A General Phenomenon of Spontaneous Amplification of Optical Purity under Achiral Chromatographic Conditions

 , ,
, , 

Abstract
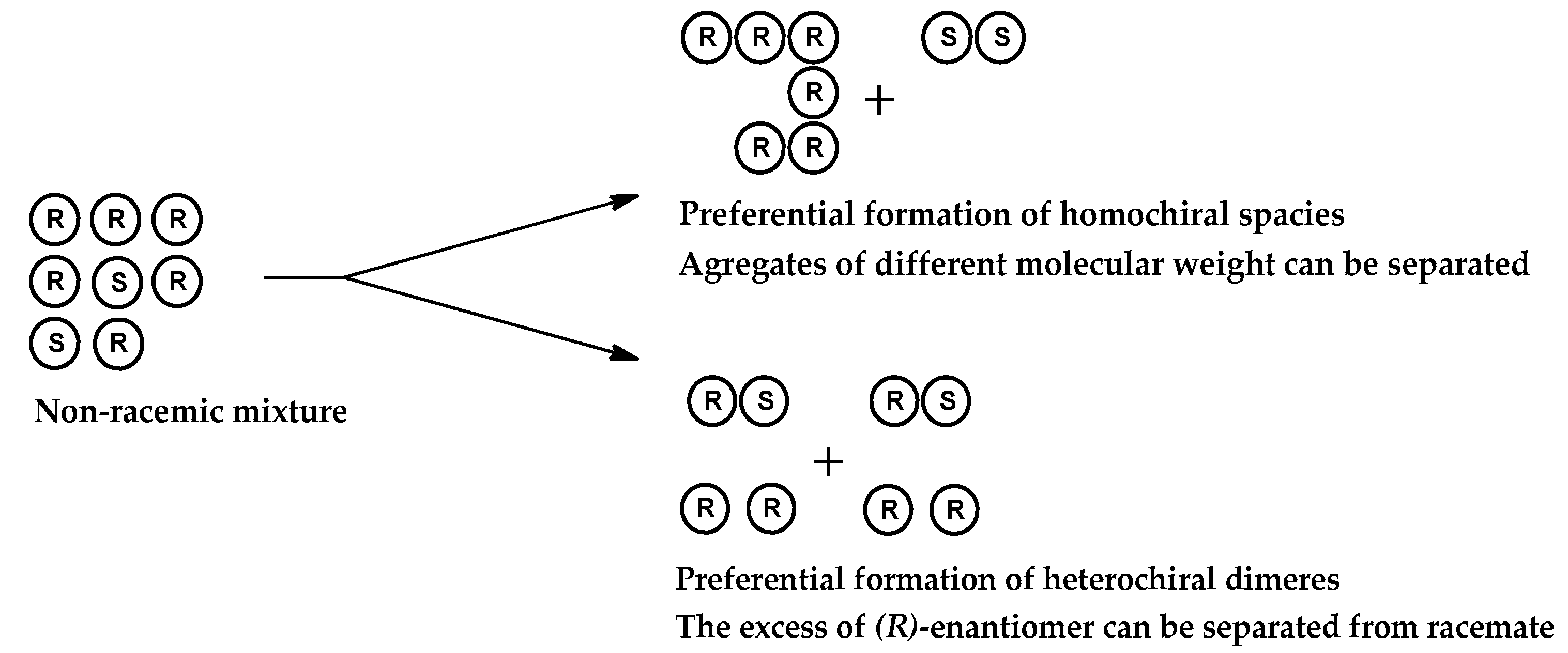
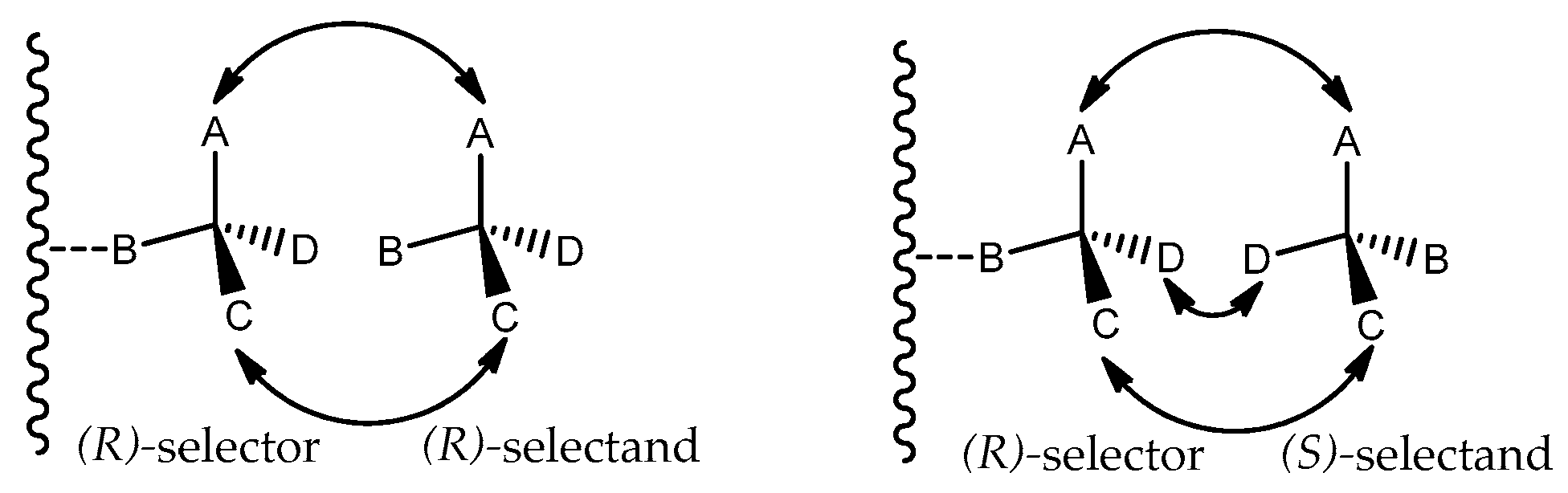
1. Introduction
2. Materials and Methods
3. Results and Discussion.
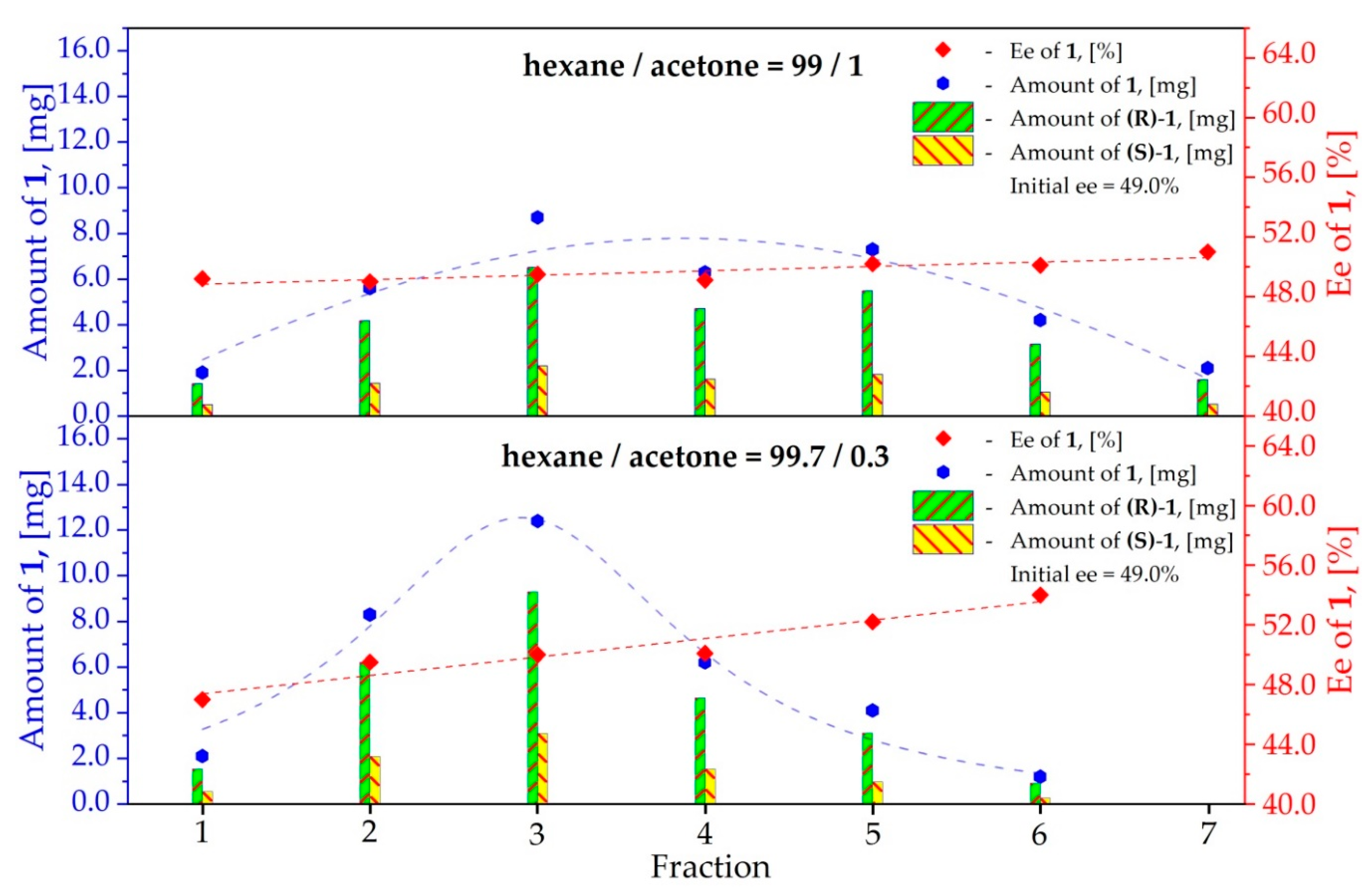
3.1. Study on the SDE of Dicarboxylic Acid Esters: (R)-Dimethyl [(2E)-1,3-Diphenyl-2-Propen-1-yl] Malonate (1)
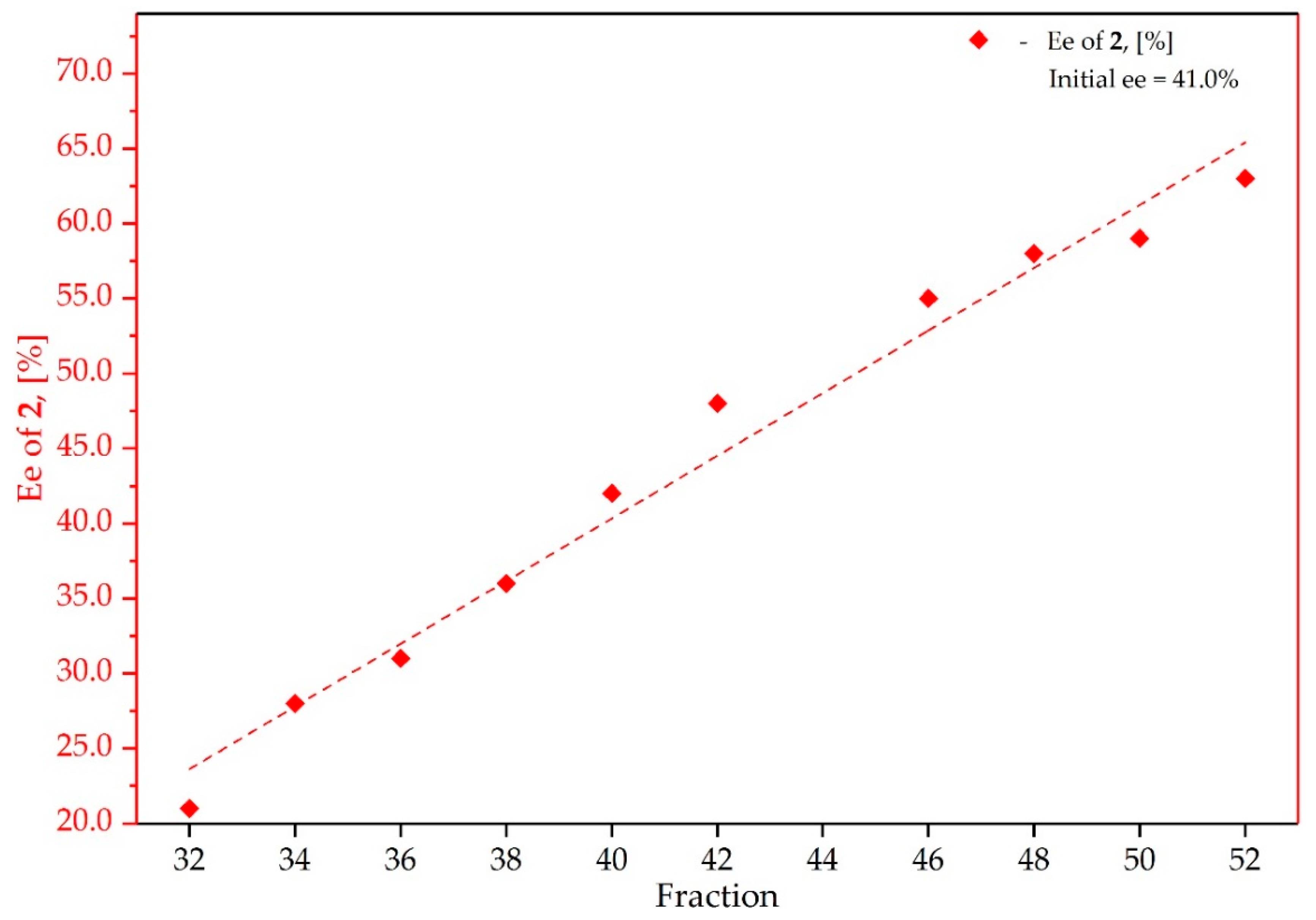
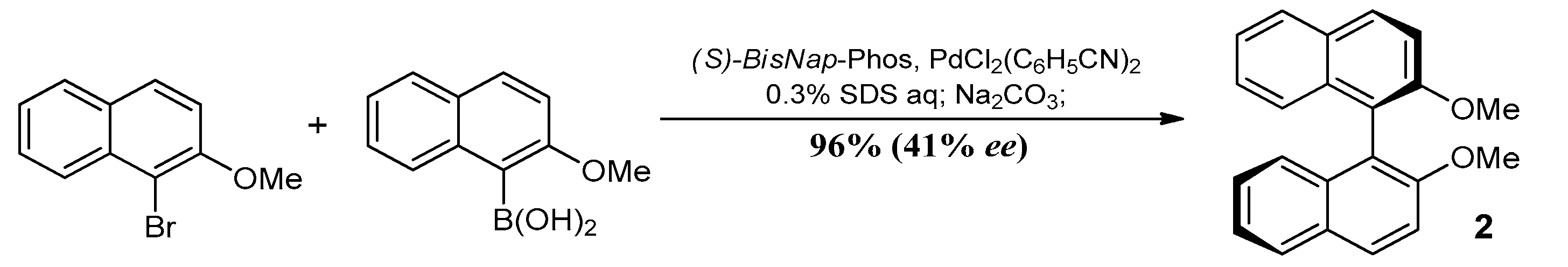
3.2. Study on the SDE of Atropisomeric Biaryl: 2,2′-Dimethoxy-1,1′-Binaphthyl (2)
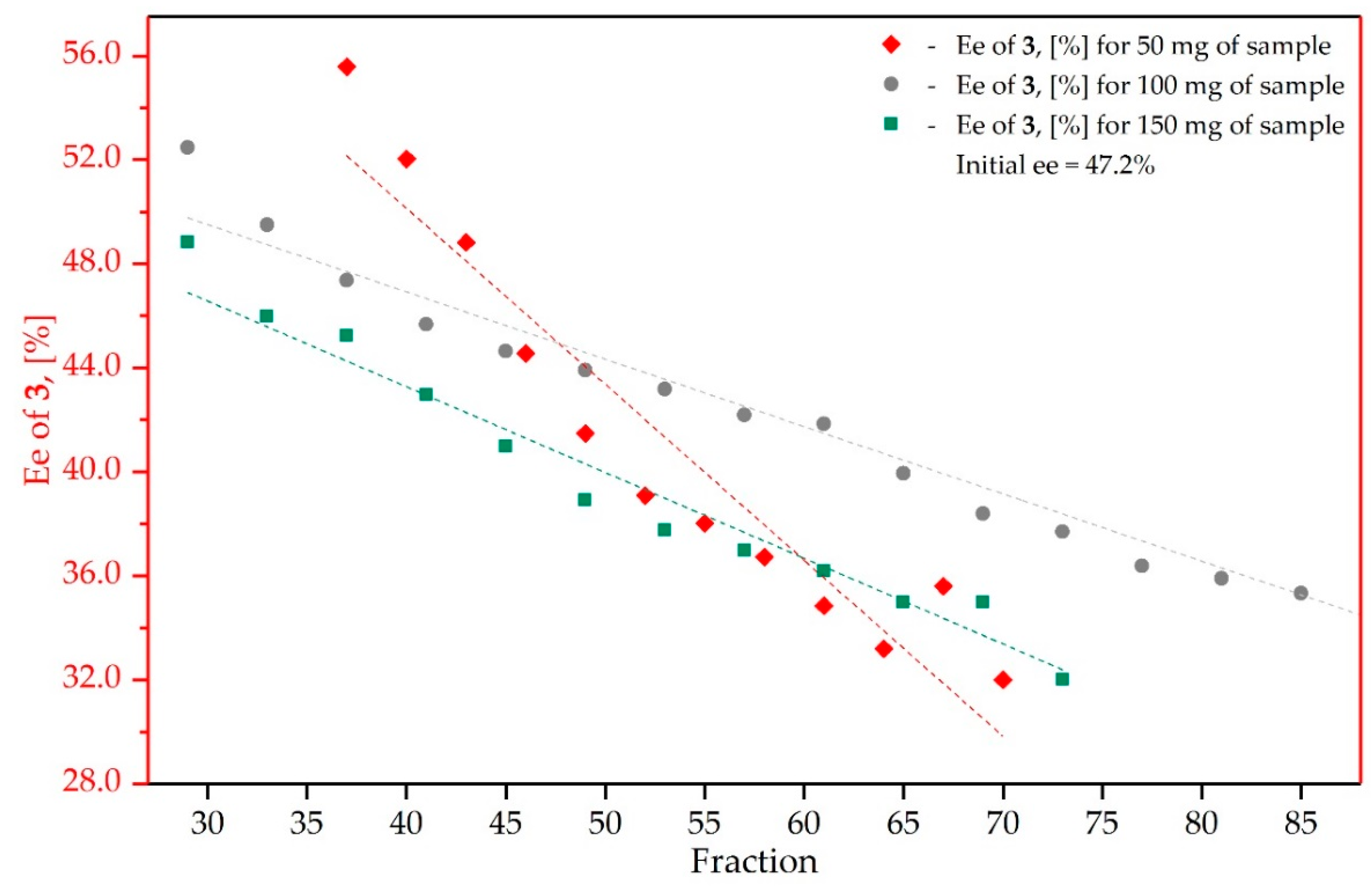
3.3. Study on the SDE of β-Hydroxy Ketone: (4R)-(4-nitrophenyl)-4-Hydroxy-2-Butanone (3)
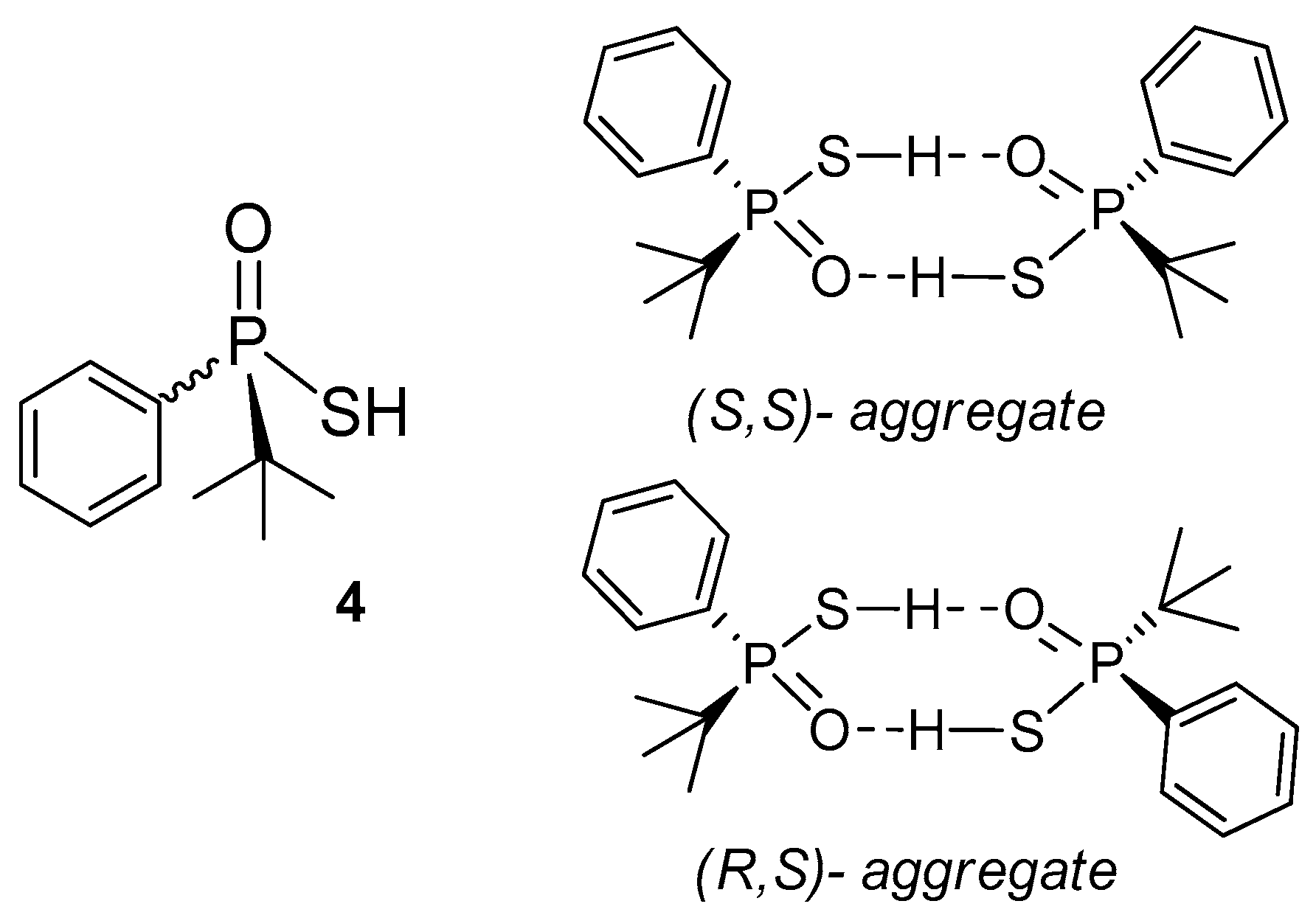
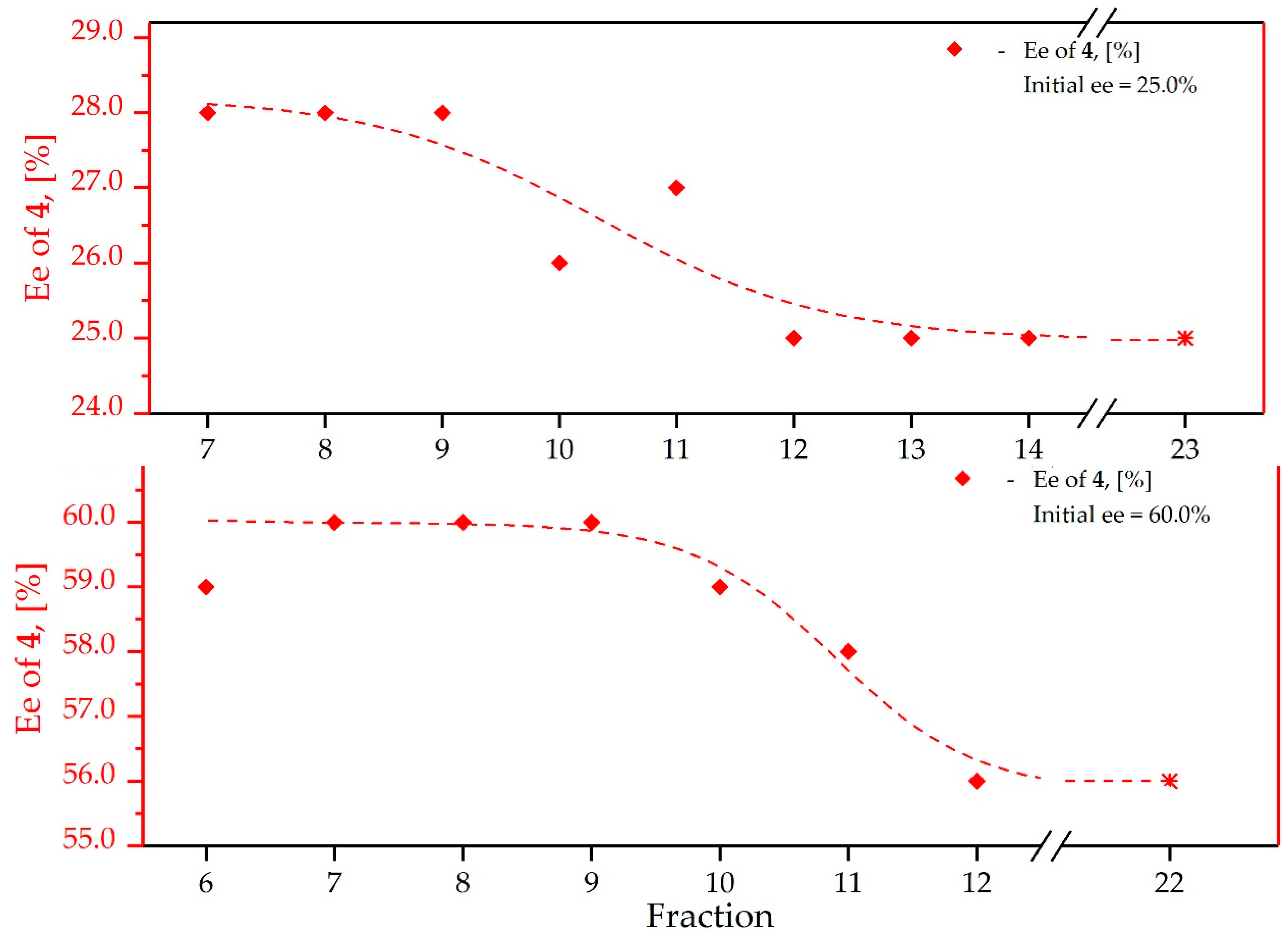
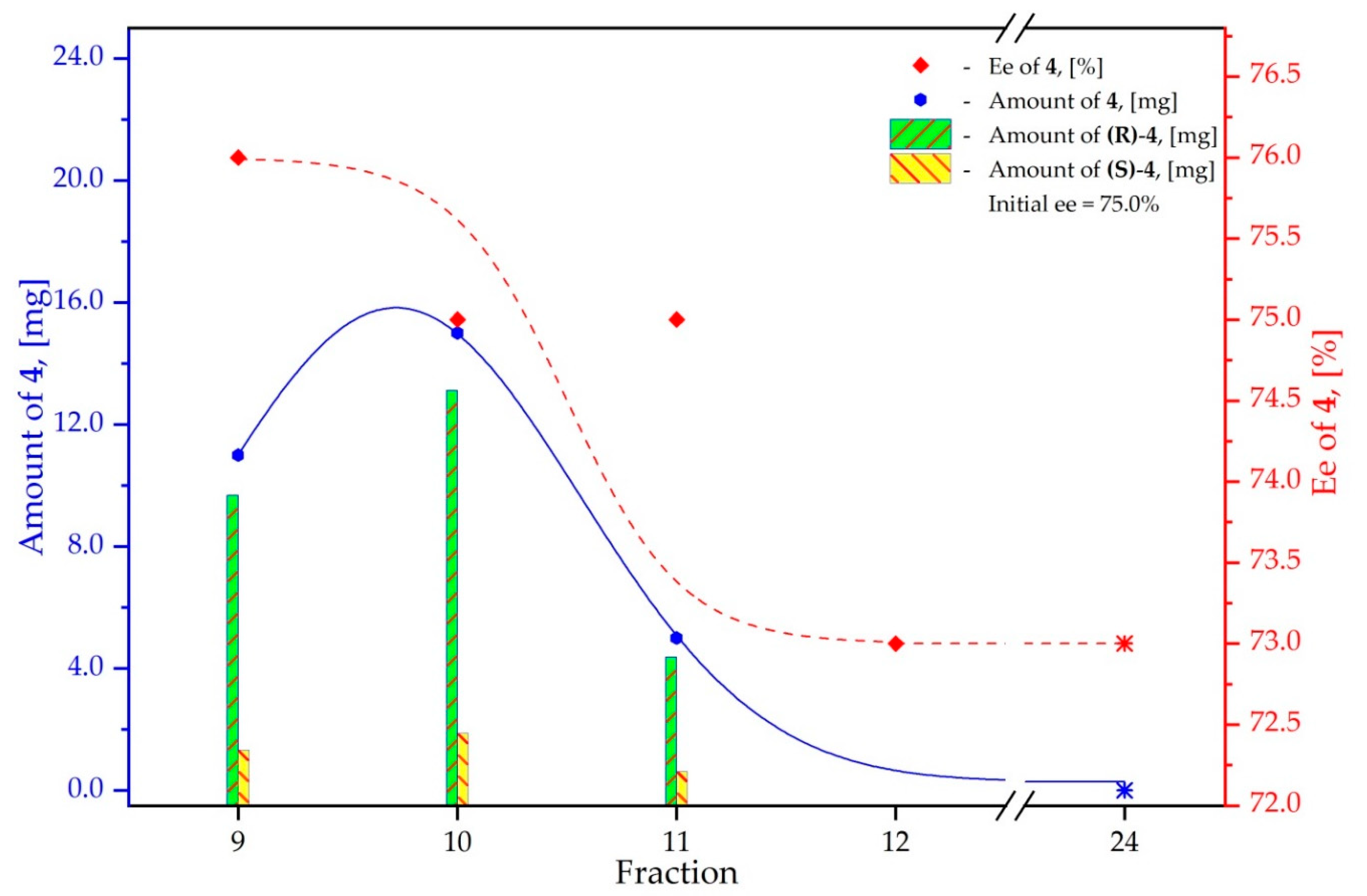
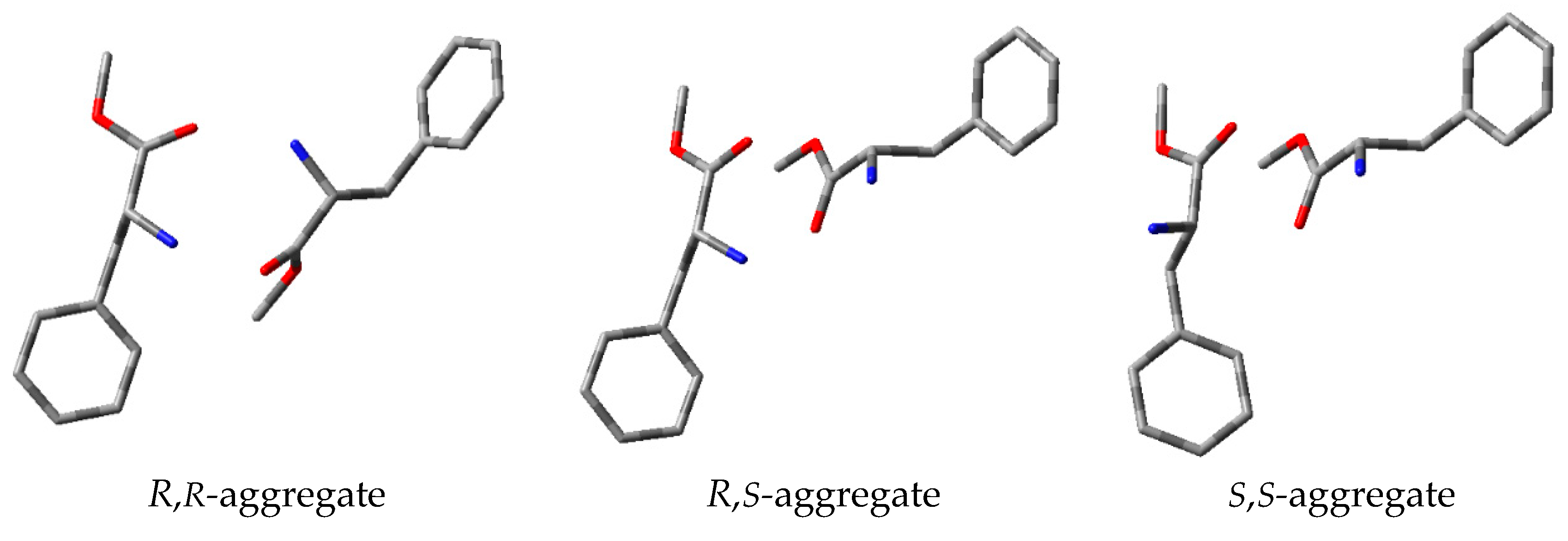
3.4. Study on the SDE of P-Chiral Compounds: tert-Butylphenylphosphinothioic acid (4)
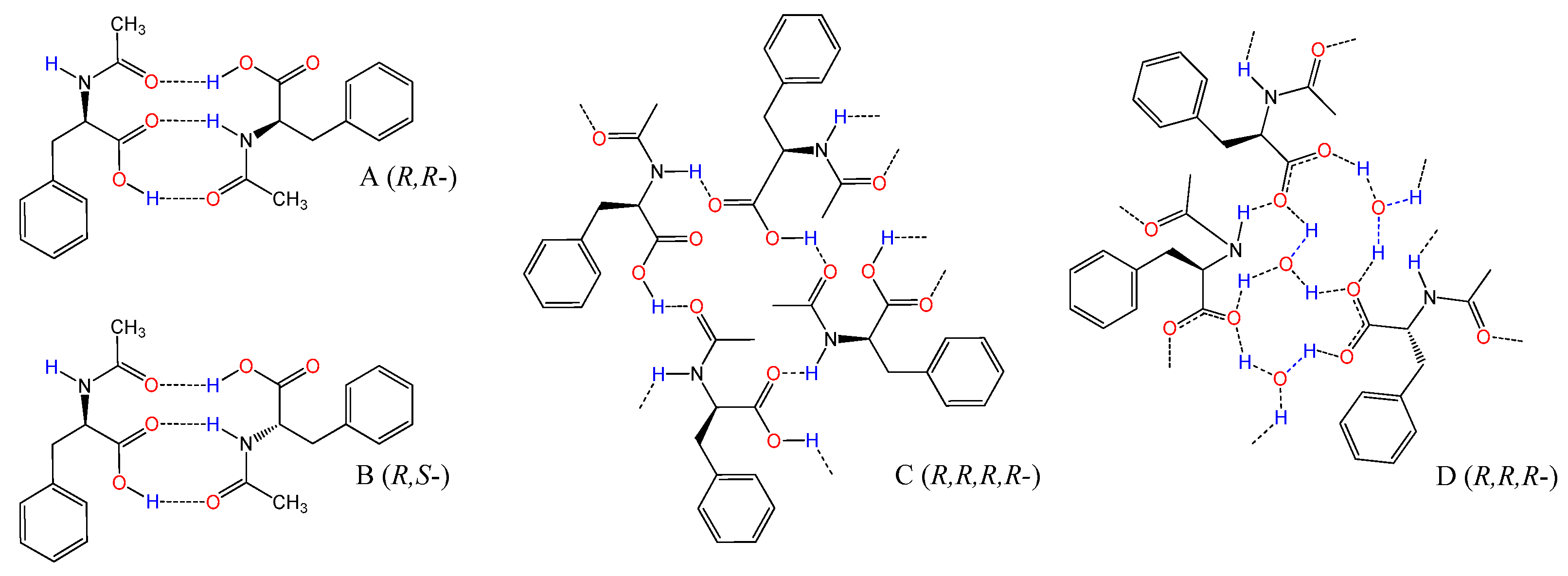
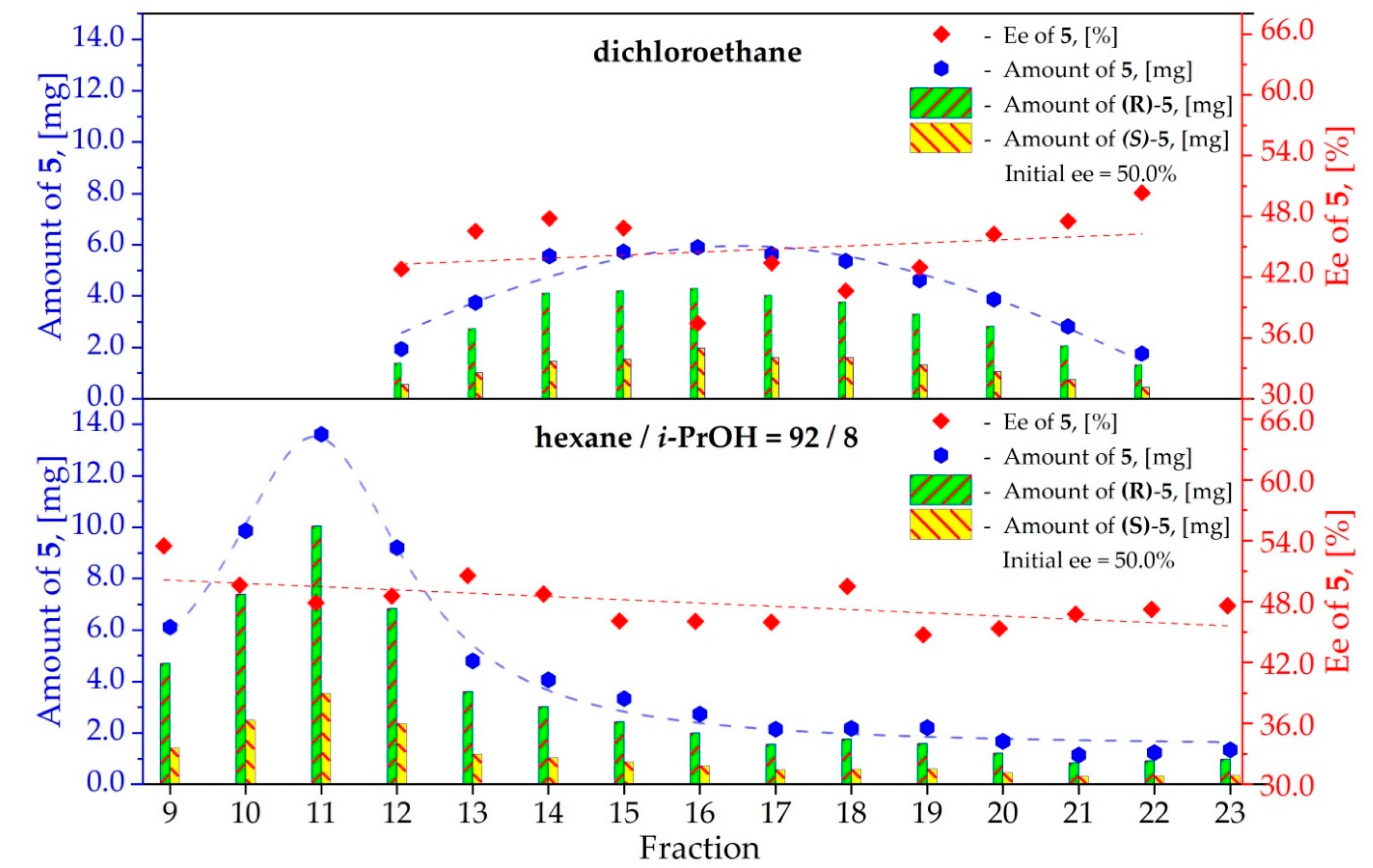
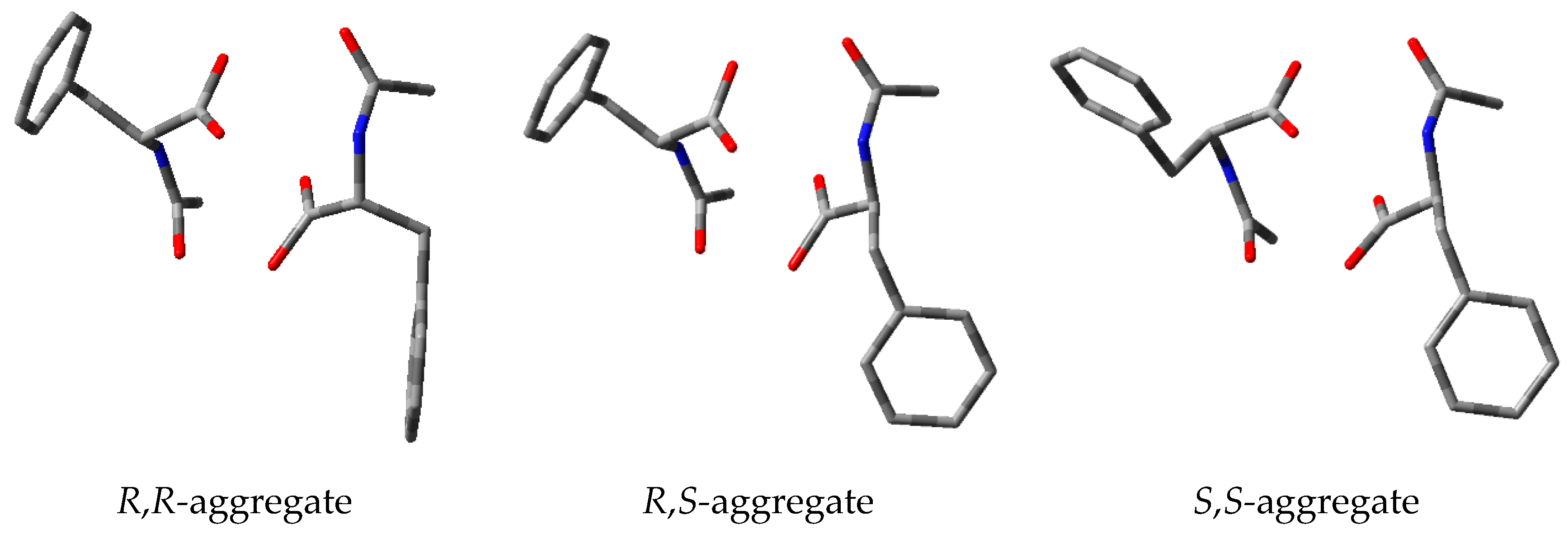
3.5. Study on the SDE of Amino Acid Derivatives: N-Acetyl-Phenylalanine (5)
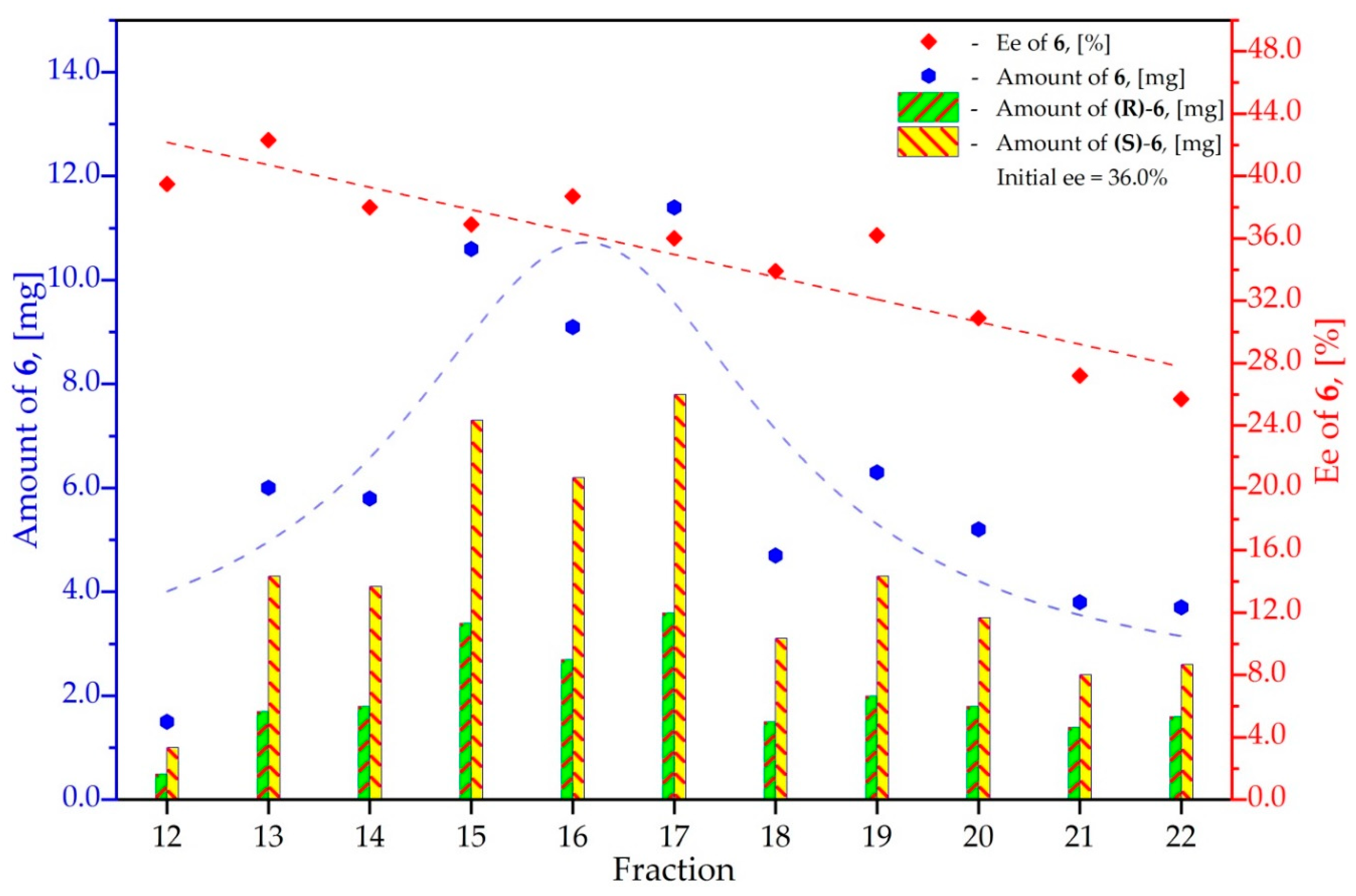
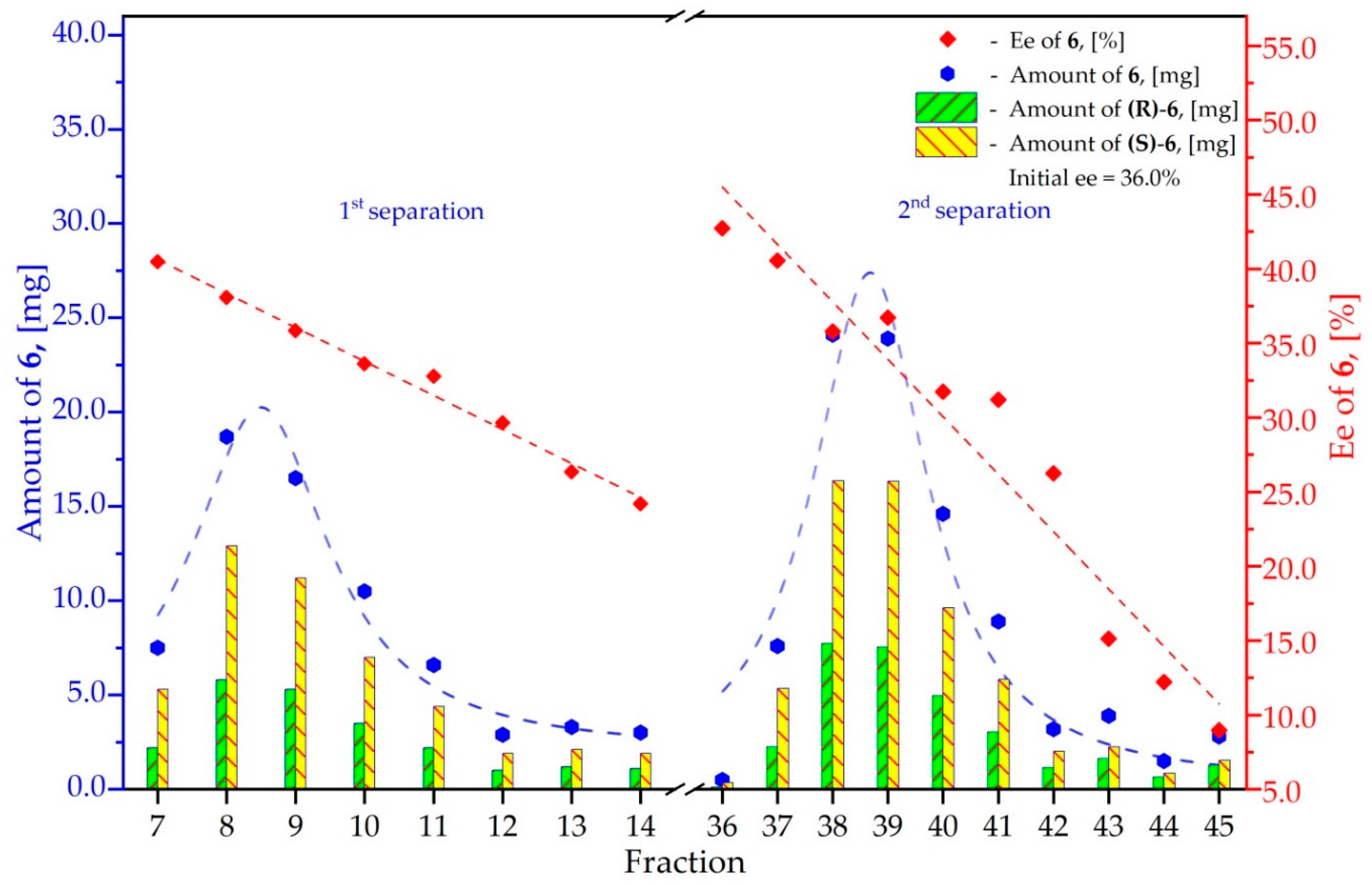
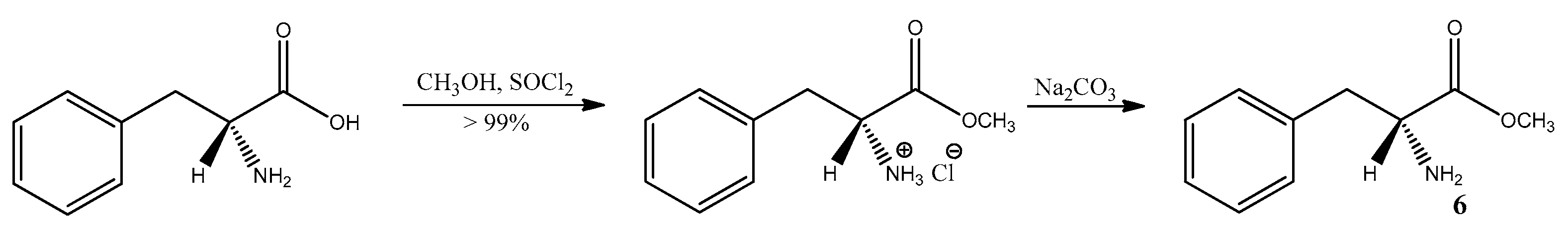
3.6. Study on the SDE of Aamino Acid Derivatives: Methyl Phenylalaninate (6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Carreira, E.M.; Yamamoto, H. Comprehensive Chirality, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2012; ISBN 978-008-095-167-6. [Google Scholar]
- Hegstrom, R.A.; Kondepudi, D.K. The Handedness of the Universe. Sci. Am. 1990, 262, 108–115. [Google Scholar] [CrossRef]
- Lubec, G.; Rosenthal, G.A. Amino Acids, Chemistry, Biology and Medicine; ESCOM: Leiden, The Netherlands, 1990; ISBN 9072199049. [Google Scholar]
- Ernst, B.; Hart, G.W.; Sinay, P. Biosynthesis and Degradation of Glycoconjugates; Wiley-VCH: Chichester, UK, 2000; ISBN 352-729-511-9. [Google Scholar]
- Gawley, R.E.; Aubé, J. Principles of Asymmetric Synthesis, 2nd ed.; Elsevier: Oxford, UK, 2012; ISBN 978-008-044-860-2. [Google Scholar]
- Cohen, J. Getting all turned around over the origins of life on earth. Science 1995, 267, 1265–1266. [Google Scholar] [CrossRef]
- McCombs, C. The Extraterrestrial Search for the Origin of Homochirality. Creat. Res. Soc. Q. 2014, 51, 5–13. [Google Scholar]
- Rauchfuss, H.; Mitchell, T.N. Chemical Evolution and the Origin of Life by Horst Rauchfuss; Springer: Berlin/Heidelberg, Germany, 2008; ISBN 978-354-078-823-2. [Google Scholar]
- McGuire, B.A.; Carroll, P.B.; Loomis, R.A.; Finneran, I.A.; Jewell, P.R.; Remijan, A.J.; Blake, G.A. Discovery of the interstellar chiral molecule propylene oxide (CH3CHCH2O). Science 2016, 352, 1449–1452. [Google Scholar] [CrossRef]
- Cooper, G.; Reed, C.; Nguyen, D.; Carter, M.; Wang, Y. Detection and formation scenario of citric acid, pyruvic acid, and other possible metabolism precursors in carbonaceous meteorites. Proc. Natl. Acad. Sci. USA 2011, 108, 14015–14020. [Google Scholar] [CrossRef] [PubMed]
- Angel, J.R.P.; Illing, R.; Martin, P.G. Circular polarization of twilight. Nature 1972, 238, 389–390. [Google Scholar] [CrossRef]
- Wolstencroft, R.D. Terrestiral and Astronomical Sources of Circular Polarisation: A fresh look at the origin of Homochirality on Earth. Bioastron. Life Stars 2004, 213, 154–158. [Google Scholar]
- Garay, A.S.; Ahigren-Beckendorf, J.A. Differential interaction of chiral β-particles with enantiomers. Nature 1990, 346, 451–453. [Google Scholar] [CrossRef]
- Rikken, G.L.J.A.; Raupach, E. Enantioselective magnetochiral photochemistry. Nature 2000, 405, 932–935. [Google Scholar] [CrossRef]
- Soai, K.; Osanai, S.; Kadowaki, K.; Yonekubo, S.; Shibata, T.; Sato, I. d- and l-Quartz-Promoted Highly Enantioselective Synthesis of a Chiral Organic Compound. J. Am. Chem. Soc. 1999, 121, 11235–11236. [Google Scholar] [CrossRef]
- Tang, L.; Shi, L.; Bonneau, C.; Sun, J.; Yue, H.; Ojuva, A.; Lee, B.L.; Kritikos, M.; Bell, R.G.; Bacsik, Z.; et al. A zeolite family with chiral and achiral structures built from the same building layer. Nat. Mater. 2008, 7, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Nógrádi, M.; Fogassy, E.; Pálovics, E.; Schindler, J. Resolution of Enantiomers by Non-Conventional Methods. Synthesis 2005, 10, 1555–1568. [Google Scholar]
- Faigl, F.; Fogassy, E.; Nógrádi, M.; Pálovics, E.; Schindler, J. Strategies in optical resolution: A practical guide. Tetrahedron Asymmetry 2008, 19, 519–536. [Google Scholar] [CrossRef]
- Nag, A. Asymmetric Synthesis of Drugs and Natural Products; CRC Press: Boca Raton, FL, USA, 2018; ISBN 978-113-803-361-0. [Google Scholar]
- Lough, W.J.; Wainer, I.W. Chirality in Natural and Applied Science; CRC Press: Boca Raton, FL, USA, 2002; ISBN 084-932-434-3. [Google Scholar]
- Kurihara, N.; Miyamoto, J. Chirality in Agrochemicals; Wiley: New York, NY, USA, 1998; ISBN 047-198-121-4. [Google Scholar]
- Kitzerow, H.-S.; Bahr, C. Chirality in Liquid Crystals; Springer: New York, NY, USA, 2001; ISBN 038-798-679-0. [Google Scholar]
- Li, Q. Photoactive Functional Soft Materials: Preparation, Properties, and Applications; Wiley: Blackwell, UK, 2019; ISBN 978-352-781-676-7. [Google Scholar]
- Mason, S.F. Optical rotatory power. Q. Rev. Chem. Soc. 1963, 17, 20–66. [Google Scholar] [CrossRef]
- Schellman, J.A. Circular dichroism and optical rotation. Chem. Rev. 1975, 75, 323–331. [Google Scholar] [CrossRef]
- Lloyd, D.K.; Goodall, D.M. Polarimetric detection in high-performance liquid chromatography. Chirality 1989, 1, 251–264. [Google Scholar] [CrossRef]
- Roussel, C.; Del Rio, A.; Pierrot-Sanders, J.; Piras, P.; Vanthuyne, N.C. Chiral liquid chromatography contribution to the determination of the absolute configuration of enantiomers. J. Chromatogr. A 2004, 1037, 311–328. [Google Scholar] [CrossRef]
- Pakulski, Z.; Demchuk, O.M.; Kwiatosz, R.; Osiński, P.W.; Świerczyńska, W.; Pietrusiewicz, K.M. The classical Kagan’s amides are still practical NMR chiral shift reagents: Determination of enantiomeric purity of P-chirogenic phospholene oxides. Tetrahedron Asymmetry 2003, 14, 1459–1462. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Świerczynska, W.; Pietrusiewicz, K.M.; Woźnica, M.; Wójcik, D.; Frelek, J. A convenient application of the NMR and CD methodologies for the determination of enantiomeric ratio and absolute configuration of chiral atropoisomeric phosphine oxides. Tetrahedron Asymmetry 2008, 19, 2339–2345. [Google Scholar] [CrossRef]
- Goering, H.L.; Eikenberry, J.N.; Koermer, G.S. Tris[3-(trifluoromethylhydroxymethylene)-d-camphorato]europium(III) a chiral shift reagent for direct determination of enantiomeric compositions. J. Am. Chem. Soc. 1971, 3, 5913–5914. [Google Scholar] [CrossRef]
- McCreary, M.D.; Lewis, D.W.; Wernick, D.L.; Whitesides, G.M. Determination of enantiomeric purity using chiral lanthanide shift reagents. J. Am. Chem. Soc. 1974, 96, 1038–1054. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Demchuk, O.M.; Gerschkovich, A.A. Application of the dimenthyl chlorophosphite for the chiral analysis of amines, amino acids and peptides. Tetrahedron Asymmetry 1999, 10, 1729–1732. [Google Scholar] [CrossRef]
- Seco, J.M.; Latypov, S.; Quiñoá, E.; Riguera, R. New chirality recognizing reagents for the determination of absolute stereochemistry and enantiomeric purity by NMR. Tetrahedron Lett. 1994, 18, 2921–2924. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Wilcox, J.D. Chiral reagents for the determination of enantiomeric excess and absolute configuration using NMR spectroscopy. Chirality 2003, 15, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A. Remarkable Amplification of the self-disproportionation of enantiomers on achiral-phase chromatography columns. Angew. Chem. Int. Ed. 2006, 45, 766–769. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Berbasov, D.O. Self-disproportionation of enantiomers of (R)-ethyl 3-(3,5-dinitrobenzamido)-4,4,4-trifluorobutanoate on achiral silica gel stationary phase. J. Fluorine Chem. 2006, 127, 597–603. [Google Scholar] [CrossRef]
- Cundy, K.C.; Crooks, P.A. Unexpected phenomenon in the high-performance liquid chromatographic analysis of racemic 14C-labelled nicotine: Separation of enantiomers in a totally achiral system. J. Chromatogr. A 1983, 281, 17–33. [Google Scholar] [CrossRef]
- Soloshonok, V.; Sorochinsky, A.; Aceña, J. Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2012, 45, 141–152. [Google Scholar] [CrossRef]
- Faigl, F.; Fogassy, E.; Nόgradi, M.; Palovics, E.; Schindler, J. Separation of non-racemic mixtures of enantiomers: An essential part of optical resolution. Org. Biomol. Chem. 2010, 8, 947–959. [Google Scholar] [CrossRef]
- Garin, D.L.; Greco, D.J.C.; Kelley, L. Enhancement of optical activity by fractional sublimation. An alternative to fractional crystallization and a warning. J. Org. Chem. 1977, 42, 1249–1251. [Google Scholar] [CrossRef]
- Omelańczuk, J.; Mikołajczyk, M. Chiral t-butylphenylphosphinothioic acid: A useful chiral solvating agent for direct determination of enantiomeric purity of alcohols, thiols, amines, diols, aminoalcohols and related compounds. Tetrahedron Asymmetry 1996, 7, 2687–2694. [Google Scholar] [CrossRef]
- Tsai, T.L.; Herman, K.; Hug, E.; Rohde, B.; Dreiding, A.S. Enantiomer-Differentiation Induced by an Enantiomeric Excess during Chromatography with Achiral Phases. Helv. Chem. Acta 1985, 68, 2238–2243. [Google Scholar] [CrossRef]
- Viedma, C.; Noorduin, W.L.; Ortiz, J.E.; de Torres, T.; Cintas, P. Asymmetric amplification in amino acid sublimation involving racemic compound to conglomerate conversion. Chem. Commun. 2011, 47, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.; Gil-Av, E. Self-amplification of optical activity by chromatography on an achiral adsorbent. J. Chromatogr. A 1984, 298, 516–520. [Google Scholar] [CrossRef]
- Dobashi, A.; Motoyama, Y.; Kinoshita, K.; Hara, S.; Fukasaku, N. Self-induced chiral recognition in the association of enantiomeric mixtures on silica gel chromatography. Anal. Chem. 1987, 59, 2209–2211. [Google Scholar] [CrossRef]
- Frost, C.G.; Howarth, J.; Williams, J.M.J. Selectivity in palladium catalyzed allylic substitution. Tetrahedron Asymmetry 1992, 3, 1089–1122. [Google Scholar] [CrossRef]
- Dawson, G.J.; Frost, C.G.; Williams, J.M.J. Asymmetric palladium catalysed allylic substitution using phosphorus containing oxazoline ligands. Tetrahedron Lett. 1993, 34, 3149–3150. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Kielar, K.; Pietrusiewicz, K.M. Rational design of novel ligands for environmentally benign cross-coupling reactions. Pure Appl. Chem. 2011, 3, 633–644. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Kapłon, K.; Kącka, A.; Pietrusiewicz, K.M. The utilization of chiral phosphorus ligands in atroposelective cross-coupling reactions. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 180–200. [Google Scholar] [CrossRef]
- Jasiński, R.; Demchuk, O.M.; Babyuk, D. A Quantum-Chemical DFT Approach to Elucidation of the Chirality Transfer Mechanism of the Enantioselective Suzuki–Miyaura Cross-Coupling Reaction. J. Chem. 2017, 2017, 3617527. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Q. Palladium catalyzed asymmetric Suzuki–Miyaura coupling reactions to axially chiral biaryl compounds: Chiral ligands and recent advances. Coord. Chem. Rev. 2015, 286, 1–16. [Google Scholar] [CrossRef]
- Sawai, K.; Tatumi, R.; Nakahodo, T.; Fijihara, H. Asymmetric Suzuki–Miyaura coupling reactions catalyzed by chiral palladium nanoparticles at room temperature. Angew. Chem. 2008, 120, 7023–7025. [Google Scholar] [CrossRef]
- Braun, M.; Devant, R. (R)- and (S)-2-acetoxy-1,1,2-triphenylethanol—Effective synthetic equivalents of a chiral acetate enolate. Tetrahedron Lett. 1984, 25, 5031–5034. [Google Scholar] [CrossRef]
- Tempkin, O.; Abel, S.; Chen, C.P.; Underwood, R.; Prasad, K.; Chen, K.M.; Repic, O.; Blacklock, T.J. Asymmetric synthesis of 3,5-dihydroxy-6(E)-heptenoate-containing HMG-CoA reductase inhibitors. Tetrahedron 1997, 53, 10659–10670. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, UK, 2010. [Google Scholar]
- Demchuk, O.M.; Jasiński, R.; Pietrusiewicz, K.M. New Insights into the Mechanism of Reduction of Tertiary Phosphine Oxides by Means of Phenylsilane. Heteroatom Chem. 2015, 26, 441–448. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.; Szwaczko, K.; Mirosław, B.; Dybała, I.; Jasiński, R.; Demchuk, O.M. New Rigid Polycyclic Bis(phosphane) for Asymmetric Catalysis. Molecules 2019, 24, 571. [Google Scholar] [CrossRef]
- Mirosław, B.; Babyuk, D.; Łapczuk-Krygier, A.; Kącka-Zych, A.; Demchuk, O.M.; Jasiński, R. Regiospecific formation of the nitromethyl-substituted 3-phenyl-4,5-dihydroisoxazole via [3 + 2]cycloaddition. Monatshefte für Chemie Chem. Mon. 2018, 149, 1877–1884. [Google Scholar] [CrossRef]
- Jin, M.-J.; Takale, V.B.; Sarkar, M.S.; Kim, Y.-M. Highly enantioselective Pd-catalyzed allylic alkylation using new chiral ferrocenylphosphinoimidazolidine ligands. Chem. Commun. 2006, 6, 663–664. [Google Scholar] [CrossRef]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Justyniak, I.; Miroslaw, B.; Jasinski, R. 2-Methoxynaphthylnaphthoquinone and its solvate: Synthesis and structure-properties relationship. J. Phys. Organ. Chem. 2014, 27, 66–73. [Google Scholar] [CrossRef]
- Nakashima, E.; Yamamoto, H. Asymmetric Aldol Synthesis: Choice of Organocatalyst and Conditions. Chem. Asian J. 2017, 12, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C.F. Amino Acid Catalyzed Direct Asymmetric Aldol Reactions: A Bioorganic Approach to Catalytic Asymmetric Carbon−Carbon Bond-Forming Reactions. J. Am. Chem. Soc. 2001, 123, 5260–5267. [Google Scholar] [CrossRef]
- Chrzanowski, J.; Krasowska, D.; Urbaniak, M.; Sieroń, L.; Pokora-Sobczak, P.; Demchuk, O.M.; Drabowicz, J. Synthesis of Enantioenriched Aryl-tert-Butylphenylphosphine Oxides via Cross-Coupling Reactions of tert-Butylphenylphosphine Oxide with Aryl Halides. Eur. J. Organ. Chem. 2018, 2018, 4614–4627. [Google Scholar] [CrossRef]
- Harger, M.J.P. Chemical shift non-equivalence of enantiomers in the proton magnetic resonance spectra of partly resolved phosphinothioic acids. J. Chem. Soc. Perkin II 1978, 4, 326–331. [Google Scholar] [CrossRef]
- Dressen, M.H.C.L.; van de Kruijs, B.H.P.; Meuldijk, J.; Vekemans, J.A.J.M.; Hulshof, L.A. From Batch to Flow Processing: Racemization of N-Acetylamino Acids under Microwave Heating. Organ. Process Res. Dev. 2009, 13, 888–895. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Arlt, D.; Jasiński, R.; Pietrusiewicz, K.M. Relationship between structure and efficiency of atropisomeric phosphine ligands in homogeneous catalytic asymmetric hydrogenation. J. Phys. Organ. Chem. 2012, 25, 1006–1011. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A. Self-disproportionation of enantiomers via achiral gravity-driven column chromatography: A case study of N-acyl-α-phenylethylamines. J. Chromatogr. A 2016, 1467, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Wzorek, A.; Kamizela, A.; Sato, A.; Soloshonok, V.A. Self-Disproportionation of Enantiomers (SDE) via achiral gravity-driven column chromatography of N-fluoroacyl-1-phenylethylamines. J. Fluorine Chem. 2017, 196, 37–43. [Google Scholar] [CrossRef]
- Suzuki, Y.; Han, J.; Kitagawa, O.; AceÇa, J.L.; Klika, K.D.; Soloshonok, V.A. Comprehensive examination of the self-disproportionation of enantiomers (SDE) of chiral amides via achiral, laboratory-routine, gravity-driven column chromatography. RCS Adv. 2015, 5, 2988–2993. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Enantiomeric enrichments via the self-disproportionation of enantiomers (SDE) by achiral, gravity-driven column chromatography: A case study using N-(1-Phenylethyl)acetamide for optimizing the enantiomerically pure yield and magnitude of the SDE. Helv. Chem. Acta 2015, 98, 1147–1159. [Google Scholar] [CrossRef]
- Mori, M.; Deodato, D.; Kasula, M.; Ferraris, D.M.; Sanna, A.; De Logu, A.; Rizzi, M.; Botta, M. Design, synthesis, SAR and biological investigation of 3-(carboxymethyl)rhodanine and aminothiazole inhibitors of Mycobacterium tuberculosis Zmp1. Bioorg. Med. Chem. Lett. 2018, 28, 637–641. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Pochapsky, T.C. Considerations of chiral recognition relevant to the liquid chromatography separation of enantiomers. Chem. Rev. 1989, 89, 347–362. [Google Scholar] [CrossRef]
- Polavarapu, P.L. Chiral Analysis—Advances in Spectroscopy, Chromatography and Emerging Methods, 2nd ed.; Elsevier Science: Atlanta, GA, USA, 2018; ISBN 978-044-464-028-4. [Google Scholar]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
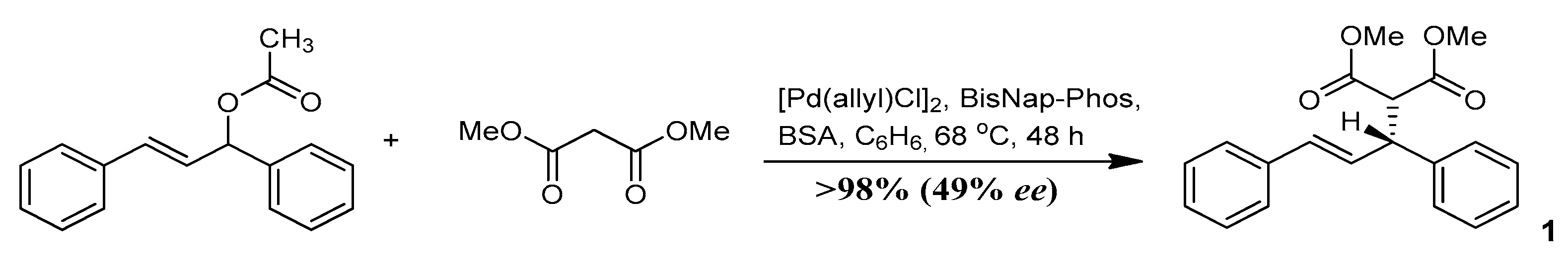{kind=link}
{kind=link}
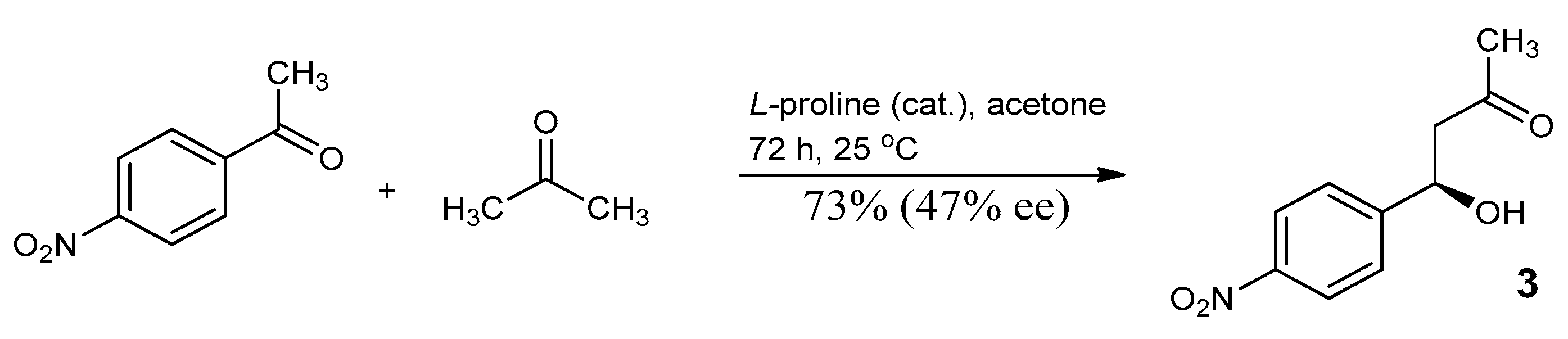{kind=link}
{kind=link}
| Transition | ΔG [kcal/mol] |
|---|---|
| (R,S) → (S,S) | 4.0 |
| (R,S) → (R,R) | 2.5 |
| Transition | ΔG [kcal/mol] |
|---|---|
| (R,R) → (R,S) | 2.2 |
| (S,S) → (R,S) | 3.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrusiewicz, K.M.; Borkowski, M.; Strzelecka, D.; Kielar, K.; Kicińska, W.; Karevych, S.; Jasiński, R.; Demchuk, O.M. A General Phenomenon of Spontaneous Amplification of Optical Purity under Achiral Chromatographic Conditions. Symmetry 2019, 11, 680. https://doi.org/10.3390/sym11050680
Pietrusiewicz KM, Borkowski M, Strzelecka D, Kielar K, Kicińska W, Karevych S, Jasiński R, Demchuk OM. A General Phenomenon of Spontaneous Amplification of Optical Purity under Achiral Chromatographic Conditions. Symmetry. 2019; 11(5):680. https://doi.org/10.3390/sym11050680
Chicago/Turabian StylePietrusiewicz, K. Michał, Mariusz Borkowski, Dorota Strzelecka, Katarzyna Kielar, Wioleta Kicińska, Sergei Karevych, Radomir Jasiński, and Oleg M. Demchuk. 2019. "A General Phenomenon of Spontaneous Amplification of Optical Purity under Achiral Chromatographic Conditions" Symmetry 11, no. 5: 680. https://doi.org/10.3390/sym11050680
APA StylePietrusiewicz, K. M., Borkowski, M., Strzelecka, D., Kielar, K., Kicińska, W., Karevych, S., Jasiński, R., & Demchuk, O. M. (2019). A General Phenomenon of Spontaneous Amplification of Optical Purity under Achiral Chromatographic Conditions. Symmetry, 11(5), 680. https://doi.org/10.3390/sym11050680