Predicting Value of Binding Constants of Organic Ligands to Beta-Cyclodextrin: Application of MARSplines and Descriptors Encoded in SMILES String



Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Descriptors
2.2. Data Pre-Treatment
2.3. Model Development Using MARSplines
3. Results and Discussion
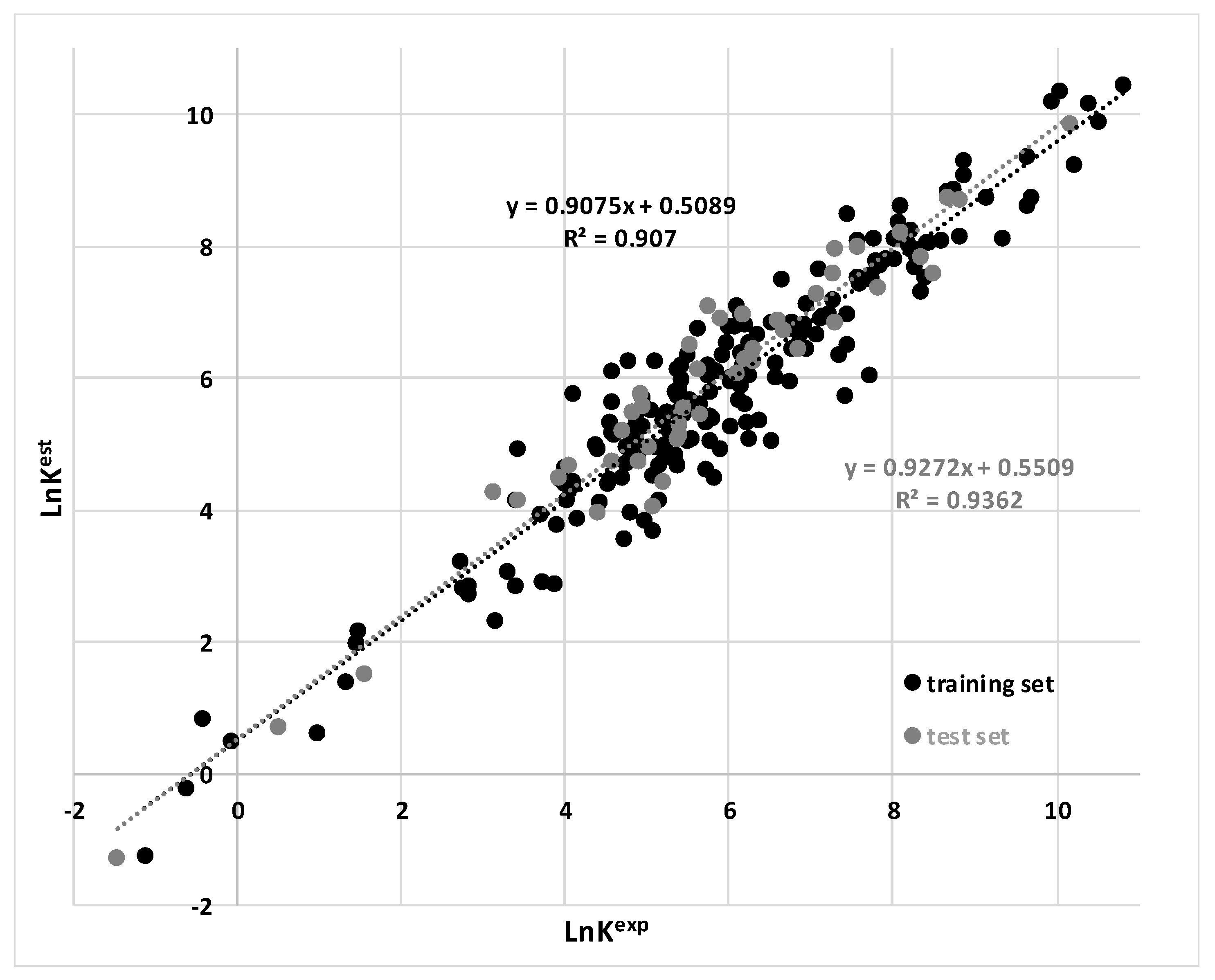
3.1. Findings
3.2. Comparison to Existing Models
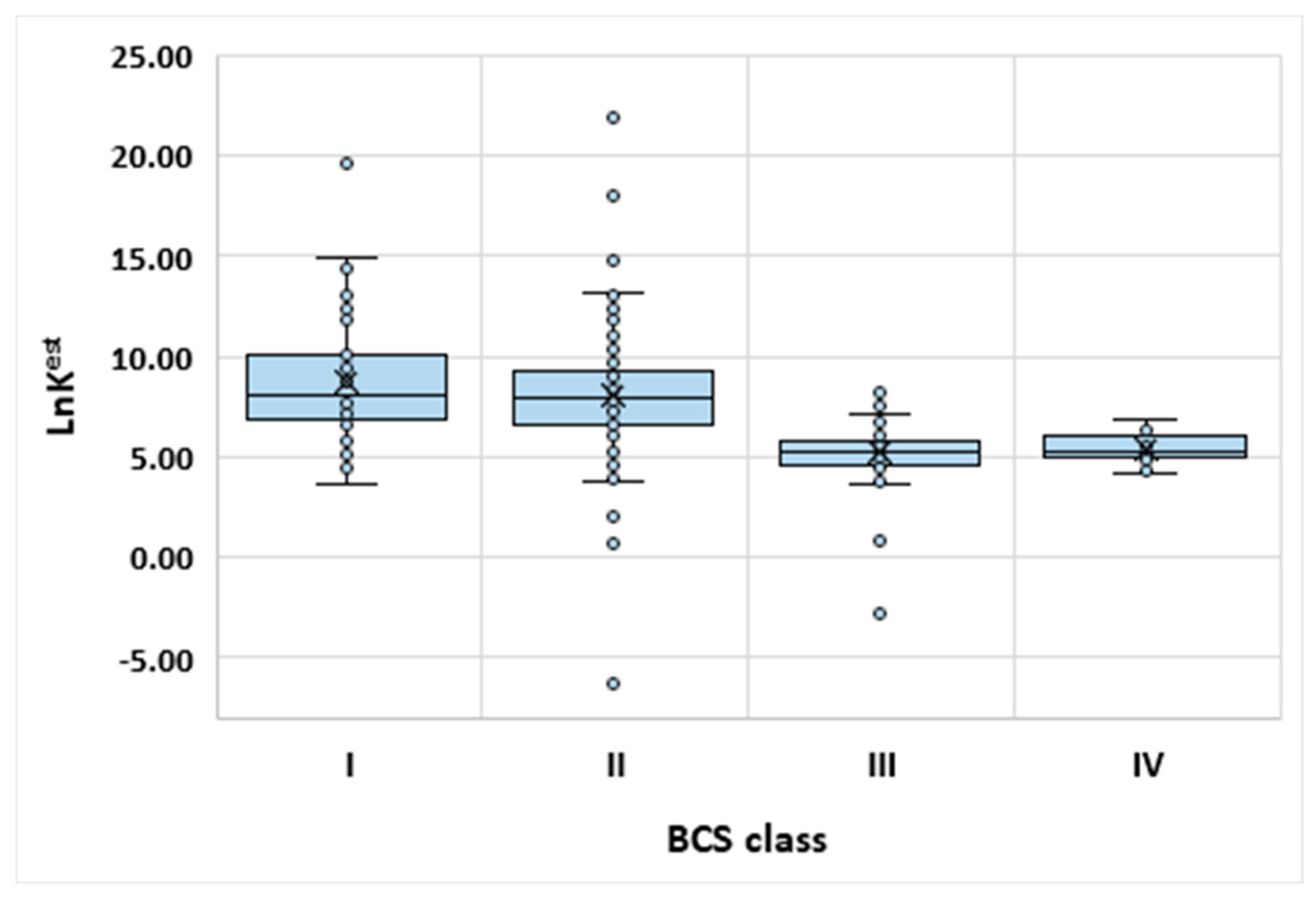
3.3. Exemplary Model Applications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, N.; Xie, C.; Hao, H.; Lu, H.; Lou, Y.; Su, W.; Guo, N. Cocrystal and its Application in the Field of Active Pharmaceutical Ingredients and Food Ingredients. Curr. Pharm. Des. 2018, 24, 2339–2348. [Google Scholar] [CrossRef]
- Korotkova, E.I.; Kratochvíl, B. Pharmaceutical Cocrystals. Procedia Chem. 2014, 10, 473–476. [Google Scholar] [CrossRef]
- Wang, Q.; Xue, J.; Hong, Z.; Du, Y. Pharmaceutical Cocrystal Formation of Pyrazinamide with 3-Hydroxybenzoic Acid: A Terahertz and Raman Vibrational Spectroscopies Study. Molecules 2019, 24, 488. [Google Scholar] [CrossRef] [PubMed]
- Cysewski, P.; Przybyłek, M. Selection of effective cocrystals former for dissolution rate improvement of active pharmaceutical ingredients based on lipoaffinity index. Eur. J. Pharm. Sci. 2017, 107, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Przybyłek, M.; Ziółkowska, D.; Mroczyńska, K.; Cysewski, P. Applicability of Phenolic Acids as Effective Enhancers of Cocrystal Solubility of Methylxanthines. Cryst. Growth Des. 2017, 17, 2186–2193. [Google Scholar] [CrossRef]
- Sinha, A.S.; Maguire, A.R.; Lawrence, S.E. Cocrystallization of nutraceuticals. Cryst. Growth Des. 2015, 15, 984–1009. [Google Scholar] [CrossRef]
- Yang, R.; Xiao, C.F.; Guo, Y.F.; Ye, M.; Lin, J. Inclusion complexes of GA 3 and the plant growth regulation activities. Mater. Sci. Eng. C 2018, 91, 475–485. [Google Scholar] [CrossRef]
- Koontz, J.L.; Marcy, J.E.; Barbeau, W.E.; Duncan, S.E. Stability of Natamycin and Its Cyclodextrin Inclusion Complexes in Aqueous Solution. J. Agric. Food Chem. 2003, 51, 7111–7114. [Google Scholar] [CrossRef]
- Martina, K.; Binello, A.; Lawson, D.; Jicsinszky, L.; Cravotto, G. Recent Applications of Cyclodextrins as Food Additives and in Food Processing. Curr. Nutr. Food Sci. 2013, 9, 167–179. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, H.; Wang, X.; Xu, J.; Liu, Y.; Liu, X.; Huang, H.; Sun, J. Crystal structure and explosive performance of a new CL-20/caprolactam cocrystal. J. Mol. Struct. 2013, 1048, 267–273. [Google Scholar] [CrossRef]
- Shen, J.P.; Duan, X.H.; Luo, Q.P.; Zhou, Y.; Bao, Q.; Ma, Y.J.; Pei, C.H. Preparation and characterization of a novel cocrystal explosive. Cryst. Growth Des. 2011, 11, 1759–1765. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Rai, A. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied Sci. 2010, 2, 72. [Google Scholar] [CrossRef] [PubMed]
- Archontaki, H.A.; Vertzoni, M.V.; Athanassiou-Malaki, M.H. Study on the inclusion complexes of bromazepam with β- and β-hydroxypropyl-cyclodextrins. J. Pharm. Biomed. Anal. 2002, 28, 761–769. [Google Scholar] [CrossRef]
- de Miranda, J.C.; Martins, T.E.A.; Veiga, F.; Ferraz, H.G. Cyclodextrins and ternary complexes: Technology to improve solubility of poorly soluble drugs. Brazilian J. Pharm. Sci. 2011, 47, 665–681. [Google Scholar] [CrossRef]
- Arima, H.; Yunomae, K.; Miyake, K.; Irie, T.; Hirayama, F.; Uekama, K. Comparative studies of the enhancing effects of cyclodextrins on the solubility and oral bioavailability of tacrolimus in rats. J. Pharm. Sci. 2001, 90, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Kumar C.K., A.; Sravanthi, V.V.N.S.S. Cyclodextrins as drug carrier molecule: A review. Sci. Pharm. 2008, 76, 567–598. [Google Scholar] [CrossRef]
- Arima, H.; Miyaji, T.; Irie, T.; Hirayama, F.; Uekama, K. Enhancing effect of hydroxypropyl-β-cyclodextrin on cutaneous penetration and activation of ethyl 4-biphenylyl acetate in hairless mouse skin. Eur. J. Pharm. Sci. 1998, 6, 53–59. [Google Scholar] [CrossRef]
- Shimpi, S.; Chauhan, B.; Shimpi, P. Cyclodextrins: application in different routes of drug administration. Acta Pharm. 2005, 55, 139–156. [Google Scholar] [PubMed]
- Sharma, N.; Baldi, A. Exploring versatile applications of cyclodextrins: An overview. Drug Deliv. 2016, 23, 739–757. [Google Scholar] [CrossRef]
- European Medicines Agencs. Cyclodextrins Used as Excipients Report; European Medicines Agencs: Amsterdam, The Netherlands, 2017.
- Numanoğlu, U.; Şen, T.; Tarimci, N.; Kartal, M.; Koo, O.M.Y.; Önyüksel, H. Use of cyclodextrins as a cosmetic delivery system for fragrance materials: Linalool and benzyl acetate. AAPS PharmSciTech 2008, 8, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H. Eckhard Schollmeyer Applications of cyclodextrins in cosmetic products: A review. J. Cosmet. Sci. 2002, 53, 185. [Google Scholar] [PubMed]
- Tabushi, I. Cyclodextrin Catalysis as a Model for Enzyme Action. Acc. Chem. Res. 1982, 15, 66–72. [Google Scholar] [CrossRef]
- Macaev, F.; Boldescu, V. Cyclodextrins in asymmetric and stereospecific synthesis. Symmetry 2015, 7, 1699–1720. [Google Scholar] [CrossRef]
- D’Souza, V.T. Modification of cyclodextrins for use as artificial enzymes. Supramol. Chem. 2003, 15, 221–229. [Google Scholar] [CrossRef]
- Bicchi, C.; Balbo, C.; D’Amato, A.; Manzin, V.; Schreier, P.; Rozenblum, A.; Brunerie, P. Cyclodextrin derivatives in GC separation of racemic mixtures of volatiles - Part XIV: Some applications of thick-film wide-bore columns to enantiomer GC micropreparation. Hrc-J. High Resolut. Chromatogr. 1998, 21, 103–106. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Ward, T.J.; Armstrong, R.D.; Beesley, T.E. Separation of drug stereoisomers by the formation of β-cyclodextrin inclusion complexes. Science 1986, 232, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Przybyłek, M.; Cysewski, P. Distinguishing Cocrystals from Simple Eutectic Mixtures: Phenolic Acids as Potential Pharmaceutical Coformers. Cryst. Growth Des. 2018, 18, 3524–3534. [Google Scholar] [CrossRef]
- Steffen, A.; Karasz, M.; Thiele, C.; Lengauer, T.; Kämper, A.; Wenz, G.; Apostolakis, J. Combined similarity and QSPR virtual screening for guest molecules of β-cyclodextrin. New J. Chem. 2007, 31, 1941–1949. [Google Scholar] [CrossRef]
- Linden, L.; Goss, K.U.; Endo, S. 3D-QSAR predictions for α-cyclodextrin binding constants using quantum mechanically based descriptors. Chemosphere 2017, 169, 693–699. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Fara, D.C.; Yang, H.; Karelson, M.; Suzuki, T.; Solov’ev, V.P.; Varnek, A. Quantitative Structure−Property Relationship Modeling of β -Cyclodextrin Complexation Free Energies. J. Chem. Inf. Comput. Sci. 2004, 44, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Prakasvudhisarn, C.; Wolschann, P.; Lawtrakul, L. Predicting complexation thermodynamic parameters of β-cyclodextrin with chiral guests by using swarm intelligence and support vector machines. Int. J. Mol. Sci. 2009, 10, 2107–2121. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Garrido, A.; Helguera, A.M.; Cordeiro, M.N.D.S.; Escudero, A.G. QSPR modelling with the topological substructural molecular design approach: β-cyclodextrin complexation. J. Pharm. Sci. 2009, 98, 4557–4576. [Google Scholar] [CrossRef] [PubMed]
- Rama Krishna, G.; Ukrainczyk, M.; Zeglinski, J.; Rasmuson, Å.C. Prediction of Solid State Properties of Cocrystals Using Artificial Neural Network Modeling. Cryst. Growth Des. 2018, 18, 133–144. [Google Scholar] [CrossRef]
- Zhokhova, N.I.; Bobkov, E.V.; Baskin, I.I.; Palyulin, V.A.; Zefirov, A.N.; Zefirov, N.S. Calculation of the stability of β-cyclodextrin complexes of organic compounds using the QSPR approach. Moscow Univ. Chem. Bull. 2007, 62, 269–272. [Google Scholar] [CrossRef]
- Blanford, W.J.; Gao, H.; Dutta, M.; Ledesma, E.B. Solubility enhancement and QSPR correlations for polycyclic aromatic hydrocarbons complexation with α, β, and γ cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2014, 78, 415–427. [Google Scholar] [CrossRef]
- Mirrahimi, F.; Salahinejad, M.; Ghasemi, J.B. QSPR approaches to elucidate the stability constants between β-cyclodextrin and some organic compounds: Docking based 3D conformer. J. Mol. Liq. 2016, 219, 1036–1043. [Google Scholar] [CrossRef]
- Veselinović, A.M.; Veselinović, J.B.; Toropov, A.A.; Toropova, A.P.; Nikolić, G.M. In silico prediction of the β-cyclodextrin complexation based on Monte Carlo method. Int. J. Pharm. 2015, 495, 404–409. [Google Scholar] [CrossRef]
- Friedman, J.H. Multivariate Adaptive Regression Splines. Ann. Stat. 1991, 19, 1–67. [Google Scholar] [CrossRef]
- Przybyłek, M.; Recki, Ł.; Mroczyńska, K.; Jeliński, T.; Cysewski, P. Experimental and theoretical solubility advantage screening of bi-component solid curcumin formulations. J. Drug Deliv. Sci. Technol. 2019, 50, 125–135. [Google Scholar] [CrossRef]
- Przybyłek, M.; Jeliński, T.; Cysewski, P. Application of Multivariate Adaptive Regression Splines (MARSplines) for Predicting Hansen Solubility Parameters Based on 1D and 2D Molecular Descriptors Computed from SMILES String. J. Chem. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Suzuki, T. A Nonlinear Group Contribution Method for Predicting the Free Energies of Inclusion Complexation of Organic Molecules with α- and β-Cyclodextrins. J. Chem. Inf. Comput. Sci. 2001, 41, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Yao, Z.J.; Wen, M.; Zhu, M.F.; Wang, N.N.; Miao, H.Y.; Lu, A.P.; Zeng, W.B.; Cao, D.S. BioTriangle: A web-accessible platform for generating various molecular representations for chemicals, proteins, DNAs/RNAs and their interactions. J. Cheminform. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- ChemDes. Available online: http://www.scbdd.com/chemdes/ (accessed on 1 June 2019).
- BioTriangle. Available online: http://biotriangle.scbdd.com (accessed on 1 June 2019).
- Ballabio, D.; Consonni, V.; Mauri, A.; Claeys-Bruno, M.; Sergent, M.; Todeschini, R. A novel variable reduction method adapted from space-filling designs. Chemom. Intell. Lab. Syst. 2014, 136, 147–154. [Google Scholar] [CrossRef]
- Ambure, P.; Aher, R.B.; Gajewicz, A.; Puzyn, T.; Roy, K. “NanoBRIDGES” software: Open access tools to perform QSAR and nano-QSAR modeling. Chemom. Intell. Lab. Syst. 2015, 147, 1–13. [Google Scholar] [CrossRef]
- Kennard, R.W.; Stone, L.A. Computer Aided Design of Experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Martin, T.M.; Harten, P.; Young, D.M.; Muratov, E.N.; Golbraikh, A.; Zhu, H.; Tropsha, A. Does rational selection of training and test sets improve the outcome of QSAR modeling? J. Chem. Inf. Model. 2012, 52, 2570–2578. [Google Scholar] [CrossRef]
- QSAR Model Development Using DTC Lab. Software Tools. Available online: http://teqip.jdvu.ac.in/QSAR_Tools/ (accessed on 1 June 2019).
- Statsoft. Statistica; Version 12; StatSoft: Tulsa, OK, USA, 2012. [Google Scholar]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- QSAR Research Unit in Environmental Chemistry and Ecotoxicology. Available online: http://www.qsar.it/ (accessed on 1 June 2019).
- Przybyłek, M.; Jeliński, T.; Słabuszewska, J.; Ziółkowska, D.; Mroczyńska, K.; Cysewski, P. Application of Multivariate Adaptive Regression Splines (MARSplines) Methodology for Screening of Dicarboxylic Acid Cocrystal Using 1D and 2D Molecular Descriptors. Cryst. Growth Des. 2019. [Google Scholar] [CrossRef]
- Todeschini, R. Data correlation, number of significant principal components and shape of molecules. The K correlation index. Anal. Chim. Acta 1997, 348, 419–430. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V.; Maiocchi, A. The K correlation index: Theory development and its application in chemometrics. Chemom. Intell. Lab. Syst. 1999, 46, 13–29. [Google Scholar] [CrossRef]
- Stanton, D.T.; Jurs, P.C. Development and Use of Charged Partial Surface Area Structural Descriptors in Computer-Assisted Quantitative Structure-Property Relationship Studies. Anal. Chem. 1990, 62, 2323–2329. [Google Scholar] [CrossRef]
- Labute, P. A widely applicable set of descriptors. J. Mol. Graph. Model. 2000, 18, 464–477. [Google Scholar] [CrossRef]
- Hall, L.H.; Mohney, B.; Kier, L.B. The Electrotopological State: Structure Information at the Atomic Level for Molecular Graphs. J. Chem. Inf. Comput. Sci. 1991, 31, 76–82. [Google Scholar] [CrossRef]
- Hall, L.H.; Kier, L.B. The Molecular Connectivity Chi Indexes and Kappa Shape Indexes in Structure-Property Modeling. Rev. Comput. Chem. 1991, 2, 367–422. [Google Scholar]
- Noolvi, M.N.; Patel, H.M. A comparative QSAR analysis and molecular docking studies of quinazoline derivatives as tyrosine kinase (EGFR) inhibitors: A rational approach to anticancer drug design. J. Saudi Chem. Soc. 2013, 17, 361–379. [Google Scholar] [CrossRef]
- Ji, H.F.; Kong, D.X.; Shen, L.; Chen, L.L.; Ma, B.G.; Zhang, H.Y. Distribution patterns of small-molecule ligands in the protein universe and implications for origin of life and drug discovery. Genome Biol. 2007, 8. [Google Scholar] [CrossRef]
- Bhatiya, R.; Vaidya, A.; Kashaw, S.K.; Jain, A.K.; Agrawal, R.K. QSAR analysis of furanone derivatives as potential COX-2 inhibitors: kNN MFA approach. J. Saudi Chem. Soc. 2014, 18, 977–984. [Google Scholar] [CrossRef]
- Veerasamy, R.; Subramaniam, D.K.; Chean, O.C.; Ying, N.M. Designing hypothesis of substituted benzoxazinones as HIV-1 reverse transcriptase inhibitors: QSAR approach. J. Enzyme Inhib. Med. Chem. 2012, 27, 693–707. [Google Scholar] [CrossRef]
- Ajmani, S.; Janardhan, S.; Viswanadhan, V.N. Toward a general predictive QSAR model for gamma-secretase inhibitors. Mol. Divers. 2013, 17, 421–434. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics: Volume 1&2; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; ISBN 978-3-527-31852-0. [Google Scholar]
- Wen, X.; Tan, F.; Jing, Z.; Liu, Z. Preparation and study the 1:2 inclusion complex of carvedilol with β-cyclodextrin. J. Pharm. Biomed. Anal. 2004, 34, 517–523. [Google Scholar] [CrossRef]
- Kano, K.; Nishiyabu, R.; Asada, T.; Kuroda, Y. Static and dynamic behavior of 2:1 inclusion complexes of cyclodextrins and charged porphyrins in aqueous organic media. J. Am. Chem. Soc. 2002, 124, 9937–9944. [Google Scholar] [CrossRef] [PubMed]
- Frixa, C.; Scobie, M.; Black, S.J.; Thompson, A.S.; Threadgill, M.D. Formation of a remarkably robust 2:1 complex between β-cyclodextrin and a phenyl-substituted icosahedral carborane. Chem. Commun. 2002, 2, 2876–2877. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration The Biopharmaceutics Classification System (BCS) Guidance. Available online: https://www.fda.gov/ (accessed on 1 June 2019).
- Loftsson, T. Cyclodextrins and the biopharmaceutics classification system of drugs. J. Incl. Phenom. 2002, 44, 63–67. [Google Scholar] [CrossRef]
- Loftsson, T. Drug permeation through biomembranes: Cyclodextrins and the unstirred water layer. Pharmazie 2012, 67, 363–370. [Google Scholar]
- Dahan, A.; Wolk, O.; Kim, Y.H.; Ramachandran, C.; Crippen, G.M.; Takagi, T.; Bermejo, M.; Amidon, G.L. Purely in silico BCS classification: Science based quality standards for the world’s drugs. Mol. Pharm. 2013, 10, 4378–4390. [Google Scholar] [CrossRef]
- Xiao, D.; Pham-Huy, L.A.; Pham-Huy, C.; Dramou, P.; He, H.; Zuo, P. Carbon Nanotubes: Applications in Pharmacy and Medicine. Biomed Res. Int. 2013, 2013, 1–12. [Google Scholar]
- Singh, I.; Rehni, A.K.; Kumar, P.; Kumar, M.; Aboul-Enein, H.Y. Carbon Nanotubes: Synthesis, Properties and Pharmaceutical Applications. Fullerenes Nanotub. Carbon Nanostruct. 2009, 17, 361–377. [Google Scholar] [CrossRef]
- Szefler, B. Nanotechnology, from quantum mechanical calculations up to drug delivery. Int. J. Nanomed. 2018, 13, 6143–6176. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Factor | βi | ai | Basis Functions |
|---|---|---|---|
| F0 | 5.4277 | ||
| F1 | −0.3991 | −1.2679 | max(0; 28.1940-Chiv1plusChiv1) |
| F2 | 0.5652 | 1.1090 | max(0; XLogP+0.1340) |
| F3 | 0.3772 | 1.3356 | max(0; carbonTypes.8) |
| F4 | −0.1559 | −1.4658 | max(0; 0.5620-MLFER_A) |
| F5 | −0.1613 | −1.3482 | max(0; Wlambda2.unity-1.2400) |
| F6 | −0.1391 | −0.3182 | max(0; 1.2400-Wlambda2.unity) |
| F7 | −0.2130 | −2.5385 | max(0; XLogP+0.1340)∙max(0;-15.8078-PNSA-3) |
| F8 | −0.1372 | −0.0120 | max(0; 66.0412- AATS6m)∙max(0; MLFER_A -0.5620) |
| F9 | −0.0977 | −0.0003 | max(0; PEOEVSA9*PEOEVSA9-988.3780)∙max(0; XLogP +0.1340) |
| F10 | −0.1258 | −0.0002 | max(0; 988.378- PEOEVSA9*PEOEVSA9)∙max(0; XLogP +0.1340) |
| F11 | 0.0910 | 0.0257 | max(0; Wlambda2.unity -1.2400)∙max(0; AATS4i-154.1756) |
| F12 | 0.0944 | 0.2092 | max(0; Wlambda2.unity -1.2400)∙max(0; 154.1756- AATS4i) |
| Model Description | R2 | Source | |
|---|---|---|---|
| Training Set | Test Set | ||
| MARSplines | 0.91 | 0.94 | This work |
| Molecular docking-based descriptors | 0.83 | 0.83 | [37] |
| Monte Carlo optimised topological descriptors | 0.92 | 0.93 | [38] |
| High Solubility | Low Solubility | |
|---|---|---|
| High permeability | Class I This class comprise compounds characterized by good absorption profiles. | Class II The bioavailability is directly related to the dissolution behavior. |
| Low permeability | Class III The active pharmaceutical ingredient (API) is soluble, however absorption profile is dependent on limited permeation behavior. | Class IV The API is characterized by very low bioavailability. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cysewski, P.; Przybyłek, M. Predicting Value of Binding Constants of Organic Ligands to Beta-Cyclodextrin: Application of MARSplines and Descriptors Encoded in SMILES String. Symmetry 2019, 11, 922. https://doi.org/10.3390/sym11070922
Cysewski P, Przybyłek M. Predicting Value of Binding Constants of Organic Ligands to Beta-Cyclodextrin: Application of MARSplines and Descriptors Encoded in SMILES String. Symmetry. 2019; 11(7):922. https://doi.org/10.3390/sym11070922
Chicago/Turabian StyleCysewski, Piotr, and Maciej Przybyłek. 2019. "Predicting Value of Binding Constants of Organic Ligands to Beta-Cyclodextrin: Application of MARSplines and Descriptors Encoded in SMILES String" Symmetry 11, no. 7: 922. https://doi.org/10.3390/sym11070922
APA StyleCysewski, P., & Przybyłek, M. (2019). Predicting Value of Binding Constants of Organic Ligands to Beta-Cyclodextrin: Application of MARSplines and Descriptors Encoded in SMILES String. Symmetry, 11(7), 922. https://doi.org/10.3390/sym11070922