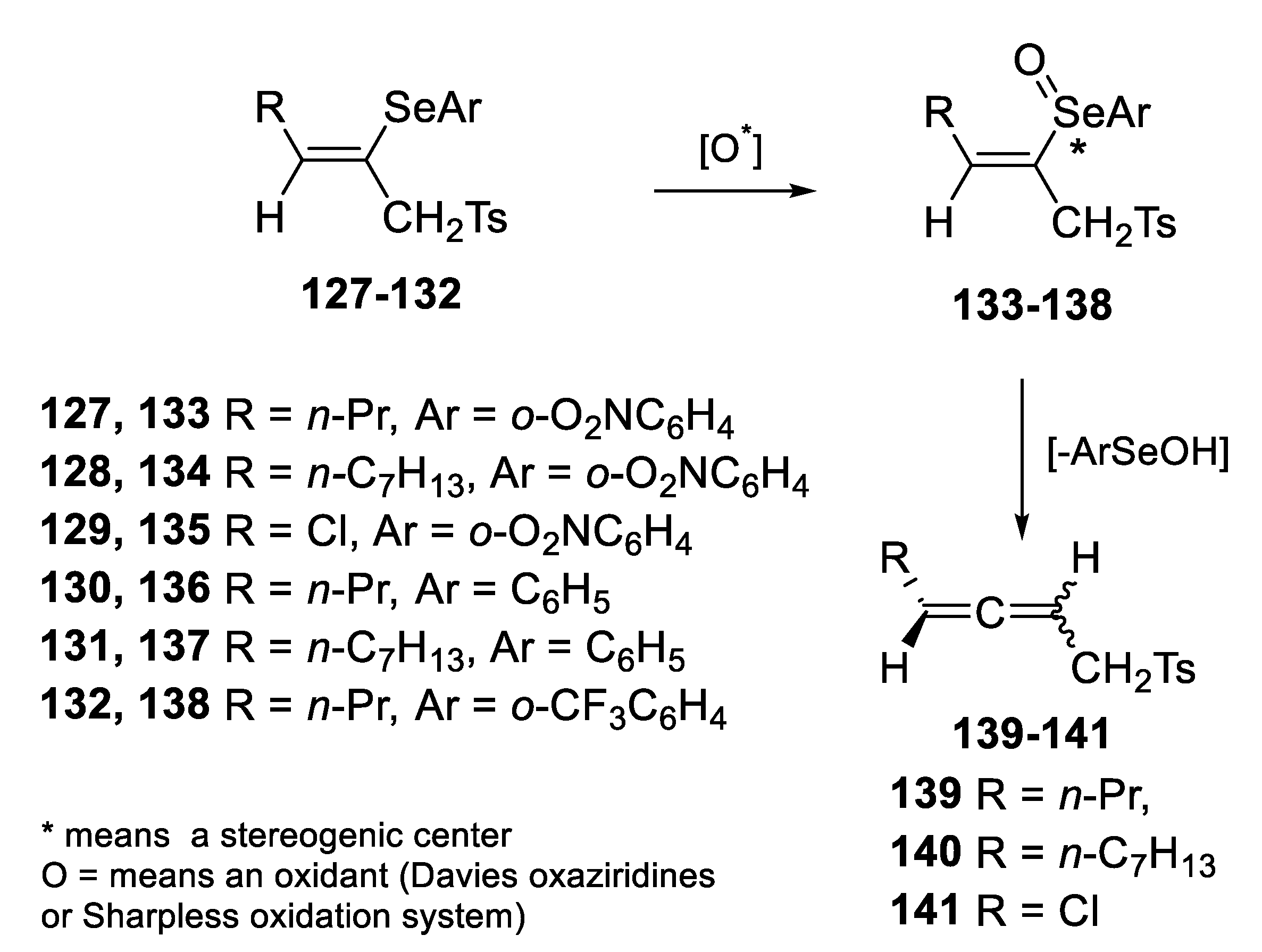
2.2.1. Asymmetric Oxidation
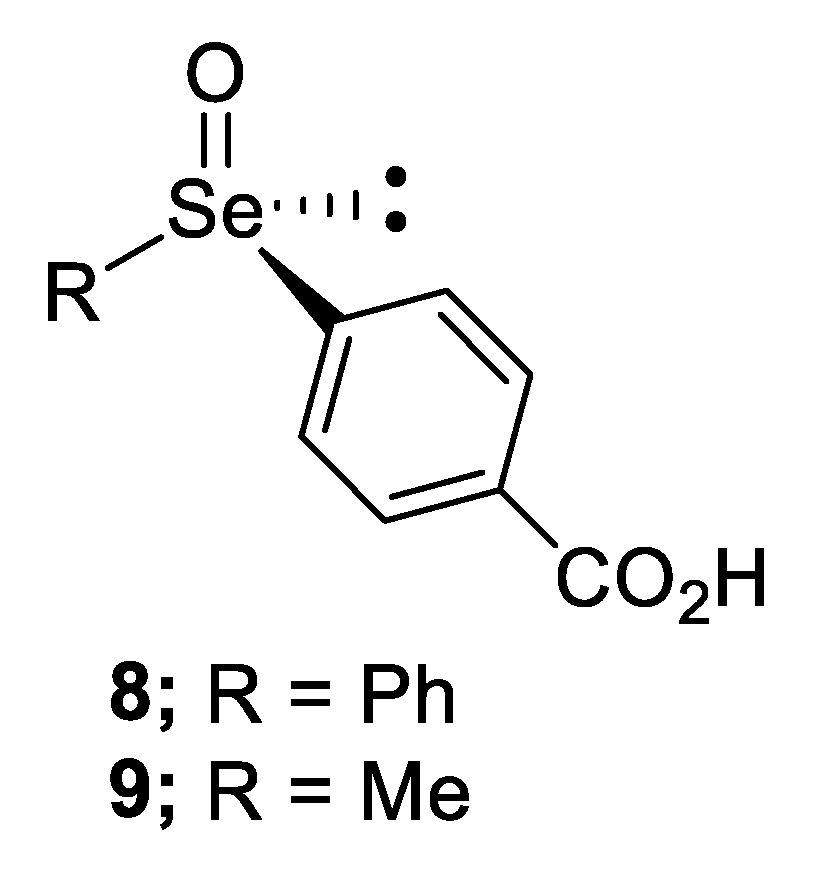
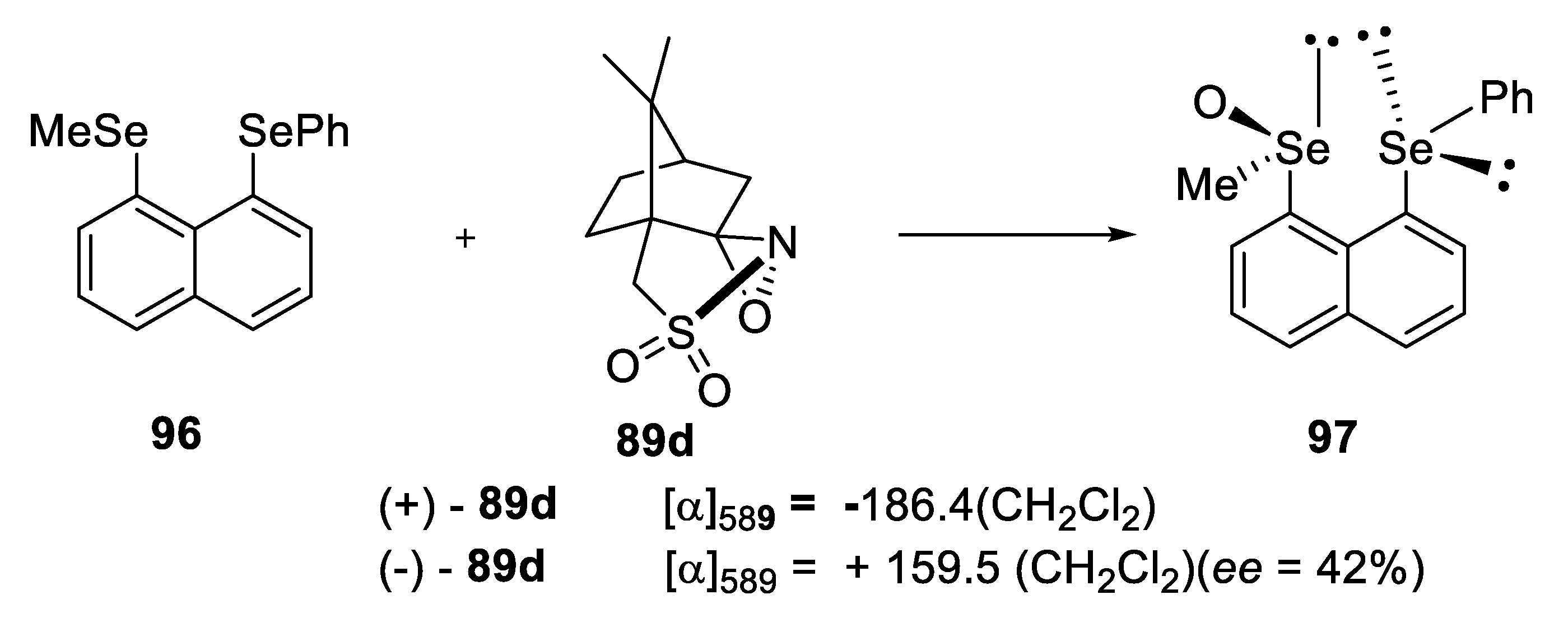
2.2.2. Chromatographic and Non-Classical Resolution of Racemates by Forming Complexes with an Optically Active Hydrogen Bond Donor
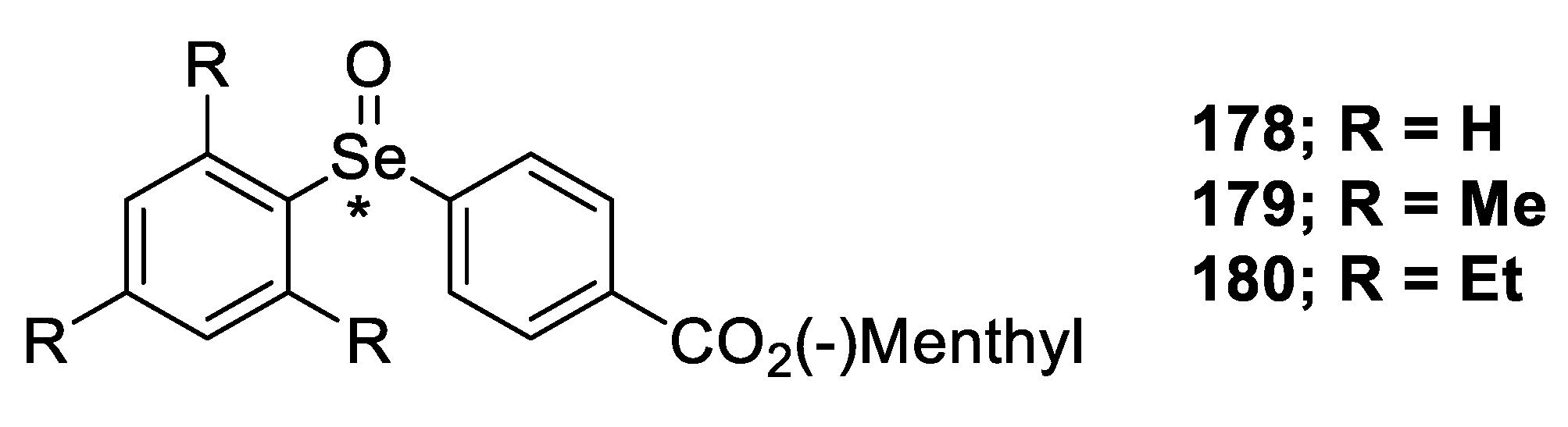
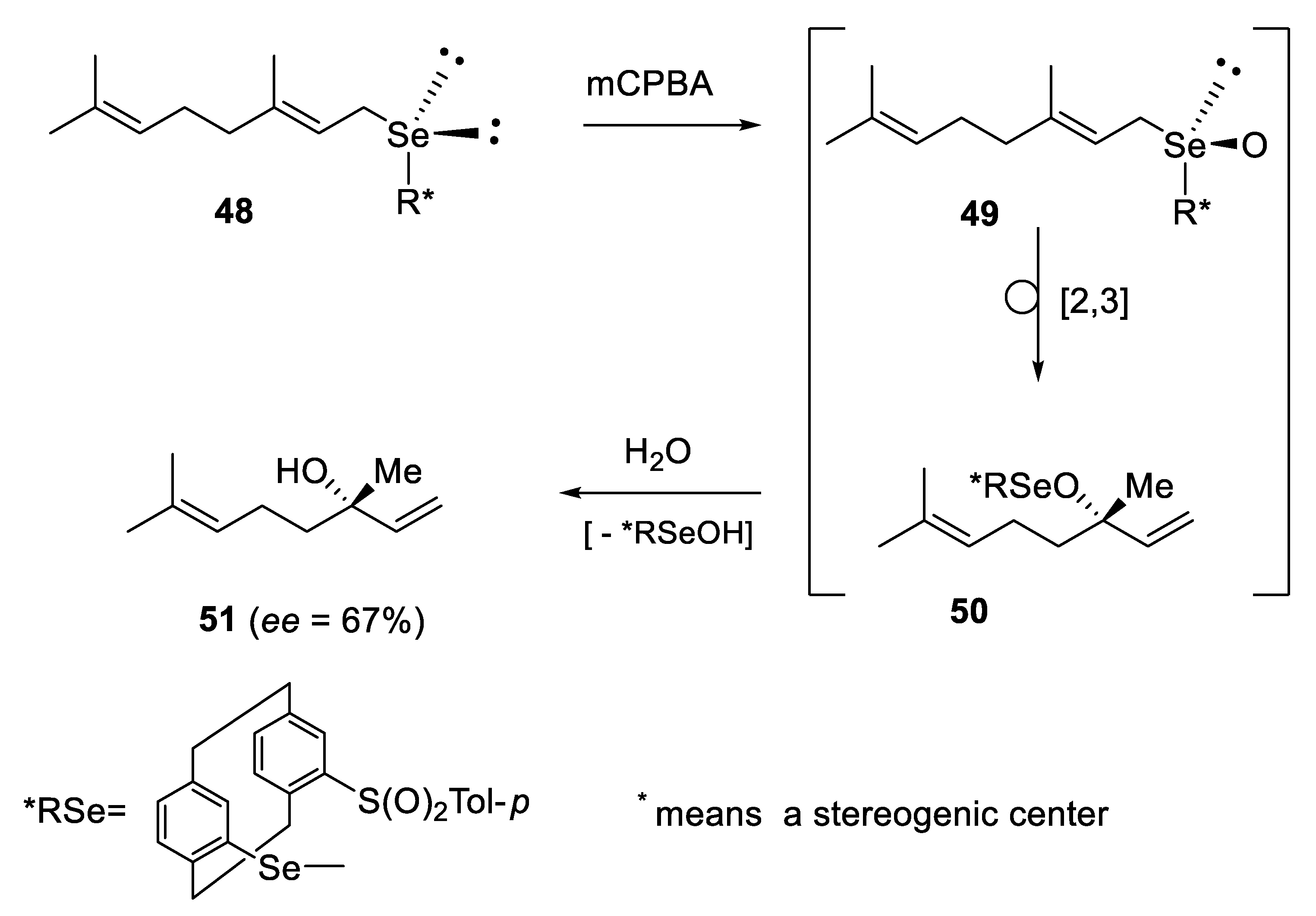
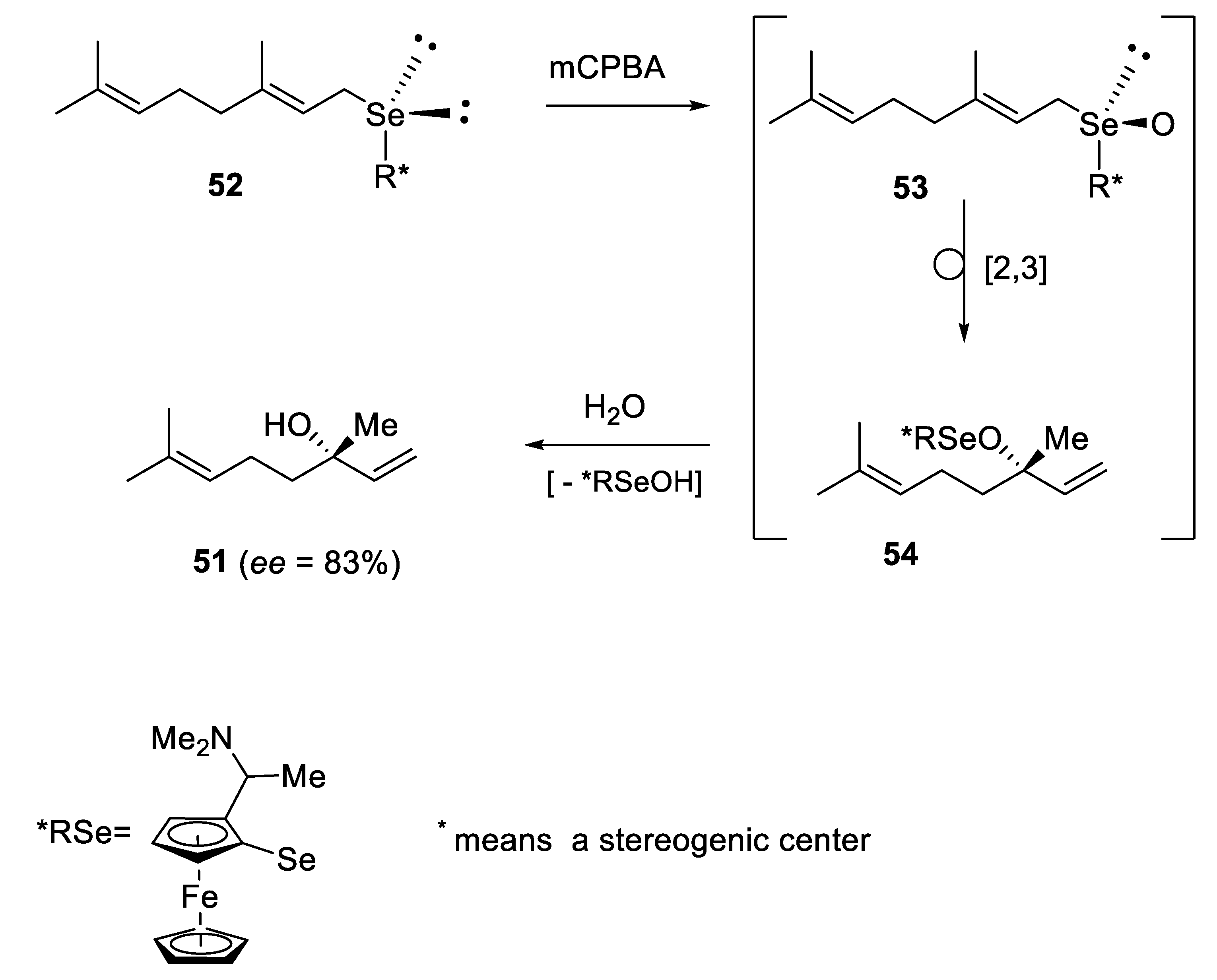
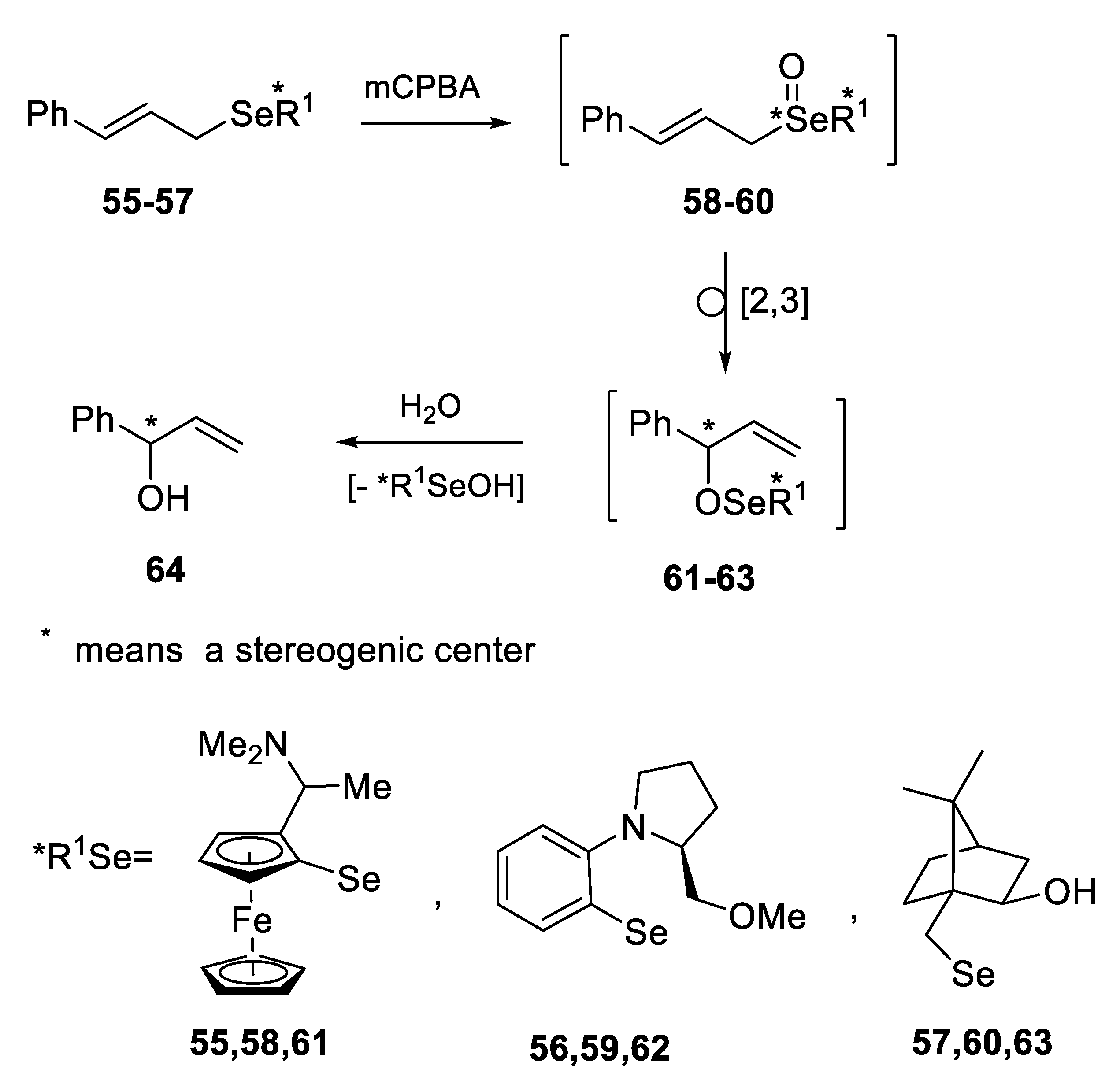
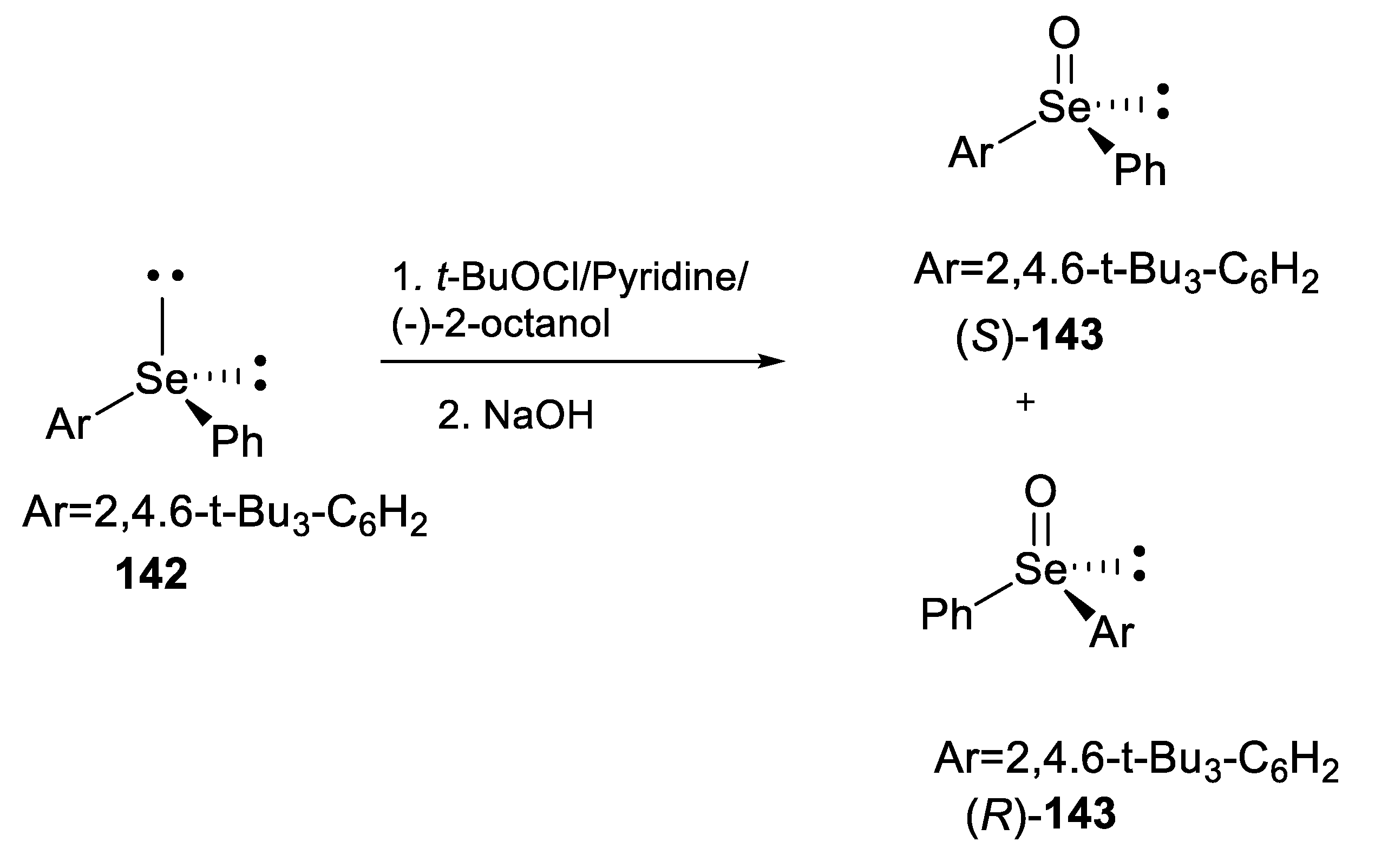
2.2.3. Kinetic Resolution of Racemates
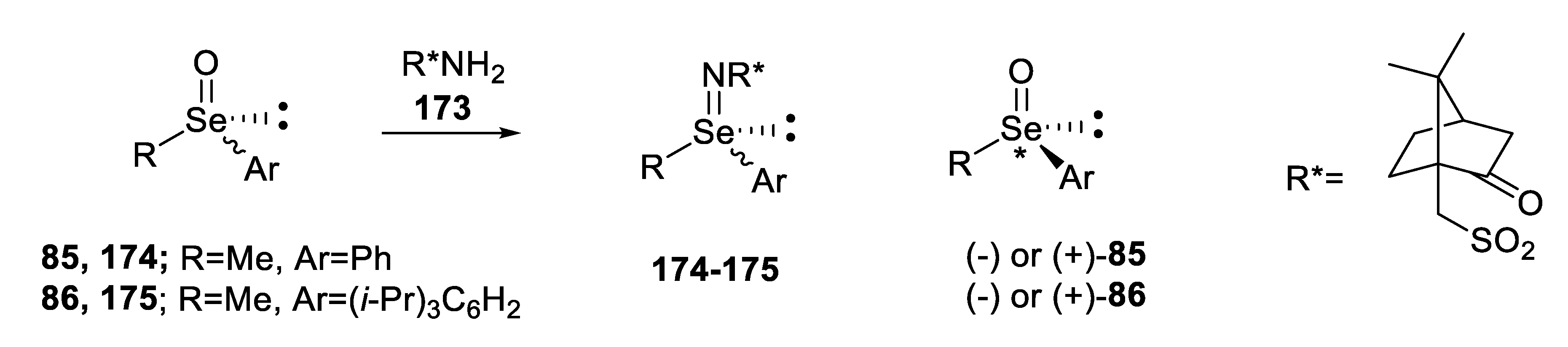
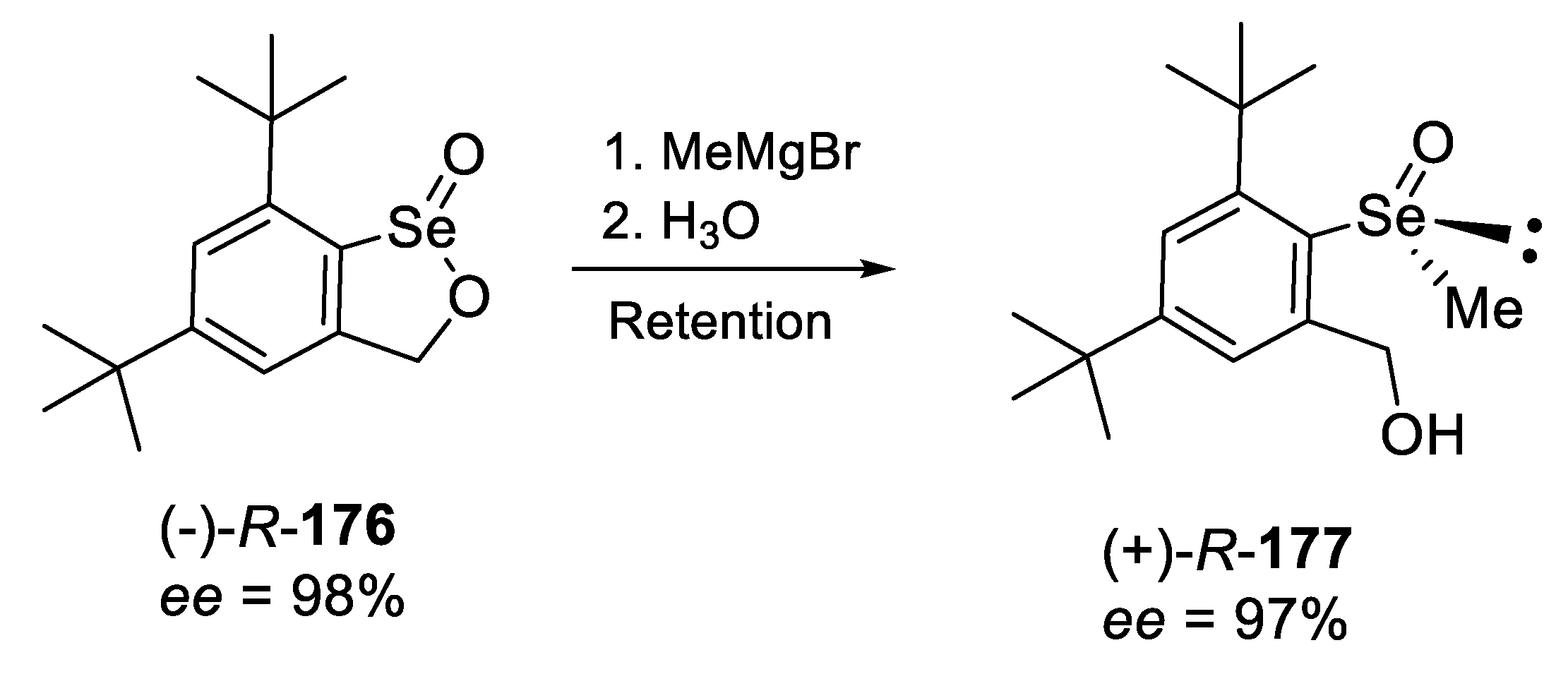
2.2.4. Reaction of Enantiopure, Cyclic Seleninic Ester with an Organometallic Reagent
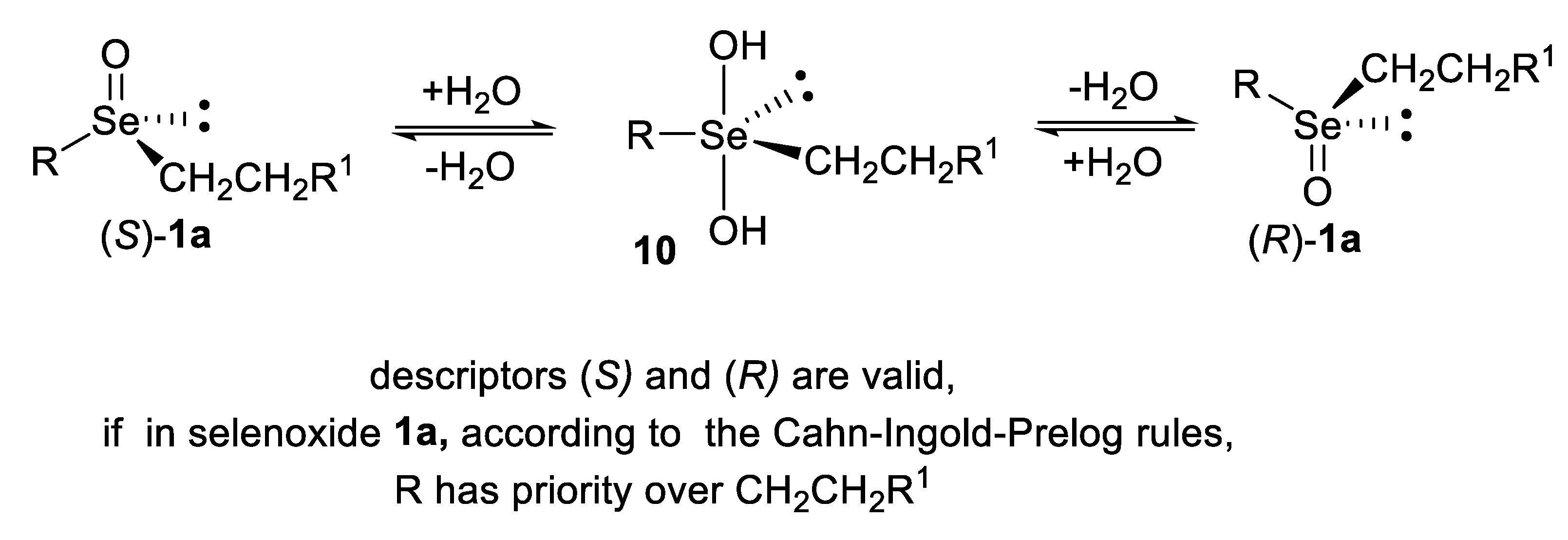
3. Absolute Configurations and Enantiomeric Excesses of Optically Active Selenoxides
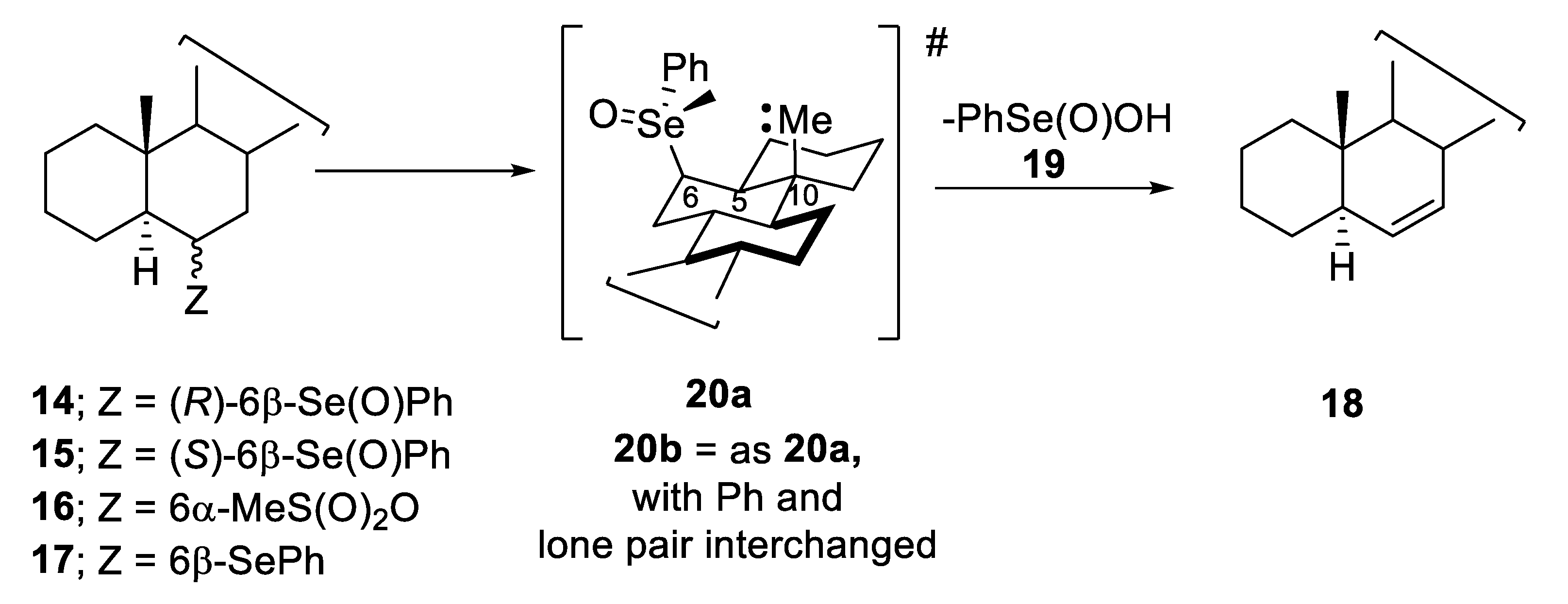
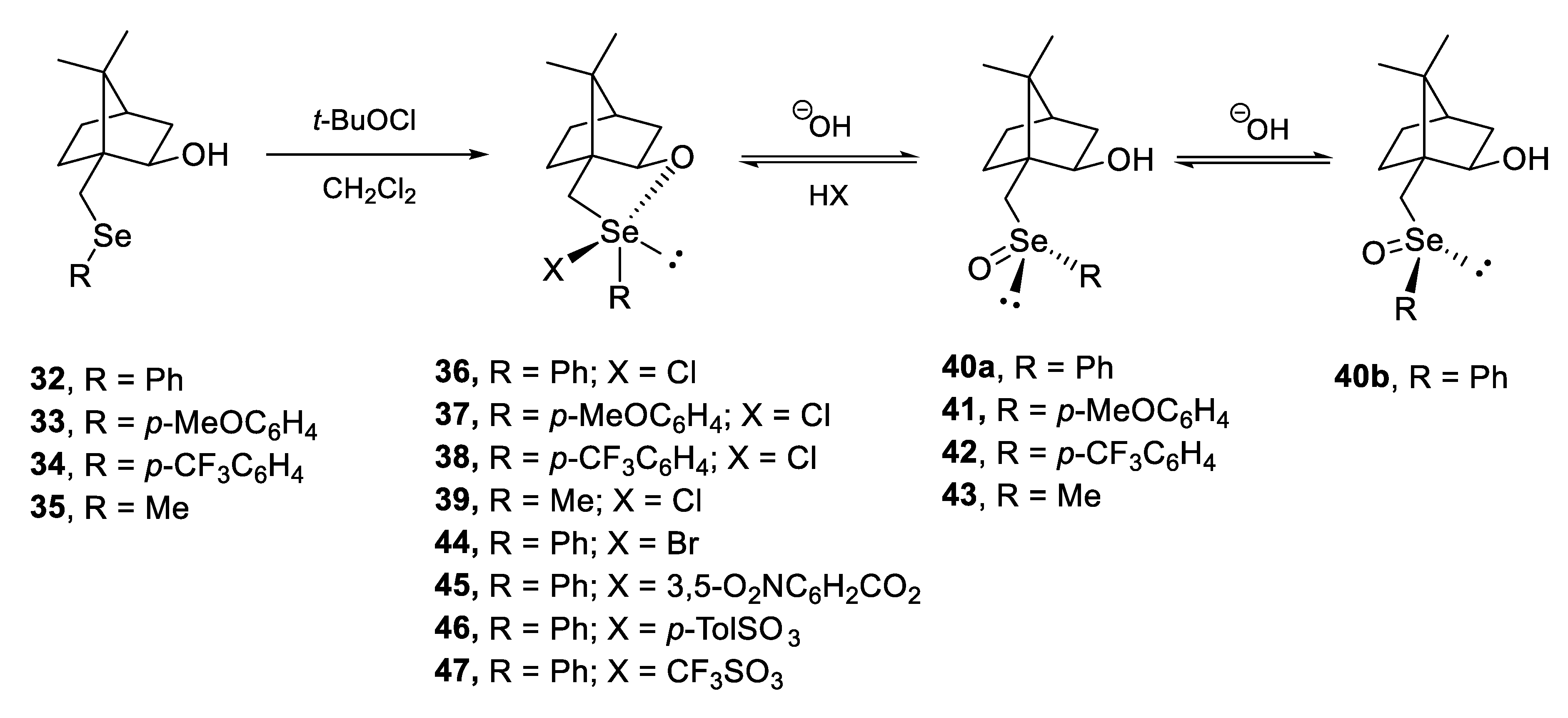
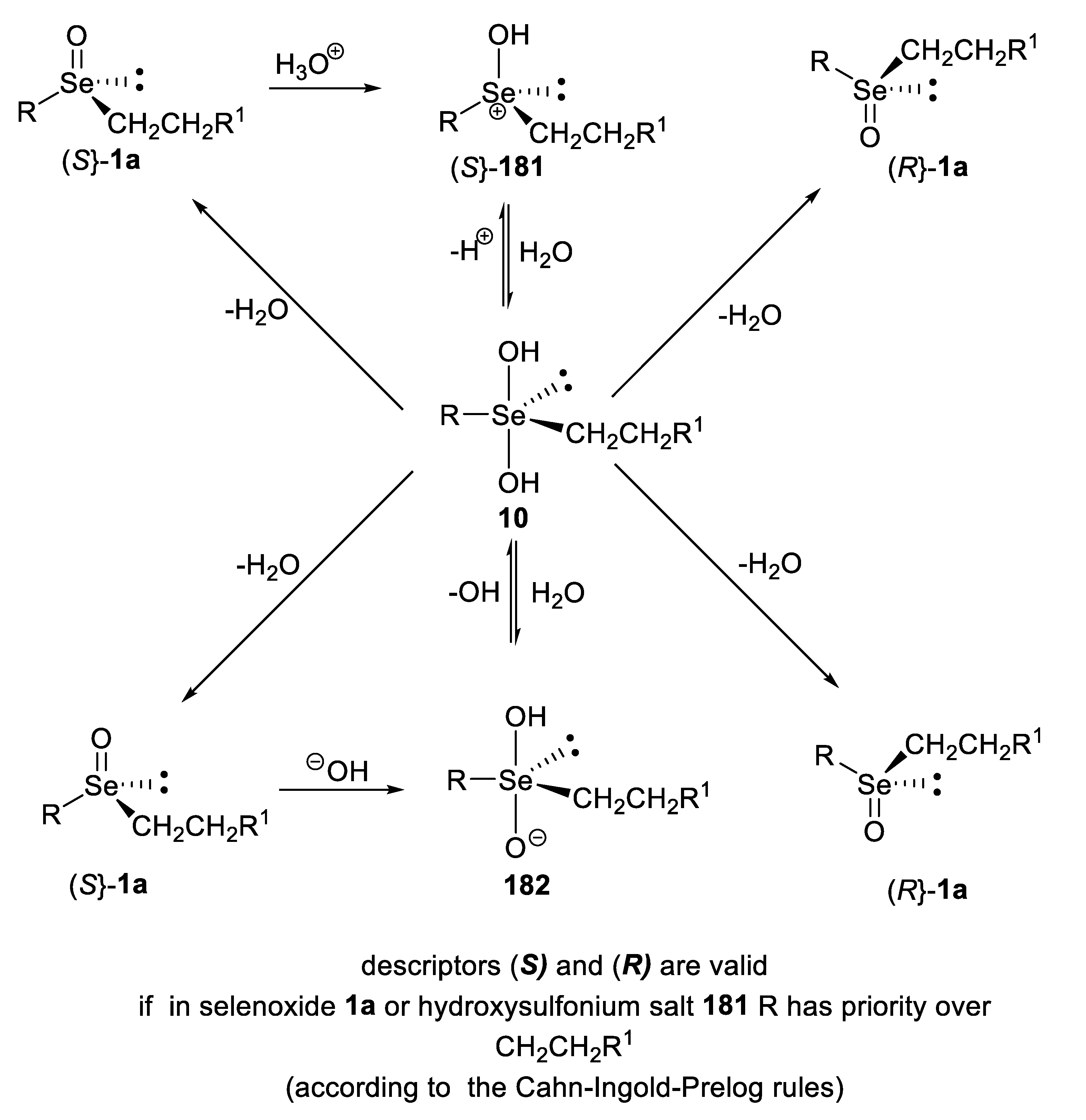
4. Configurational Stability of Optically Active Selenoxides
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Twidell, T.T. Oxidation of Alcohols by Activated Dimethyl Sulofoxide and Related Reactions. Synthesis 1990, 10, 857–870. [Google Scholar] [CrossRef]
- Młochowski, J.; Lisiak, R.; Wójtowicz-Młochowska, H. Organoselenium and Organotellurium Oxidation and Reduction. In PATAI’S Chemistry of Functional Groups; Wiley: Hoboken, NJ, USA, 2011; pp. 1083–1160. [Google Scholar]
- Ponthieux, S.; Paulmier, C. Selenium-Stabilized Carboanions. Organselenium Chem. 2000, 208, 113–142. [Google Scholar]
- Mikolajczyk, M.; Drabowicz, J.; Kielbasinski, P. Chiral Sulfur Reagents: Applications in Asymmetric and Stereoselective Synthesis; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Nishibayashi, Y. Uemura Selenoxide Elimination and [2,3]Sigmatropic Rearrangement. Organselenium Chem. 2000, 208, 201–234. [Google Scholar]
- Nomoto, A.; Ogawa, A. ChemInform Abstract: Preparative Uses of Organoselenium and Organotellurium Compounds. In The Chemistry of Organic Selenium and Tellurium Compounds; Rappoport, Z., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2012; pp. 623–688. [Google Scholar]
- Ishii, A.; Matsubayashi, T.; Takahashi, T.; Nakayama, J. Preparation of a selenenic acid and isolation of selenoseleninates. J. Org. Chem. 1999, 64, 1084–1085. [Google Scholar] [CrossRef]
- Reich, H.J.; Jasperse, C.P. Organoselenium chemistry. Preparation and reactions of 2,4,6-tri-tert-butylbenzeneselenenic acid. J. Org. Chem. 1988, 53, 2389–2390. [Google Scholar] [CrossRef]
- Goto, K.; Nagahama, M.; Mizushima, T.; Shimada, K.; Kawashima, T.; Okazaki, R. The first direct oxidative conversion of a selenol to a stable selenenic acid: Experimental demonstration of three processes included in the catalytic cycle of glutathione peroxidase. Org. Lett. 2001, 3, 3569–3572. [Google Scholar] [CrossRef]
- Penn, R.E.; Block, E.; Revelle, L.K. Methanesulfenic Acid. J. Am. Chem. Soc. 1978, 100, 3622–3624. [Google Scholar] [CrossRef]
- Goto, K.; Holler, M.; Okazaki, R. Synthesis, Structure, and Reactions of a Sulfenic Acid Bearing a Novel Bowl-Type Substituent: The First Synthesis of a Stable Sulfenic Acid by Direct Oxidation of a Thiol. J. Am. Chem. Soc. 1997, 119, 1460–1461. [Google Scholar] [CrossRef]
- Ishii, A.; Komiya, K.; Nakayama, J. Synthesis of a Stable Sulfenic Acid by Oxidation of a Sterically Hindered Thiol (Thiophenetriptycene-8-thiol)1 and Its Characterization. J. Am.Chem. Soc. 1996, 118, 12836–12837. [Google Scholar] [CrossRef]
- Rheinboldt, H.; Giesbrecht, E. The Configuration of Organic Selenoxides: Mixed Crystals of Selenoxides with Sulfoxides. J. Am. Chem. Soc. 1946, 68, 2671–2673. [Google Scholar] [CrossRef]
- Rheinboldt, H.; Giesbrecht, E. Mixed Crystals of Sulfoxides, Sulfones, Selenoxides and Selenones. J. Am. Chem. Soc. 1947, 69, 644–646. [Google Scholar] [CrossRef]
- Gaythwaite, W.R.; Kenyon, J.; Phillips, H. CCXCVII.—The quadrivalency of selenium. Part I. 4-Carboxydiphenyl and p-carboxyphenyl methyl selenoxides. J. Chem. Soc. 1928, 2280–2287. [Google Scholar] [CrossRef]
- Harrison, A.; Kenyon, J.; Phillips, J. Phillips. J. Chem. Soc. 1926, 2079–2786. [Google Scholar] [CrossRef]
- Campbell, T.W.; Walker, H.G.; Coppinger, G.M. Some Aspects of the Organic Chemistry of Selenium. Chem. Rev. 1952, 50, 279–349. [Google Scholar] [CrossRef]
- Ōki, M.; Iwamura, H. The configuration of the selenoxides. Tetrahedron Lett. 1966, 7, 2917–2920. [Google Scholar] [CrossRef]
- Burlant, W.J.; Gould, E.S. Metalation of Dibenzoselenophene. J. Am. Chem. Soc. 1954, 76, 5775–5776. [Google Scholar] [CrossRef]
- Cinquini, M.; Colonna, S.; Landini, D. Diastereomeric diselenoxides. Bol. Sci. Fac. Chim. Ind. Bol. 1969, 27, 207–209. [Google Scholar]
- Musher, J.I. The Chemistry of Hypervalent Molecules. Angew. Chem. Int. Ed. 1969, 8, 54–68. [Google Scholar] [CrossRef]
- Akiba, K. Chemistry of Hypervalent Compounds; Akiba, K., Ed.; Wiley-VCH: New York, NY, USA, 1999. [Google Scholar]
- Drabowicz, J.; Halaba, G. ChemInform Abstract: Stereochemical Aspect of the Chemistry of Hypervalent Chalcogen Compounds. Cheminform 2010, 33, 1–32. [Google Scholar] [CrossRef]
- Drabowicz, J.; Kiełbasiński, P.; Zając, A.; Pokora-Sobczak, P. Hypervalent Selenium Derivatives. In Organoselenium Chemistry between Synthesis and Biochemistry; Santi, C., Ed.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2014; pp. 120–145. [Google Scholar]
- Komatsu, N.; Uemura, S. Recent Advances in Asymmetric Synthesis Using Organochalcogen Compounds. J. Synth. Org. Chem. 1995, 53, 268–274. [Google Scholar] [CrossRef][Green Version]
- Kamigata, N.; Shimizu, T. Synthesis and Stereochemistry of Optically Active Selenoxides. J. Synth. Org. Chem. 1990, 48, 229–239. [Google Scholar] [CrossRef]
- Kobayashi, M.; Shimizu, T. Synthesis of the Optically Active Diaryl Selenoxides. Cheminform 1987, 7, 1437–1441. [Google Scholar]
- Kamigata, N. Synthesis and Stereochemistry of Chiral Selenoxides and Telluroxides. Phosphorus Sulfur Silicon Relat. Elem. 2001, 171, 207–229. [Google Scholar] [CrossRef]
- Uemura, S. Application of Asymmetric Oxidation and Imidation of Organic Selenides to Organic Transformations. Phosphorus Sulfur Silicon Relat. Elem. 2001, 171, 13–30. [Google Scholar] [CrossRef]
- Drabowicz, J.; Kiełbasiński, P.; Zajac, A. Hypervalent Derivatives of Selenium and Tellurium. In PATAI’S Chemistry of Functional Groups; Wiley: Hoboken, NJ, USA, 2011; pp. 891–948. [Google Scholar]
- Jones, D.N.; Mundy, D.; Whitehouse, R.D. Steroidal selenoxides diastereoisomeric at selenium; syn-elimination, absolute configuration, and optical rotatory dispersion characteristics. J. Chem. Soc. D 1970, 2, 86–87. [Google Scholar] [CrossRef]
- Ayrey, G.; Barnard, D.; Woodbridge, D.T. 401. The oxidation of organoselenium compounds by ozone. J. Chem. Soc. 1962, 2089–2099. [Google Scholar] [CrossRef]
- Cram, D.J. Steric Effects in Organic Chemistry; Newman, M., Ed.; Wiley: New York, NY, USA, 1956. [Google Scholar]
- Salmond, W.; Barta, M.; Cain, A.; Sobala, M. Alternative modes of decomposition of allylic selenoxides diastereoisomeric at selenium. Preparation of Δ5,7- and 5β-hydroxy-Δ6-steroiods. Tetrahedron Lett. 1977, 18, 1683–1686. [Google Scholar] [CrossRef]
- Back, T.G.; Ibrahim, N.; McPhee, D.J. Studies of enamidic. DELTA.5-4-azasteroidal selenoxides: Preparation, Pummerer reactions, configurational stability, and conversion to carbinol amides. J. Org. Chem. 1982, 47, 3283–3289. [Google Scholar] [CrossRef]
- Kurose, N.; Takahashi, T.; Koizumi, T. First synthesis of optically pure selenuranes and stereoselective alkaline hydrolysis. Their application to asymmetric [2,3]sigmatropic rearrangement of allylic selenoxides. Tetrahedron 1997, 53, 12115–12129. [Google Scholar] [CrossRef]
- Takahashi, T.; Kurose, N.; Kawanami, S.; Arai, Y.; Koizumi, T.; Shiro, M. Optically Pure Haloselenuranes. First Synthesis and Nucleophilic Substitutions. J. Org. Chem. 1994, 59, 3262–3264. [Google Scholar] [CrossRef]
- Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis; Baldwin, J.E., Ed.; Pergamon Press: Oxford, UK, 1986. [Google Scholar]
- Kamigata, N.; Shimizu, T. Reviews on Heteroatom Chemistry; Oae, S., Ed.; MYU: Tokyo, Japan, 1991. [Google Scholar]
- Tomoda, S.; Usuki, Y.; Fujita, K.; Iwaoka, M. Reviews on Heteroatom Chemistry; Oae, S., Ed.; MYU: Tokyo, Japan, 1991. [Google Scholar]
- Reich, H.J. Organoselenium Chemistry; Liotta, D., Ed.; John Wiley & Sons: New York, NY, USA, 1987. [Google Scholar]
- Reich, H.J.; Yelm, K.E. Asymmetric induction in the oxidation of [2.2]paracyclophane-substituted selenides. Application of chirality transfer in the selenoxide [2,3]sigmatropic rearrangement. J. Org. Chem. 1991, 56, 5672–5679. [Google Scholar] [CrossRef]
- Kawakami, R.; Singh, J.D.; Fukuzawa, S.-I.; Uemura, S. Synthesis of [R,S;R,S]- and [S,R;S,R]-Bis[2 -[1-(dimethylamino)ethyl]ferrocenyl] Diselenides and Their Application to Asymmetric Selenoxide Elimination and [2,3]Sigmatropic Rearrangement. J. Org. Chem. 1995, 60, 4114–4120. [Google Scholar]
- Fujita, K.-I.; Kanakubo, M.; Ushijima, H.; Oishi, A.; Ikeda, Y.; Taguchi, Y. Asymmetric [2,3]Sigmatropic Rearrangement of Optically Active Allylic Selenides. Synlett 1998, 1998, 987–988. [Google Scholar] [CrossRef]
- Shimizu, T.; Kikuchi, K.; Ishikawa, Y.; Ikemoto, I.; Kobayashi, M.; Kamigata, N. Synthesis and absolute configuration of optically active selenoxides. X-Ray molecular structure of (–) se -4-menthyloxycarbonylphenyl 2,4,6-tri-isopropylphenyl selenoxide. J. Chem. Soc. Perkin Trans. 1989, 1, 597–602. [Google Scholar] [CrossRef]
- Shimizu, T.; Seki, N.; Taka, H.; Kamigata, N. Stereoselective Transformation of Optically Active Selenoxide into Optically Active Selenonium Imide. J. Org. Chem. 1996, 61, 6013–6014. [Google Scholar] [CrossRef]
- Davis, F.A.; Stringer, O.D.; McCauley, J.P. Asymmetric oxidation of achiral selenides to optically active selenoxides. Tetrahedron 1985, 41, 4747–4757. [Google Scholar] [CrossRef]
- Davis, F.A.; ThimmaReddy, R.; Weismiller, M.C. (-)-.alpha.,.alpha.-Dichlorocamphorsulfonyloxaziridine: A superior reagent for the asymmetric oxidation of sulfides to sulfoxides. J. Am. Chem. Soc. 1989, 111, 5964–5965. [Google Scholar] [CrossRef]
- Davis, F.A.; Reddy, R.T. Asymmetric oxidation of simple selenides to selenoxides in high enantiopurity. Stereochemical aspects of the allyl selenoxide/allyl selenenate rearrangement. J. Org. Chem. 1992, 57, 2599–2606. [Google Scholar] [CrossRef]
- Davis, F.A.; Towson, J.C.; Weismiller, M.C.; Lal, S.; Carroll, P.J. Chemistry of oxaziridines. 11. (Camphorylsulfonyl)oxaziridine: Synthesis and properties. J. Am. Chem. Soc. 1988, 110, 8477–8482. [Google Scholar] [CrossRef]
- Drabowicz, J.; Nakanishi, W.; Haiashi, S. Unpublished preliminary results, manuscript in preparation. 2020. [Google Scholar]
- Shimizu, T.; Kobayashi, M.; Kamigata, N. Asymmetric oxidation of alkyl aryl selenides under Sharpless oxidation conditions. Bull. Chem. Soc. Jpn. 1989, 62, 2099–2101. [Google Scholar] [CrossRef]
- Tiecco, M.; Tingoli, M.; Testaferri, L.; Bartoli, D. Asymmetric oxidation of prochiral selenides to optically active selenoxides. Tetrahedron Lett. 1987, 28, 3849–3852. [Google Scholar] [CrossRef]
- Komatsu, N.; Matsunaga, S.; Sugita, T.; Uemura, S. Asymmetric selenoxide elimination leading to chiral alkyl and aryl cyclohexylidenemethyl ketones. J. Am. Chem. Soc. 1993, 115, 5847–5848. [Google Scholar] [CrossRef]
- Komatsu, N.; Murakami, T.; Kawakami, R.; Sugita, T.; Uemura, S. Asymmetric selenoxide elimination leading to chiral allenic sulfones. J. Org. Chem. 1993, 58, 3697–3702. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ohkubo, H.; Shimizu, T. Synthesis of selenoxides by oxidation of selenides with t-butyl hypochlorite, and its application for synthesis of optically active selenoxide. Bull. Chem. Soc. Jpn. 1986, 59, 503–506. [Google Scholar] [CrossRef]
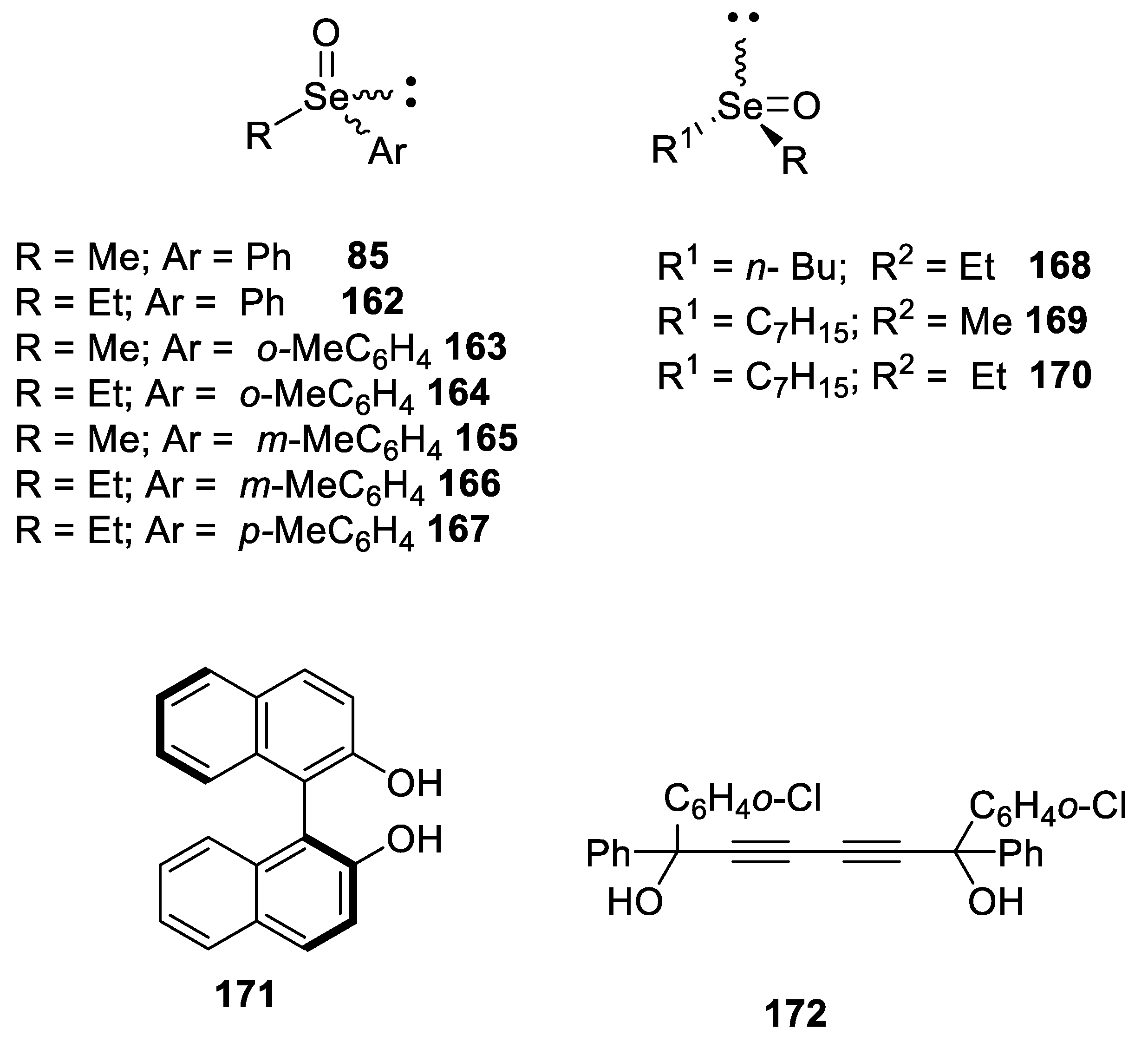
- Shimizu, T.; Kobayashi, M. Optically active selenoxides: Chromatographic separation and absolute configuration. J. Org. Chem. 1987, 52, 3399–3403. [Google Scholar] [CrossRef]
- Shimizu, T.; Kobayashi, M. Optical resolution of asymmetric selenoxides by high-performance liquid chromatography using optically active column. Bull. Chem. Soc. Jpn. 1986, 59, 2654–2656. [Google Scholar] [CrossRef]
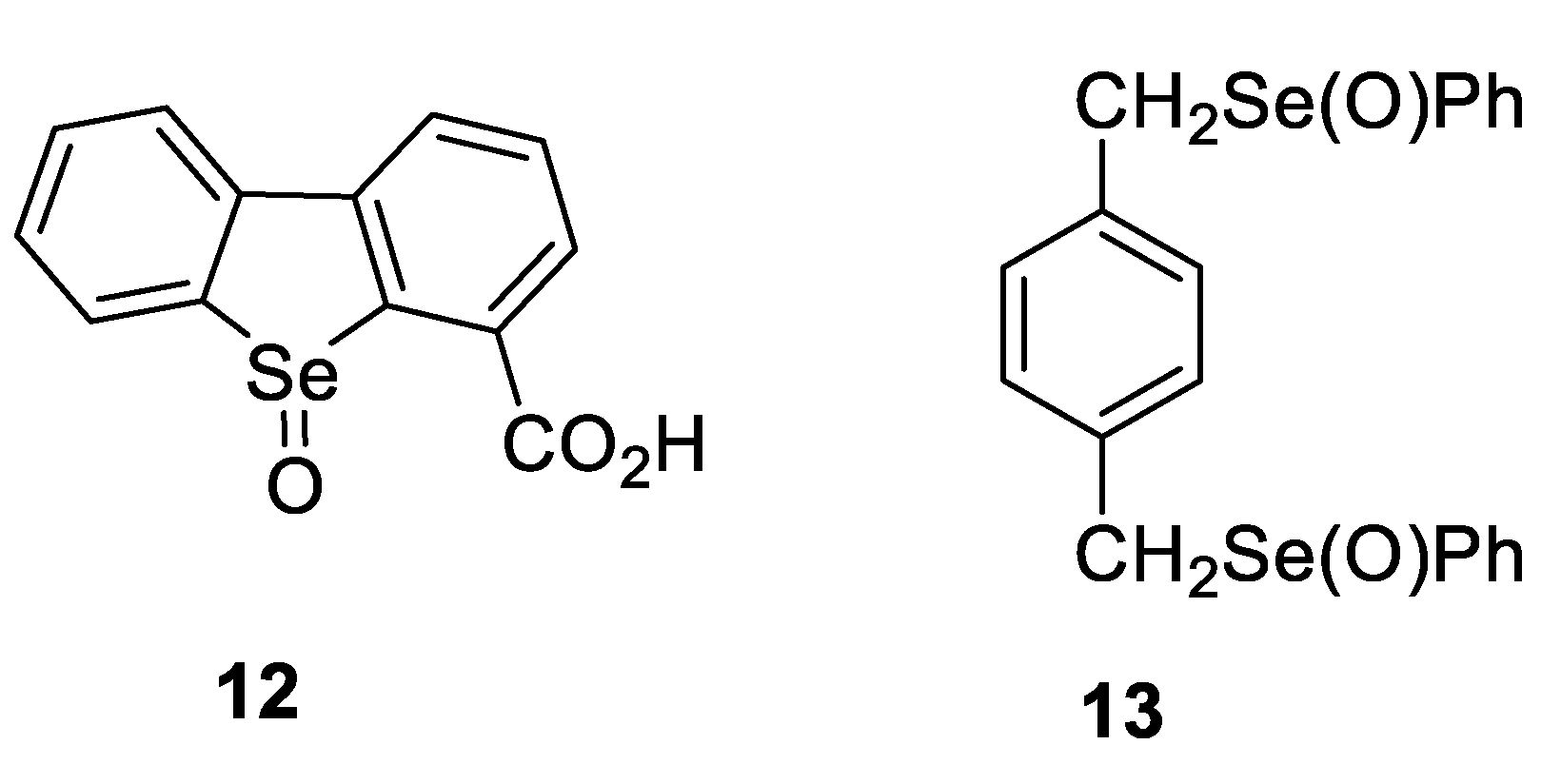
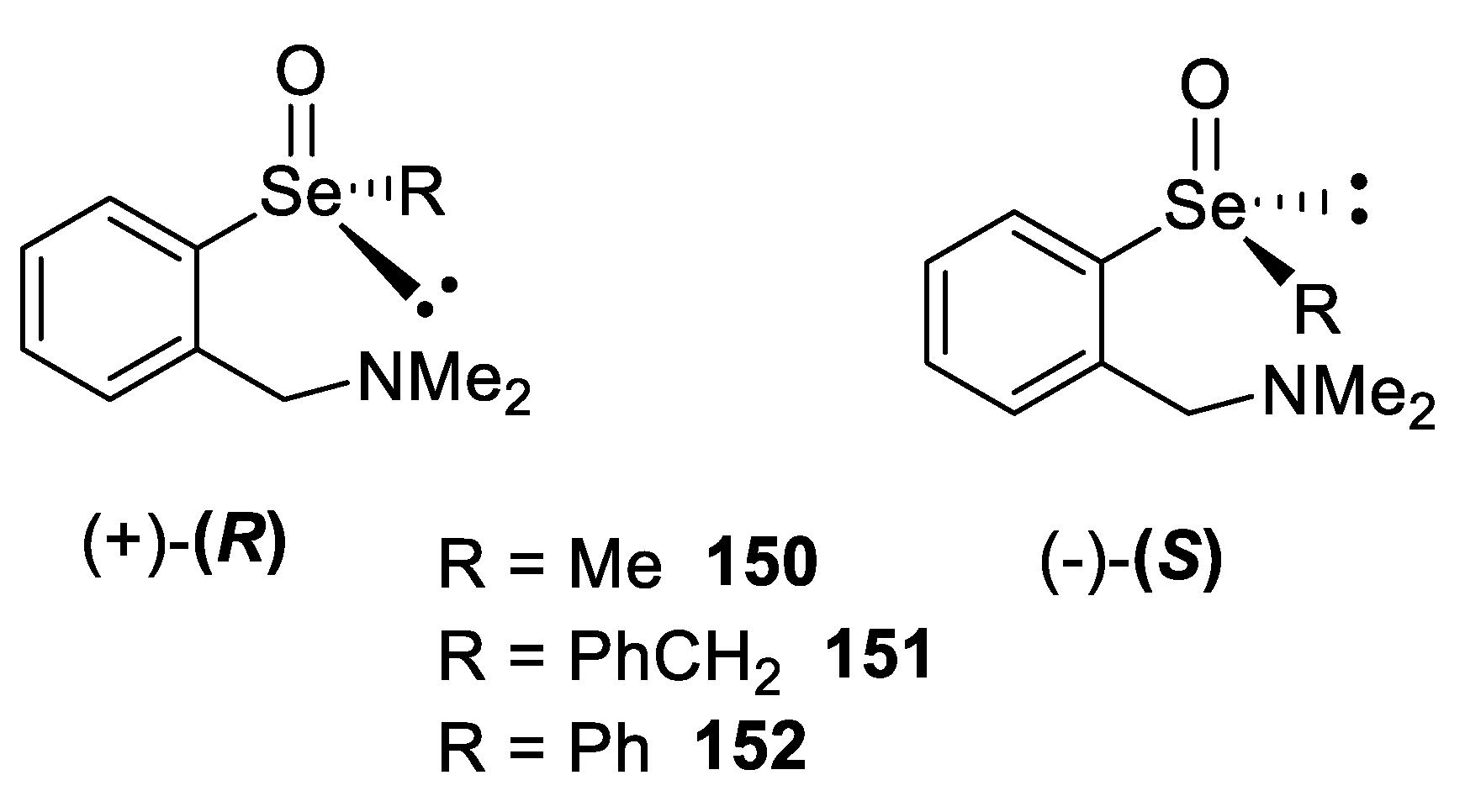
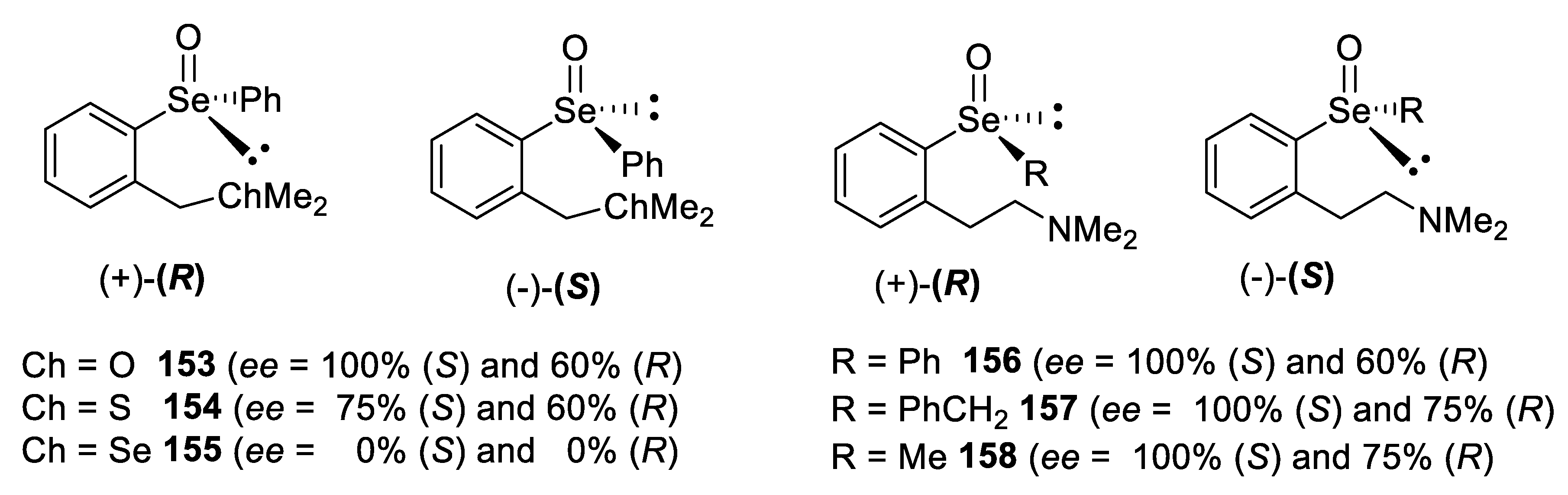
- Shimizu, T.; Enomoto, M.; Taka, H.; Kamigata, N. Optical Resolution and Configurational Stability of Selenoxides Stabilized by Intramolecular Coordination. J. Org. Chem. 1999, 64, 8242–8247. [Google Scholar] [CrossRef]
- Soma, T.; Shimizu, T.; Hirabayashi, K.; Kamigata, N. Stabilizing effect of intramolecular lewis base toward racemization of optically active selenoxides. Heteroat. Chem. 2007, 18, 301–311. [Google Scholar] [CrossRef]
- Taka, H.; Matsumoto, A.; Shimizu, T.; Kamigata, N. Thermodynamically stabilized chiral chalcogen oxides: Optical resolution and stereochemistry. Heteroat. Chem. 2001, 12, 227–237. [Google Scholar] [CrossRef]
- Taka, H.; Matsumoto, A.; Shimizu, T.; Kamigata, N. Optical Resolution of Thermodynamically Stabilized Selenoxides by 8-(Dimethylamino)-1-naphthyl Group. Chem. Lett. 2000, 29, 726–727. [Google Scholar] [CrossRef]
- Toda, F.; Mori, K. Optical resolution of selenoxides by complexation with optically active 2,2?-dihydroxy-1,1?-binaphthol or 1,6-di(o-chlorophenyl)-1,6-diphenylhexa-2,4-diyne-1,6-diol. J. Chem. Soc., Chem. Commun. 1986, 1357–1359. [Google Scholar] [CrossRef]
- Davis, F.A.; Billmers, J.M.; Stringer, O.D. First synthesis of simple optically active selenoxides. Tetrahedron Lett. 1983, 24, 3191–3194. [Google Scholar] [CrossRef]
- Nakashima, Y.; Shimizu, T.; Hirabayashi, K.; Iwasaki, F.; Yamasaki, M.; Kamigata, N. Optically Active Seleninate Esters: Isolation, Absolute Configuration, Racemization Mechanism, and Transformation into Chiral Selenoxide. J. Org. Chem. 2005, 70, 5020–5027. [Google Scholar] [CrossRef] [PubMed]
- Rayner, D.R.; Gordon, A.J.; Mislow, K. Thermal racemization of diaryl, alkyl aryl, and dialkyl sulfoxides by pyramidal inversion. J. Am. Chem. Soc. 1968, 90, 4854–4860. [Google Scholar] [CrossRef]
- Andersen, K.K. Sulphones and Sulphoxides (1988); Patai, S., Rappoport, Z., Stirling, C., Eds.; John Wiley & Sons: Chichester, UK, 1988. [Google Scholar]
- Mikołajczyk, M.; Drabowicz, J.; Mikoajczyk, M. Chiral Organosulfur Compounds. In Topics in Stereochemistry; Wiley: Hoboken, NJ, USA, 2007; Volume 13, pp. 333–468. [Google Scholar]
- Oae, S.; Yokoyama, M.; Kise, M. Oxygen Exchange and Racemization Reactions of Sulfoxides in Acetic and Chloroacetic Acid. Bull. Chem. Soc. Jpn. 1968, 41, 1221–1224. [Google Scholar] [CrossRef]
- Shimizu, T.; Yoshida, M.; Kobayashi, M. An estimation of the configurational stability of diaryl selenoxides by means of 77Se NMR spectroscopy. Bull. Chem. Soc. Jpn. 1987, 60, 1555–1557. [Google Scholar] [CrossRef]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. SDE-Phoric functional groups. Chiral Sulfoxides: Asymmetric preparation and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Jasiak, A.; Mielniczak, G.; Owsianik, K.; Koprowski, M.; Krasowska, D.; Drabowicz, J. Solvent-Free Michaelis−Arbuzov Rearrangement under Flow Conditions. J. Org. Chem. 2019, 84, 2619–2625. [Google Scholar] [CrossRef]



































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasowska, D.; Sancineto, L.; Deska, M.; Drabowicz, J. Optically Active Selenoxides: Structural and Synthetic Aspects. Symmetry 2020, 12, 349. https://doi.org/10.3390/sym12030349
Krasowska D, Sancineto L, Deska M, Drabowicz J. Optically Active Selenoxides: Structural and Synthetic Aspects. Symmetry. 2020; 12(3):349. https://doi.org/10.3390/sym12030349
Chicago/Turabian StyleKrasowska, Dorota, Luca Sancineto, Małgorzata Deska, and Józef Drabowicz. 2020. "Optically Active Selenoxides: Structural and Synthetic Aspects" Symmetry 12, no. 3: 349. https://doi.org/10.3390/sym12030349
APA StyleKrasowska, D., Sancineto, L., Deska, M., & Drabowicz, J. (2020). Optically Active Selenoxides: Structural and Synthetic Aspects. Symmetry, 12(3), 349. https://doi.org/10.3390/sym12030349