Chirality: The Backbone of Chemistry as a Natural Science


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
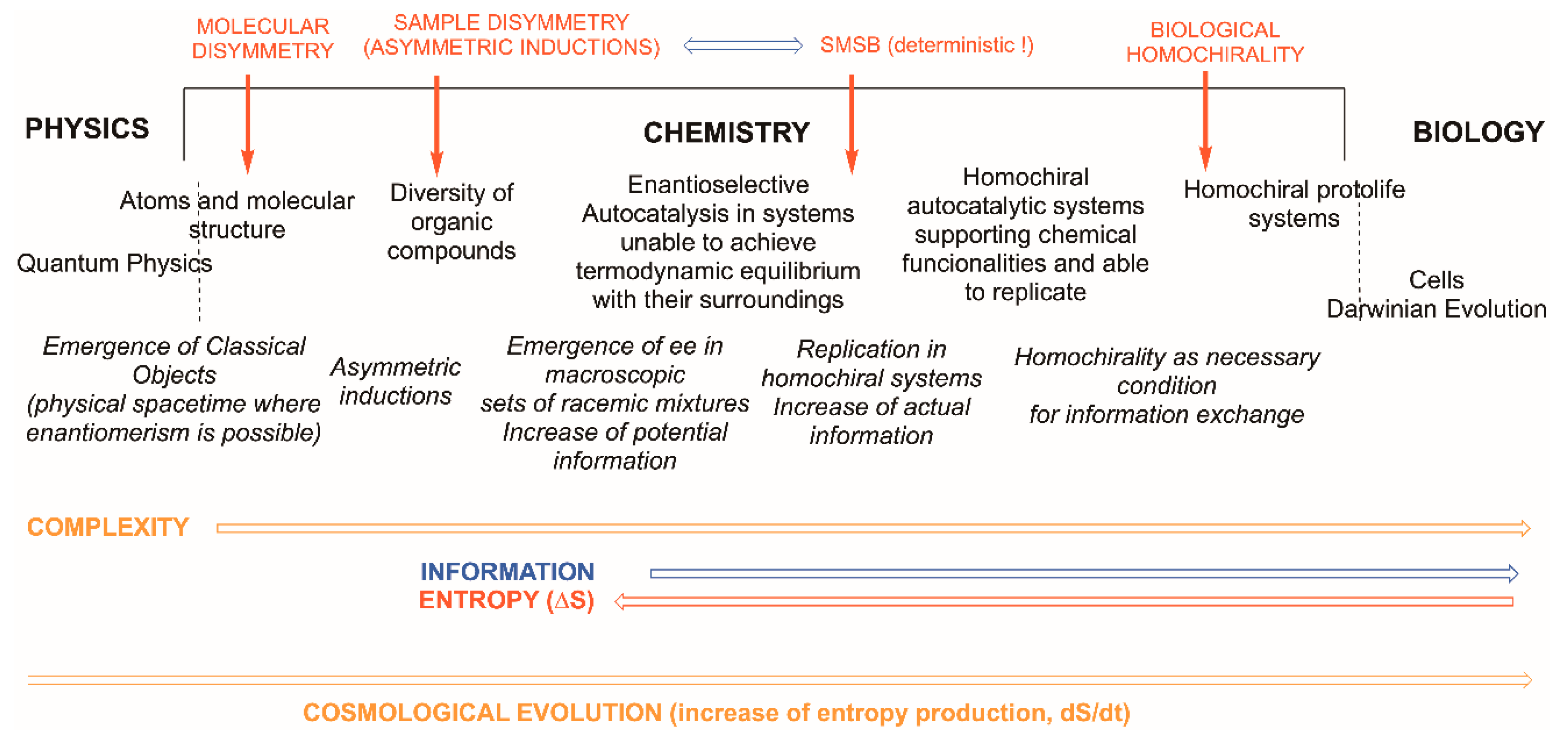
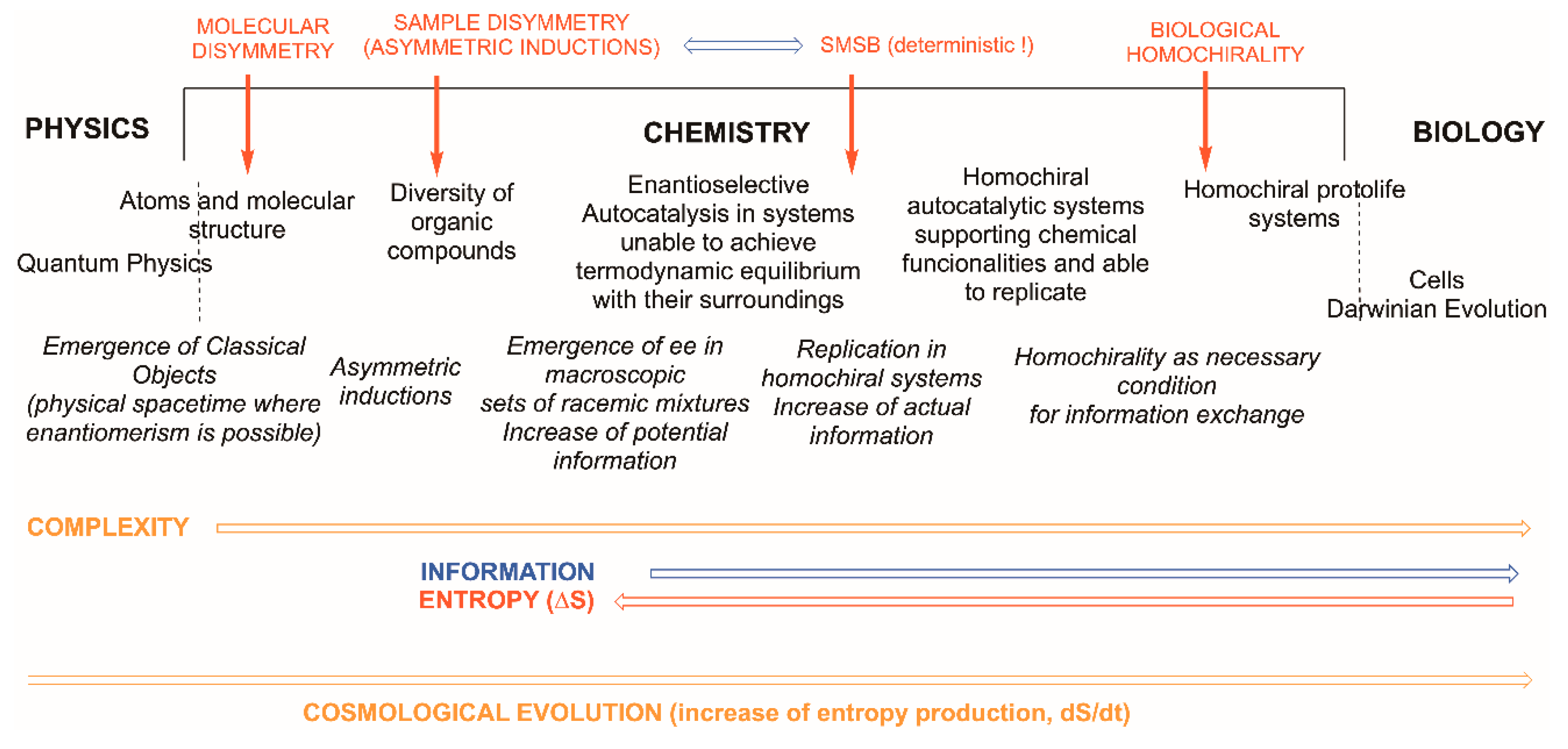
:1. Introduction
- (i)
- Consideration of atoms and molecules as quasi-classical objects. In fact, chemistry teaching mostly extrapolated them to atoms as robust classical objects. Atoms and molecules correspond to the frontier, or boundary, of chemistry at its lower level of complexity, that is, to the emergence of the classical from the quantum physics world. This lower frontier of chemistry is well understood by physicists and chemical physicists [10,11] but is generally ignored by chemists.
- (ii)
- Application of the constraints originated by the thermodynamic principles and the description of the behaviour of chemical substances and materials as very large sets of atoms and molecules, i.e., of real chemical samples. Complex chemical systems working in open systems to matter and energy exchange, whose stability derives from the dissipation of energy, are those that represent the upper boundary between biology and chemistry and are still poorly understood.
- (a)
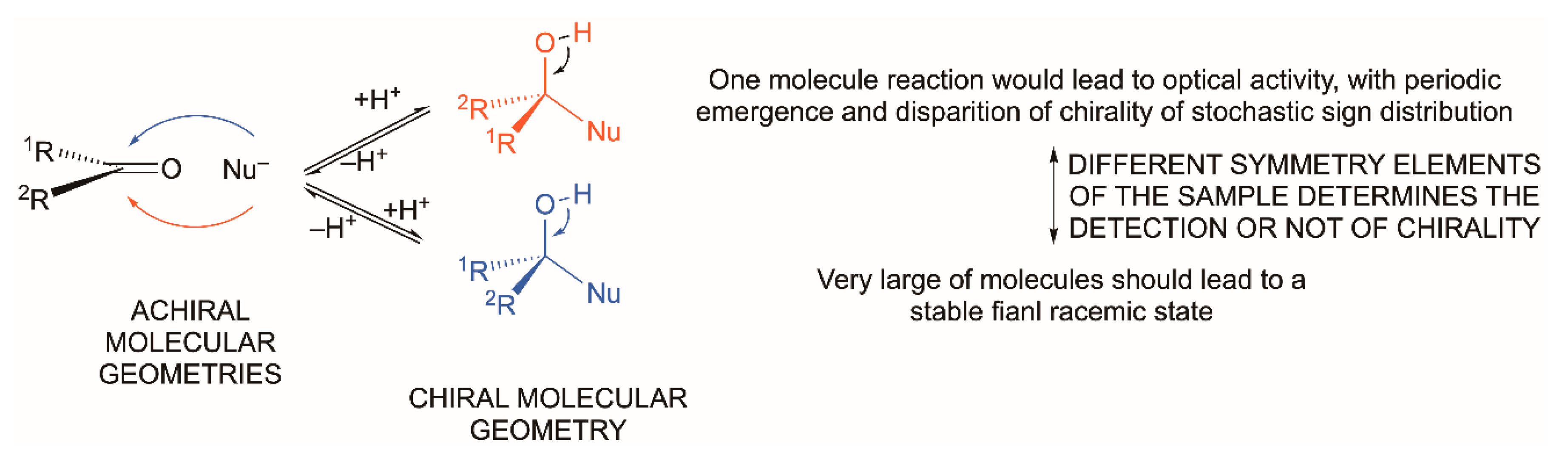
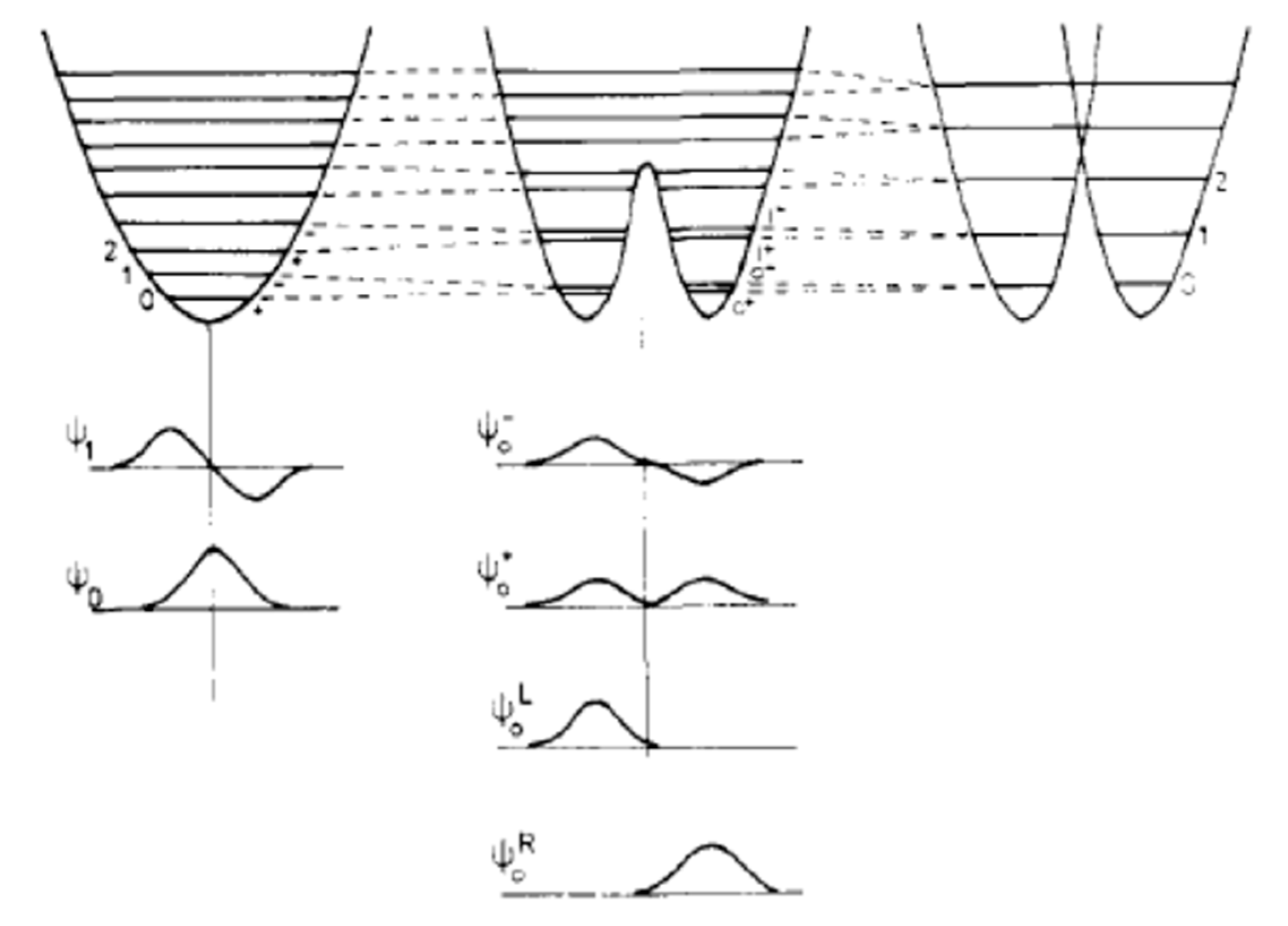
- At the molecular level there is the question of enantiomerism; that chemical curricula study chirality as a part of stereochemistry is only related to the existence of structures that break parity, and parity breaking is the consequence of the symmetry violations—time the primordial one—implied in the cosmological emergence of space–time, already dissymmetric [27], and matter. The metaphysics of nature has long ago observed, and been aware of, the chirality phenomenon within the topic of the nature of space [28], well before chemistry was established as a science (see ref. [29] and cites herein).
- (b)
- At the frontier with biology, the dramatic experimental evidence of biological homochirality, overcoming the racemic mixture (racemate), suggests the advantage of chirality for sustaining the information related to the chemical functionalities of biological macromolecules and supramolecular systems implied in replication and in Darwinian evolution, i.e., in the properties that characterize the phenomenon of life.
2. Chemical Chirality: Molecular Structures and Chemical Materials
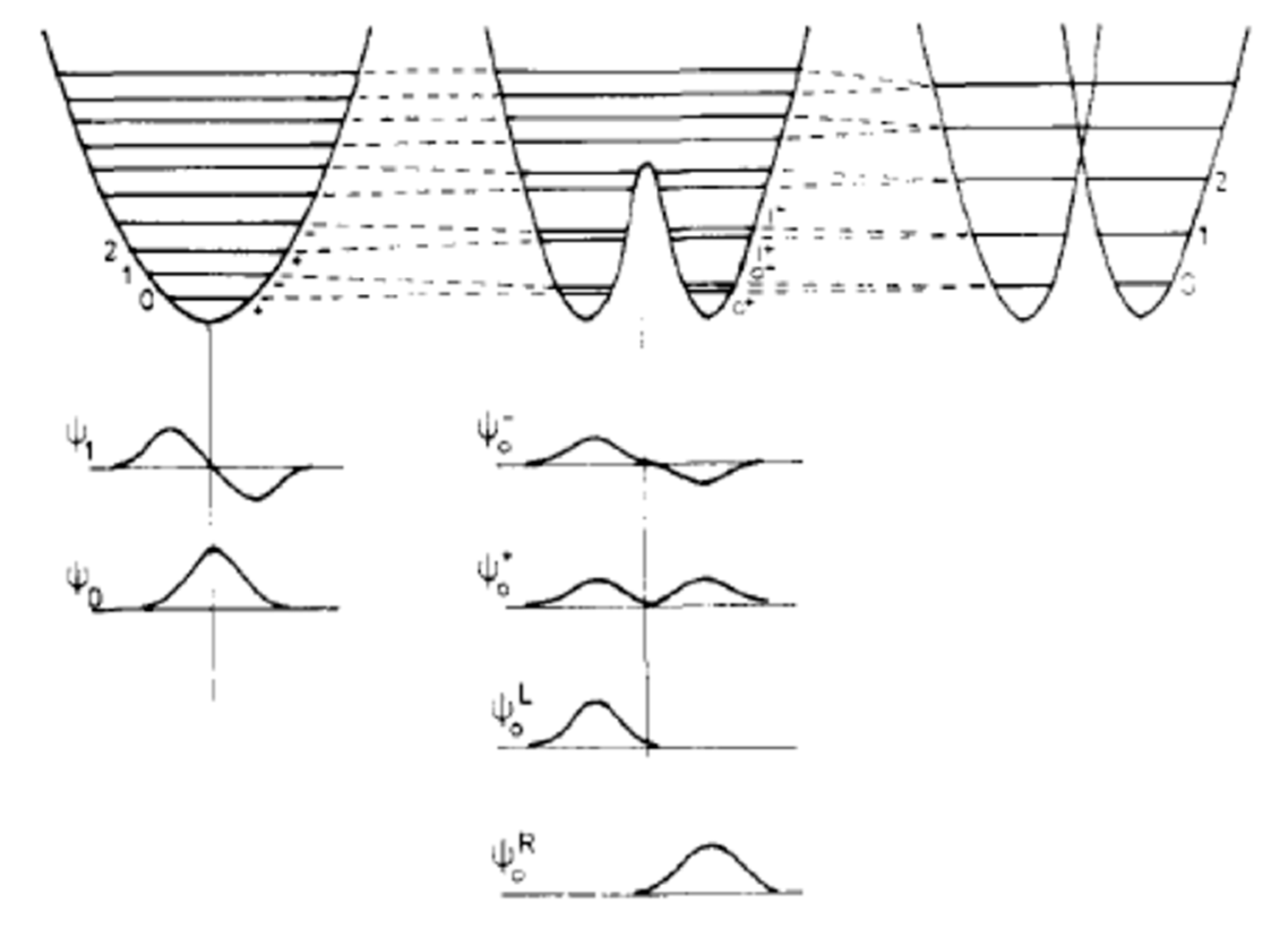
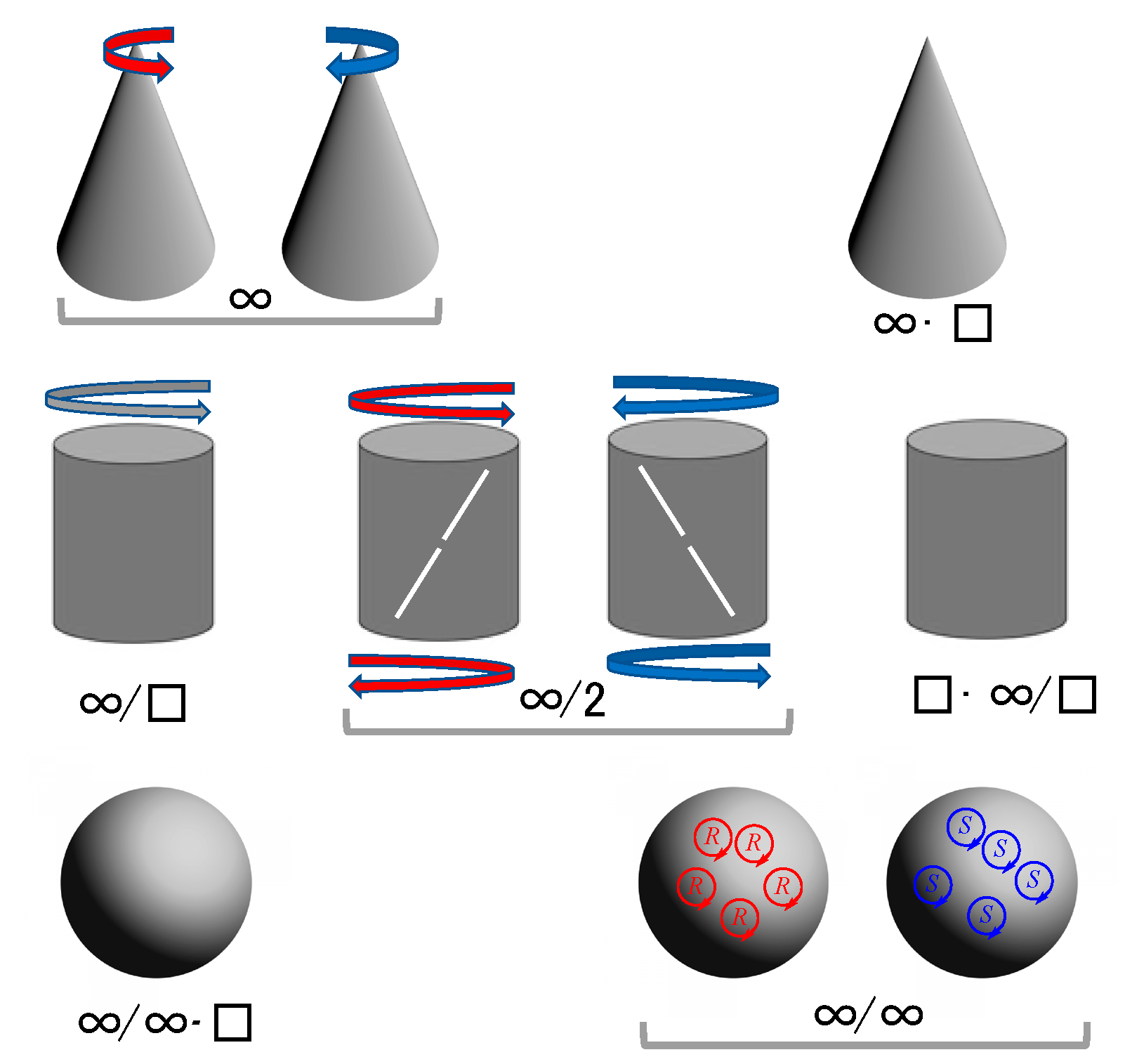
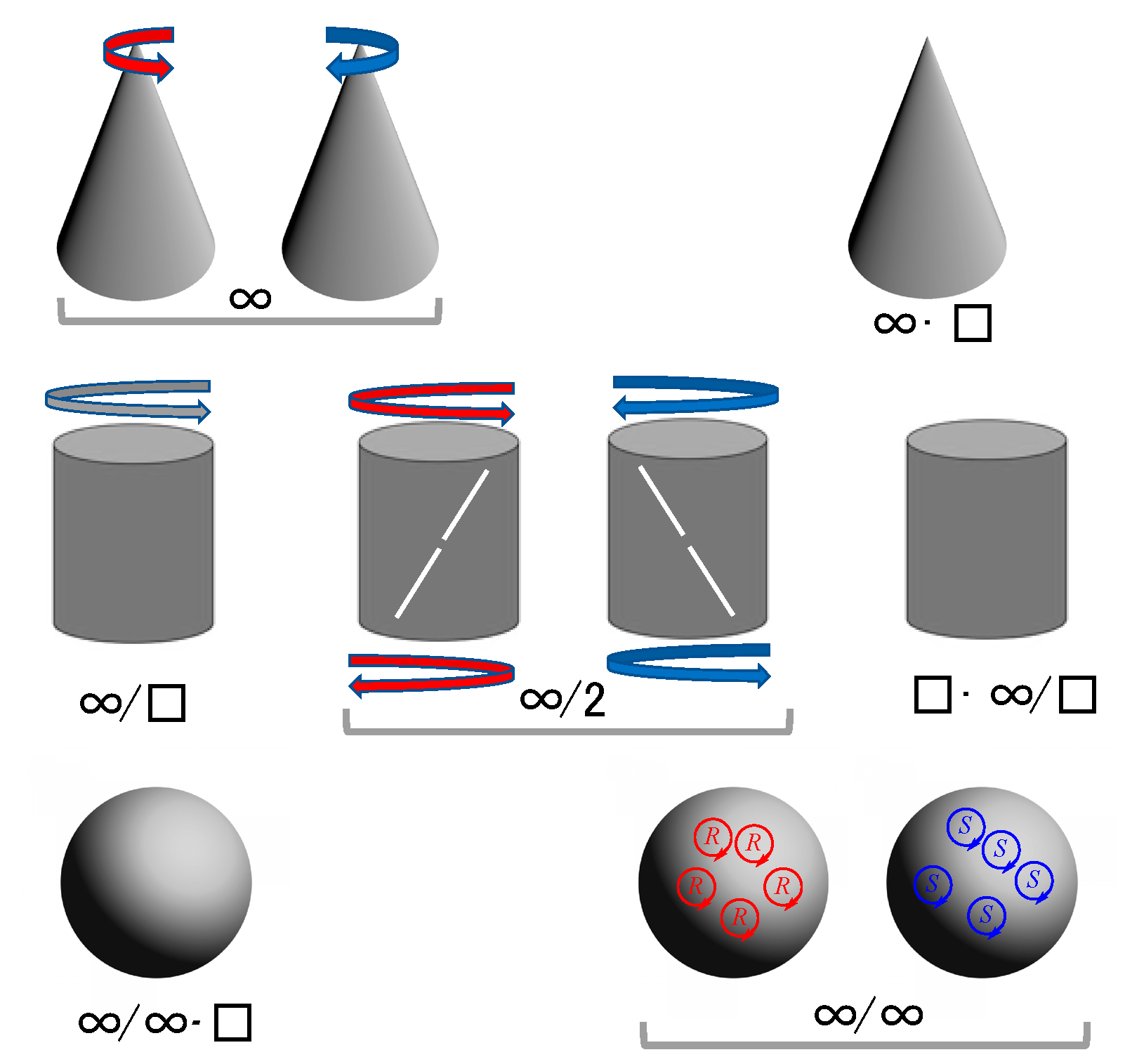
The Question of Distinguishability between Enantiomers
3. Molecular Chirality vs. Chirality of Materials
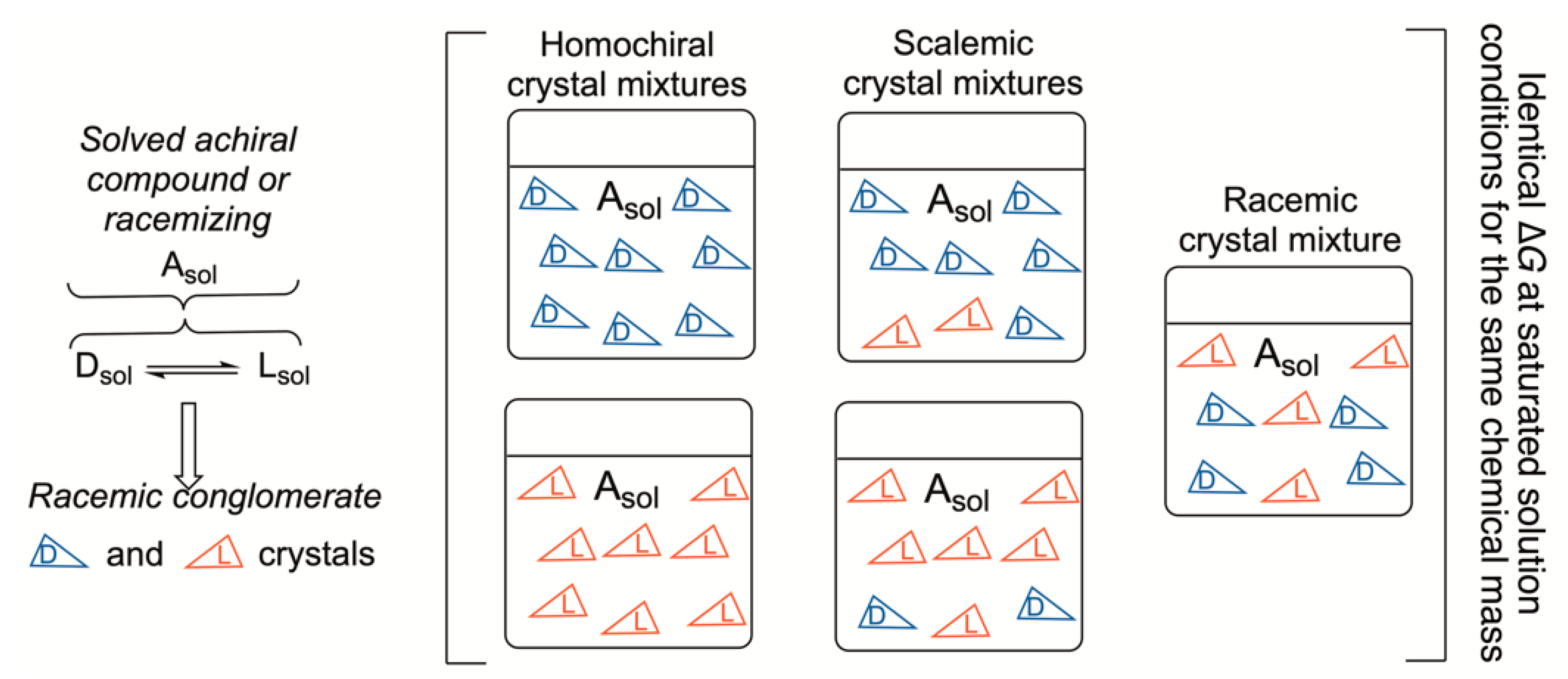
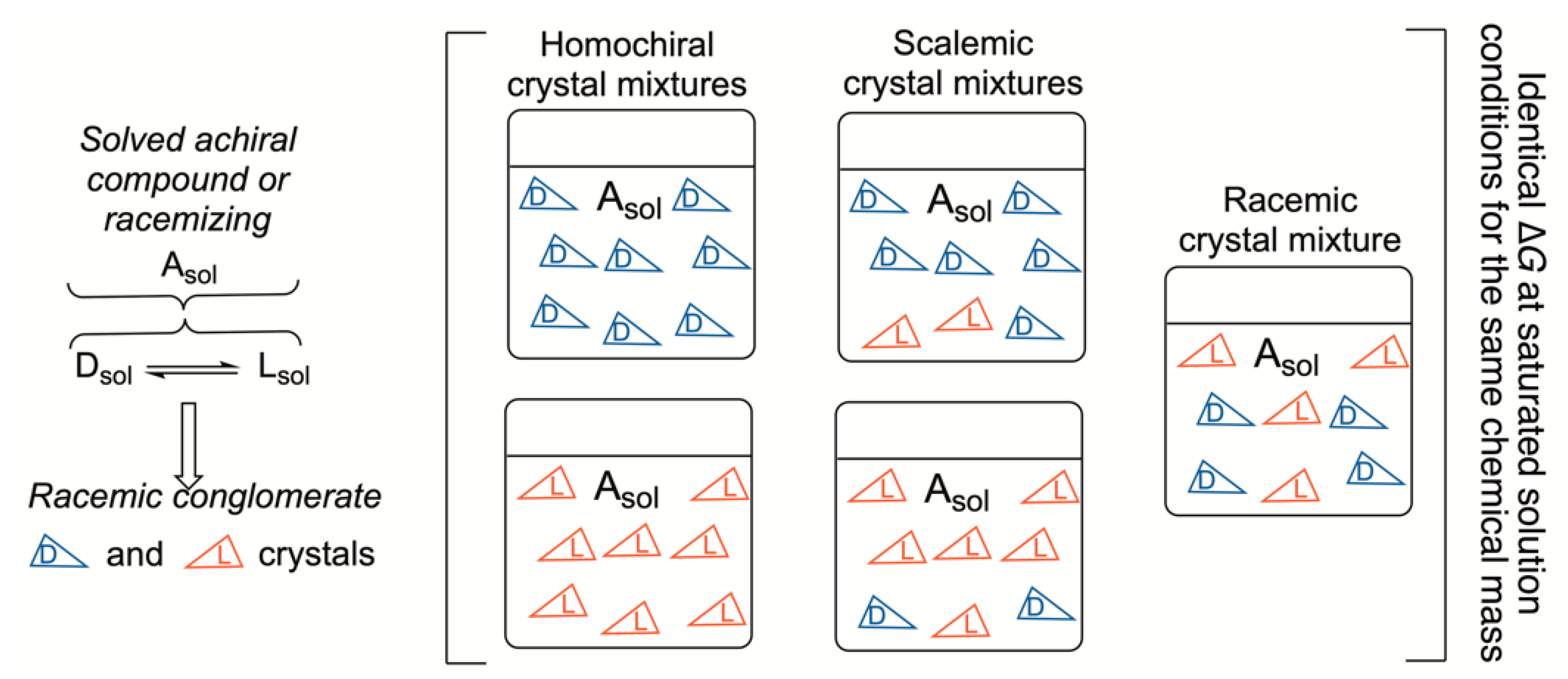
4. Racemates vs. Scalemic Mixtures in Chemical Reactions and Phase Transitions
Kinetically and Thermodynamically Controlled Reactions
5. Emergence of Biological Homochirality: The Boundary between Systems Chemistry and Systems Biology
Is Dissymmetry an Advantage for Chemical Evolution?
- (1)
- Electron transport is necessarily a one-electron process, but the redox processes of organic compounds (closed shell) are two-electron redox reactions. The enzymatic machinery of life solves this problem using transition metal prosthetic groups of open shell configuration that receive the single electrons, and, mediated by substrates, is able to show one-electron redox transformations intermediate to achieve the two-electron processes of the redox processes of closed shell organic molecules. In the case of DNA damage, the noxious free radical at the nucleic acid simply reacts with the carried electron to a closed shell configuration.
- (2)
- In regard to the former point, since living systems do not involve solid state reactions, reactions are forbidden when the total electronic spin numbers of the reactants and products are not the same. Therefore, in the case of the primordial one-electron reactions, only free electrons showing the spin sign that agrees with that of prosthetic metal group or of the free radical can participate in the reaction.
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Gell-Mann, M. The Quark and the Jaguar: Adventures in the Simple and the Complex; Henry Holt: New York, NY, USA, 1994; ISBN 10 0-8050-7253-5. [Google Scholar]
- Bak, P. How Nature Works. The Sicence of Self-Organized Criticality; Springer: New York, NY, USA, 1996; ISBN 978-1-4757-5426-1. [Google Scholar]
- Barron, L.D. Symmetry and molecular chirality. Chem. Soc. Rev. 1986, 15, 189–223. [Google Scholar] [CrossRef]
- Kosmann-Schwarz, Y. The Norther Theorems: Invariance and Conservation Laws in the Twentieth Century; Springer: New York, NY, USA, 2010; ISBN 978-0-387-87867-6. [Google Scholar]
- Feynman, P.; Leighton, R.B.; Sands, M. Feynman Lectures on Physics. Available online: https://www.feynmanlectures.caltech.edu (accessed on 29 November 2020).
- Rowe, M.A.; Kielpinski, D.; Meyer, V.; Sackett, C.A.; Itano, W.M.; Monroe, C.; Wineland, D.J. Experimental violation of a Bell’s inequality with efficient detection. Nature 2001, 409, 791–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einstein, A.; Podolsky, B.; Rosen, N. Can Quantum-Mechanical Description of Physical Reality Be Considered Complete? Phys. Rev. 1935, 47, 777–780. [Google Scholar] [CrossRef] [Green Version]
- Glansdorf, P.; Prigogine, I. Thermodynamic Theory of Structure, Stbility and Fluctuations; Wiley-Interscience: London, UK, 1971; ISBN 0-471-30280-5. [Google Scholar]
- Kondepudi, D.; Prigogine, I. Modern Thermodynamics: From Heat Engines to Dissipative Structures, 2nd ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2014; ISBN 9781118698723. [Google Scholar]
- Fein, Y.Y.; Geyer, P.; Zwick, P.; Kiałka, F.; Pedalino, S.; Mayor, M.; Gerlich, S.; Arndt, M. Quantum superposition of molecules beyond 25 kDa. Nat. Phys. 2019, 15, 1242–1245. [Google Scholar] [CrossRef]
- Schätti, J.; Köhler, V.; Mayor, M.; Fein, Y.Y.; Geyer, P.; Mairhofer, L.; Gerlich, S.; Arndt, M. Matter–wave interference and deflection of tripeptides decorated with fluorinated alkyl chains. J. Mass Spectrom. 2020, 55, e4514. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann, S.A. The Origin of Order. Self-Organization and Selction in Evolution; Oxford Univesity Press: New York, NY, USA, 1993; ISBN 0-19-505811-9/0-19-507951-5. [Google Scholar]
- Kauffmann, S.A.; Johnsen, S. Coevolution to the edge of chaos: Coupled fitness landscapes, poised states, and coevolutionary avalanches. J. Theor. Biol. 1991, 149, 467–505. [Google Scholar] [CrossRef]
- Eigen, M.; Schuster, P. A principle of natural self-organization. Naturwissenschaften 1977, 64, 541–565. [Google Scholar] [CrossRef]
- Eigen, M.; Schuster, P. The Hypercycle—A principle of natural self-organization Part B: The abstract hypercycle. Naturwissenschaften 1978, 65, 7–41. [Google Scholar] [CrossRef]
- Tibor, G. The principles of life (selections). In The Nature of Life: Classical and Contemporary Perspectives from Philosophy and Science; Cambridge University Press: Cambridge, UK, 2010; pp. 102–112. ISBN 9780511730191. [Google Scholar]
- Krishnamurthy, R. Life’s Biological Chemistry: A Destiny or Destination Starting from Prebiotic Chemistry? Chem. A Eur. J. 2018, 24, 16708–16715. [Google Scholar] [CrossRef]
- Krishnamurthy, R. Giving Rise to Life: Transition from Prebiotic Chemistry to Protobiology. Acc. Chem. Res. 2017, 50, 455–459. [Google Scholar] [CrossRef]
- Islam, S.; Bučar, D.-K.; Powner, M.W. Prebiotic selection and assembly of proteinogenic amino acids and natural nucleotides from complex mixtures. Nat. Chem. 2017, 9, 584–589. [Google Scholar] [CrossRef]
- Benner, S.A.; Kim, H.-J.; Carrigan, M.A. Asphalt, Water, and the Prebiotic Synthesis of Ribose, Ribonucleosides, and RNA. Acc. Chem. Res. 2012, 45, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, D.; Tremmel, P.; Pallan, P.S.; Egli, M.; Richert, C. The Enzyme-Free Release of Nucleotides from Phosphoramidates Depends Strongly on the Amino Acid. Angew. Chem. Int. Ed. 2020, 59, 20154–20160. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Semenov, S.N.; Kraft, L.J.; Ainla, A.; Zhao, M.; Baghbanzadeh, M.; Campbell, V.E.; Kang, K.; Fox, J.M.; Whitesides, G.M. Autocatalytic, bistable, oscillatory networks of biologically relevant organic reactions. Nature 2016, 537, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, C.; Bai, X.; Tian, F.; Hu, G.; Sun, J. Enantiomorphic Microvortex-Enabled Supramolecular Sensing of Racemic Amino Acids by Using Achiral Building Blocks. Angew. Chem. Int. Ed. 2020, 59, 3486–3490. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, X.; Khan, T.; Ji, C.; Asghar, M.A.; Zhao, S.; Li, L.; Hong, M.; Luo, J. A Photoferroelectric Perovskite-Type Organometallic Halide with Exceptional Anisotropy of Bulk Photovoltaic Effects. Angew. Chem. Int. Ed. 2016, 55, 6545. [Google Scholar] [CrossRef]
- Primas, H. Chemistry, Quantum Mechanics and Reductionism; Springer: Berlin, Germany, 1981; ISBN 0-387-10696-0. [Google Scholar]
- Petitjean, M. About Chirality in Minkowski Spacetime. Symmetry 2019, 11, 1320. [Google Scholar] [CrossRef] [Green Version]
- Wehrli, H. Metaphysics. Chirality as the Basic Principle of Physics; Copyright Hans Wehrli 2008: Zürich, Switzerland, 2008; ISBN 05212567719780521256773. [Google Scholar]
- van Cleve, J.; Frederick, R. (Eds.) The Philosophy of Right and Left; Kluwer Academi Publishers: Dordrecht, The Netherlands, 1991; ISBN 0-7923-0844-1. [Google Scholar]
- Mislow, K.; Raban, M. Stereoisomeric relationships of groups in molecules. In Topics in Stereochemistry, Vol 1; Allinger, N.L., Eliel, E.L., Eds.; Intersience, John Wiley: New York, NY, USA, 1967. [Google Scholar]
- Varela, F.G.; Maturana, H.R.; Uribe, R. Autopoiesis: The organization of living systems, its characterization and a model. Biosystems 1974, 5, 187–196. [Google Scholar] [CrossRef]
- Luisi, P.L. The Emergence of Life; Cambridge University Press: Cambridge, UK, 2006; ISBN 10 0-521-82117-7. [Google Scholar]
- van Brakel, J. On the Neglect of the Philosophy of Chemistry. Found. Chem. 1999, 1, 111–174. [Google Scholar] [CrossRef] [Green Version]
- Amann, A. The Gestalt problem in quantum theory: Generation of molecular shape by the environment. Synthese 1993, 97, 125–156. [Google Scholar] [CrossRef]
- Bishop, R.C.; Atmanspacher, H. Contextual Emergence in the Description of Properties. Found. Phys. 2006, 36, 1753–1777. [Google Scholar] [CrossRef]
- Joos, E.; Zeh, H.D.; Kiefer, C.; Giulini, D.; Kupsch, J. Decoherence and the Appearance of a Classical World in Quantum Theory; Springer: Berlin/Heidelberg, Germany, 2003; ISBN 978-3-642-055576-8. [Google Scholar]
- Woolley, R.G. Quantum theory and molecular structure. Adv. Phys. 1976, 25, 27–52. [Google Scholar] [CrossRef]
- von Weizsäcker, C.F. Aufbau der Physik; Carl Hanser Verlag: München, Germany, 1985; ISBN 3-446-14142-1. [Google Scholar]
- Pauling, L. General Chemistry; Dover Publications: Mineola, NY, USA, 1988; ISBN 10 0486656225. [Google Scholar]
- Ernst, K.-H.; Wild, F.R.W.P.; Blacque, O.; Berke, H. Alfred Werner’s Coordination Chemistry: New Insights from Old Samples. Angew. Chem. Int. Ed. 2011, 50, 10780–10787. [Google Scholar] [CrossRef]
- Barron, L.D. Optical activity as a mixed parity phenomenon. J. Am. Chem. Soc. 1979, 101, 269–270. [Google Scholar] [CrossRef]
- Hund, F. Zur Deutung der Molekelspektren. III. Z. Phys. 1927, 43, 805–826. [Google Scholar] [CrossRef]
- Trost, J.; Hornberger, K. Hund’s Paradox and the Collisional Stabilization of Chiral Molecules. Phys. Rev. Lett. 2009, 103, 23202. [Google Scholar] [CrossRef] [Green Version]
- Woolley, G. Natural optical activity and the molecular hypothesis. In Structures vs. Special Properties. Sturcture and Bonding; Springer: Berlin/Heidelberg, Germany, 1982; ISBN 978-3-540-11781-0. [Google Scholar]
- Harris, R.A.; Stodolsky, L. Quantum beats in optical activity and weak interactions. Phys. Lett. B 1978, 78, 313–317. [Google Scholar] [CrossRef]
- Amann, A. Chirality: A superselection rule generated by the molecular environment? J. Math. Chem. 1991, 6, 1–15. [Google Scholar] [CrossRef]
- Shubnikov, A.V. On the works of Pierre Curie on symmetry. Comput. Math. Appl. 1988, 16, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Avnir, D. On Left and Right: Chirality from Molecules to Galaxies to Rembrandt. Available online: Chem.ch.huji.ac.il/presentations.html (accessed on 29 November 2020).
- Lifschitz, E.M.; Pitayesvski, L.P. Physical Kinetics. In Course of Theoretical Physics; Butterworth-Heinemann Ltd.: Oxford, UK, 1981; Volume 10. [Google Scholar]
- Dieks, D. The Gibbs paradox revisited. In Explanation, prediction and confirmation. The phylosophy of science in a European perspective. In Explanation, Prediciion and Confirmation. The Phylosophy of Science in a European Perspective; Dieks, D., Gonzalez, W.J., Hartmann, S., Uebel, T., Weber, M., Eds.; Springer: Dordrecht, The Netherlands, 2011; pp. 376–377. [Google Scholar]
- Schrödinger, E. Isotopie and Gibbsches Paradoxon. Z. Phys. A 1921, 5, 163–166. [Google Scholar]
- Denbigh, K.G.; Denbigh, J.S. Entropy in Relation to Incomplete Knowledge; Cambridge University Press: Cambridge, UK, 1985. [Google Scholar]
- Eliel, E.L.; Wilen, S.H. Stereochemistry of Organic Compounds; Wiley: New York, NY, USA, 1994; ISBN 978-0-471-01670-0. [Google Scholar]
- Kozma, D. CRC Handbook of Optical Resolutions via Diastereomeric Salt Formation; CRC Press LLC: Boca Raton, FL, USA, 2002. [Google Scholar]
- van’t Hoff, J.H. Die Phasenlehre. Ber. Dtsch. Chem. Ges. 1902, 35, 4252–4264. [Google Scholar] [CrossRef]
- Byk, A. Zu den Ausnahmen von der Phasenregel, besonders bei optisch-aktiven Körpern. Z. Phys. Chem. 1903, 45, 465–495. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S. Enantiomers, Racemates and Resolutions; Wiley-Interscience: New York, NY, USA, 1981; ISBN 0-471-08058-6. [Google Scholar]
- Crusats, J.; Veintemillas-Verdaguer, S.; Ribó, J.M. Homochirality as a consequence of thermodynamic equilibrium? Chem. A Eur. J. 2006, 12, 7776–7781. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Arteaga, O.; Canillas, A.; Crusats, J.; Sorrenti, A.; Veintemillas-Verdaguer, S.; Ribo, J.M. Achiral-to-chiral transition in benzil solidification: Analogies with racemic conglomerates systems showing deracemization. Chirality 2013, 25, 393–399. [Google Scholar] [CrossRef]
- Viedma, C. Chiral symmetry breaking during crystallization: Complete chiral purity induced by nonlinear autocatalysis and recycling. Phys. Rev. Lett. 2005, 94, 065504. [Google Scholar] [CrossRef] [Green Version]
- Blanco, C.; Crusats, J.; El-Hachemi, Z.; Moyano, A.; Veintemillas-Verdaguer, S.; Hochberg, D.; Ribó, J.M. The Viedma deracemization of racemic conglomerate mixtures as a paradigm of spontaneous mirror symmetry breaking in aggregation and polymerization. ChemPhysChem 2013, 14, 3982–3993. [Google Scholar] [CrossRef]
- Brandmüller, J. An extension of the Neumann–Minnigerode–Curie principle. In Symmertry. Unifying Human Understanding; Hargittai, I.B.T., Ed.; Pergamon Press: New York, NY, USA; Oxford, UK; Toronto, ON, Canada, 1986; pp. 97–100. ISBN 978-0-08-033986-3. [Google Scholar]
- Newnham, R.E. Properties of Materials. Anisotropy, Symmetry, Structure; Oxford University Press: Oxford, UK, 2005; ISBN 0-19-852076-x (pbk). [Google Scholar]
- Ercolani, G.; Piguet, C.; Borkovec, M.; Hamacek, J. Symmetry Numbers and Statistical Factors in Self-Assembly and Multivalency. J. Phys. Chem. B 2007, 111, 12195–12203. [Google Scholar] [CrossRef]
- Buhse, T.; Cruz, J.-M.; Noble-Teran, M.E.; Ribó, J.M.; Crusats, J.; Micheau, J.C. Spontaneous deracemizations. Chem. Rev. 2021. accepted. [Google Scholar]
- Hochberg, D.; Bourdon García, R.D.; Ágreda Bastidas, J.A.; Ribó, J.M. Stoichiometric network analysis of spontaneous mirror symmetry breaking in chemical reactions. Phys. Chem. Chem. Phys. 2017, 19, 17618–17636. [Google Scholar] [CrossRef]
- Hochberg, D.; Ribó, J.M. Stoichiometric network analysis of entropy production in chemical reactions. Phys. Chem. Chem. Phys. 2018, 20, 23726–23739. [Google Scholar] [CrossRef] [PubMed]
- Ribó, J.M.; Hochberg, D. Spontaneous mirror symmetry breaking: An entropy production survey of the racemate instability and the emergence of stable scalemic stationary states. Phys. Chem. Chem. Phys. 2020, 22, 14013–14025. [Google Scholar] [CrossRef]
- Nicolis, G.; Prigogine, I. Self-Organization in Non-Equilibrium Systems; Wiley-Interscience: New York, NY, USA, 1977; ISBN 0-471-02401-5. [Google Scholar]
- Soai, K.; Sato, I.; Shibata, T.; Komiya, S.; Hayashi, M.; Matsueda, Y.; Imamura, H.; Hayase, T.; Morioka, H.; Tabira, H.; et al. Asymmetric synthesis of pyrimidyl alkanol without adding chiral substances by the addition of diisopropylzinc to pyrimidine-5-carbaldehyde in conjunction with asymmetric autocatalysis. Tetrahedron Asymmetry 2003, 14, 185–188. [Google Scholar] [CrossRef]
- Islas, J.R.; Lavabre, D.; Grevy, J.-M.; Lamoneda, R.H.; Cabrera, H.R.; Micheau, J.-C.; Buhse, T. Mirror-symmetry breaking in the Soai reaction: A kinetic understanding. Proc. Natl. Acad. Sci. USA 2005, 102, 13743–13748. [Google Scholar] [CrossRef] [Green Version]
- Ribó, J.M.; Blanco, C.; Crusats, J.; El-Hachemi, Z.; Hochberg, D.; Moyano, A. Absolute asymmetric synthesis in enantioselective autocatalytic reaction networks: Theoretical games, speculations on chemical evolution and perhaps a synthetic option. Chem. A Eur. J. 2014, 20, 17250–17271. [Google Scholar] [CrossRef] [PubMed]
- Kondepudi, D.K.; Nelson, G.W. Chiral-symmetry-breaking states and their sensitivity in nonequilibrium chemical systems. Phys. A Stat. Mech. Appl. 1984, 125, 465–496. [Google Scholar] [CrossRef]
- Kondepudi, D.K.; Nelson, G.W. Weak neutral currents and the origin of biomolecular chirality. Nature 1985, 314, 438–441. [Google Scholar] [CrossRef]
- Lente, G. Homogeneous chiral autocatalysis: A simple, purely stochastic kinetic model. J. Phys. Chem. A 2004, 108, 9475–9478. [Google Scholar] [CrossRef]
- Lente, G. Stochastic Kinetic Models of Chiral Autocatalysis: A General Tool for the Quantitative Interpretation of Total Asymmetric Synthesis. J. Phys. Chem. A 2005, 109, 11058–11063. [Google Scholar] [CrossRef]
- Plasson, R.; Kondepudi, D.K.; Bersini, H.; Commeyras, A.; Asakura, K. Emergence of homochirality in far-from-equilibrium systems: Mechanisms and role in prebiotic chemistry. Chirality 2007, 19, 589–600. [Google Scholar] [CrossRef]
- Frank, F.C. On spontaneous asymmetric synthesis. BBA Biochim. Biophys. Acta 1953, 11, 459–463. [Google Scholar] [CrossRef]
- Crusats, J.; Hochberg, D.; Moyano, A.; Ribó, J.M. Frank model and spontaneous emergence of chirality in closed systems. ChemPhysChem 2009, 10, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Ribó, J.M.; Crusats, J.; El-Hachemi, Z.; Moyano, A.; Hochberg, D. Spontaneous mirror symmetry breaking in heterocatalytically coupled enantioselective replicators. Chem. Sci. 2016, 8, 763–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochberg, D.; Ribó, J.M. Entropic Analysis of Mirror Symmetry Breaking in Chiral Hypercycles. Life 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochberg, D.; Sanchez Torralba, A.; Morán, F. Chaotic oscillations, dissipation and mirror symmetry breaking in a chiral catalytic network. Phys. Chemisry Chem. Phys. 2020. accepted. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, D.; Ribó, J.M. Thermodynamic evolution theorem for chemical reactions. Phys. Rev. Res. 2021. accepted. [Google Scholar]
- Soai, K.; Kawasaki, T.; Matsumoto, A. Asymmetric autocatalysis of pyrimidyl alkanol and its application to the study on the origin of homochirality. Acc. Chem. Res. 2014, 47, 3643–3654. [Google Scholar] [CrossRef]
- von Weizsäcker, C.F. Evolution und Entropie Wachstum. In Offene Systeme I; von Weizsäcker, E.U., Ed.; Ernst Klett-Verlag: Stuttgart, Germany, 1974; ISBN 3-608-93118-X. [Google Scholar]
- Kauffmann, S.A. Autocatalytic sets of proteins. J. Theor. Biol. 1986, 119, 1–24. [Google Scholar] [CrossRef]
- Stadler, P.F.; Schuster, P. Dynamics of small autocatalytic reaction networks—I. bifurcations, permanence and exclusion. Bull. Math. Biol. 1990, 52, 485–508. [Google Scholar] [CrossRef]
- Eschenmoser, A.; Volkan Kisakürek, M. Chemistry and the Origin of Life. Helv. Chim. Acta 1996, 79, 1249–1259. [Google Scholar] [CrossRef]
- Hordijk, W.; Hein, J.; Steel, M. Autocatalytic sets and the origin of life. Entropy 2010, 12, 1733–1742. [Google Scholar] [CrossRef]
- Schmitt-Kopplin, P.; Gabelica, Z.; Gougeon, R.D.; Fekete, A.; Kanawati, B.; Harir, M.; Gebefuegi, I.; Eckel, G.; Hertkorn, N. High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall. Proc. Natl. Acad. Sci. USA 2010, 107, 2763–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avetisov, V.; Goldanskii, V. Mirror symmetry breaking at the molecular level. Proc. Natl. Acad. Sci. USA 1996, 93, 11435–11442. [Google Scholar] [CrossRef] [Green Version]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef] [PubMed]
- Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Palacios, J.C.; Barron, L.D. Absolute Asymmetric Synthesis under Physical Fields: Facts and Fictions. Chem. Rev. 1998, 98, 2391–2404. [Google Scholar] [CrossRef]
- Guijarro, A.; Yus, M. The Origin of Chirality in the Molecules of Life: A Revision from Awareness to the Current Theories and Prespectives of this Unsolved Problem; Royal Society Publishing: Cambridge, UK, 2009; ISBN 978-0-85404-156-5. [Google Scholar]
- Weissbuch, I.; Lahav, M. Crystalline Architectures as Templates of Relevance to the Origins of Homochirality. Chem. Rev. 2011, 111, 3236–3267. [Google Scholar] [CrossRef]
- Ribó, J.M.; Crusats, J.; El-Hachemi, Z.; Hochberg, D.; Moyano, A. Abiotic Emergence of Biological Homochirality. In Advances in Asymmetric Autocatalysis and Related Topics; Academic Press: Salt Lake City, UT, USA, 2017; pp. 299–316. ISBN 9780128128251. [Google Scholar]
- Shanon, C.E.; Weaber, W.Q. The Mathematical Theory of Communication; University of Illinois Press: London, UK, 1949; ISBN 0252725484. [Google Scholar]
- Weber, B.H.; Depew, D.J.; Smith, J.D. (Eds.) Entropy, Information, and Evolution: New Perspectives on Physical and biological Evolution; MIT Press: Cambridge, MA, USA, 1988; ISBN 0262231328. [Google Scholar]
- Comeau, S.R.; Camacho, C.J. Predicting oligomeric assemblies: N-mers a primer. J. Struct. Biol. 2005, 150, 233–244. [Google Scholar] [CrossRef]
- Schneider, T.D. Evolution of biological information. Nucleic Acids Res. 2000, 28, 2794–2799. [Google Scholar] [CrossRef] [Green Version]
- Melkikh, A.V. Quantum information and the problem of mechanisms of biological evolution. Biosystems 2014, 115, 33–45. [Google Scholar] [CrossRef]
- Adami, C. Information-Theoretic Considerations Concerning the Origin of Life. Orig. Life Evol. Biosph. 2015, 45, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Bohme, G. Information und Verständigung. In Offene Systeme I; von Weizsäcker, E.U., Ed.; Ernst Klett-Verlag: Stuttgart, Germany, 1974; ISBN 3-608-93118-X. [Google Scholar]
- Trambouze, P. Structuring Information and Entropy: Catalyst as Information Carrier. Entropy 2006, 8, 113–130. [Google Scholar] [CrossRef]
- Roderer, J.G. Information and Its Role in the Nature; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2005; ISBN 10 3-540-23075-0. [Google Scholar]
- Regan, J.J.; Ramirez, B.E.; Winkler, J.R.; Gray, H.B.; Malmström, B.G. Pathways for Electron Tunneling in Cytochrome c Oxidase. J. Bioenerg. Biomembr. 1998, 30, 35–39. [Google Scholar] [CrossRef]
- Fink, H.-W.; Schönenberger, C. Electrical conduction through DNA molecules. Nature 1999, 398, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Barraclough, T.G.; Nee, S. Phylogenetics and speciation. Trends Ecol. Evol. 2001, 16, 391–399. [Google Scholar] [CrossRef]
- Vattay, G.; Salahub, D.; Csabai, I.; Nassimi, A.; Kaufmann, S.A. Quantum criticality at the origin of life. J. Phys. Conf. Ser. 2015, 626, 12023. [Google Scholar] [CrossRef]
- Sneath, P.H.A. Cladistic Representation of Reticulate Evolution. Syst. Zool. 1975, 24, 360–368. [Google Scholar] [CrossRef]
- Mondal, P.C.; Fontanesi, C.; Waldeck, D.H.; Naaman, R. Spin-Dependent Transport through Chiral Molecules Studied by Spin-Dependent Electrochemistry. Acc. Chem. Res. 2016, 49, 2560–2568. [Google Scholar] [CrossRef]
- Göhler, B.; Hamelbeck, V.; Markus, T.Z.; Kettner, M.; Hanne, G.F.; Vager, Z.; Naaman, R.; Zacharias, H. Spin Selectivity in Electron Transmission Through Self-Assembled Monolayers of Double-Stranded DNA. Science 2011, 331, 894–897. [Google Scholar] [CrossRef]
- Kettner, M.; Göhler, B.; Zacharias, H.; Mishra, D.; Kiran, V.; Naaman, R.; Waldeck, D.H.; Sęk, S.; Pawłowski, J.; Juhaniewicz, J. Spin Filtering in Electron Transport Through Chiral Oligopeptides. J. Phys. Chem. C 2015, 119, 14542–14547. [Google Scholar] [CrossRef]
- Yeganeh, S.; Ratner, M.A.; Medina, E.; Mujica, V. Chiral electron transport: Scattering through helical potentials. J. Chem. Phys. 2009, 131, 14707. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribó, J.M. Chirality: The Backbone of Chemistry as a Natural Science. Symmetry 2020, 12, 1982. https://doi.org/10.3390/sym12121982
Ribó JM. Chirality: The Backbone of Chemistry as a Natural Science. Symmetry. 2020; 12(12):1982. https://doi.org/10.3390/sym12121982
Chicago/Turabian StyleRibó, Josep M. 2020. "Chirality: The Backbone of Chemistry as a Natural Science" Symmetry 12, no. 12: 1982. https://doi.org/10.3390/sym12121982
APA StyleRibó, J. M. (2020). Chirality: The Backbone of Chemistry as a Natural Science. Symmetry, 12(12), 1982. https://doi.org/10.3390/sym12121982