In Vitro Bioactivity and Cell Biocompatibility of a Hypereutectic Bioceramic

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.2. Ceramics Characterization
2.3. SBF Test
2.4. Biocompatibility, Adhesion, and Proliferation Assays
2.5. Cell Viability Assay
3. Results
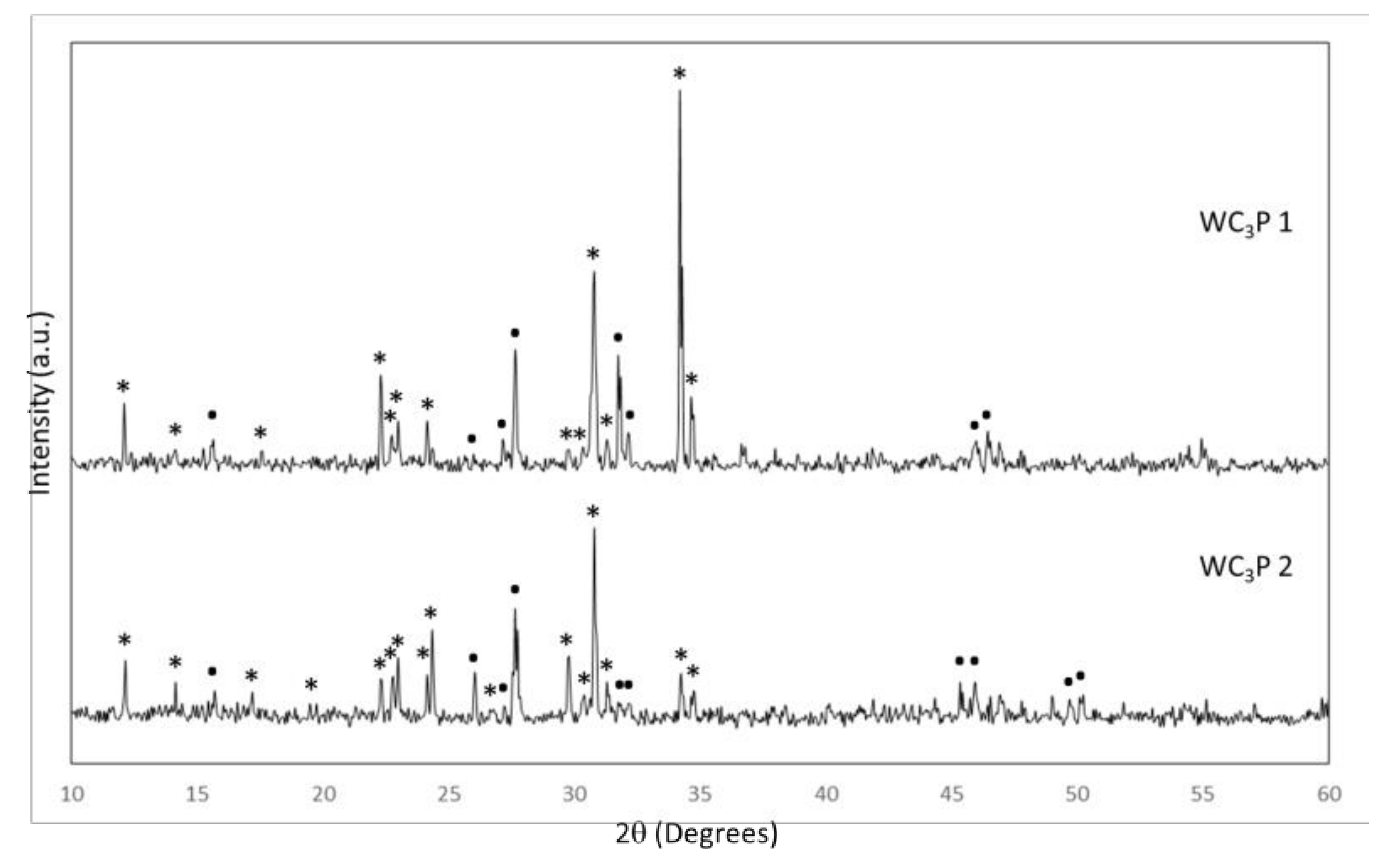
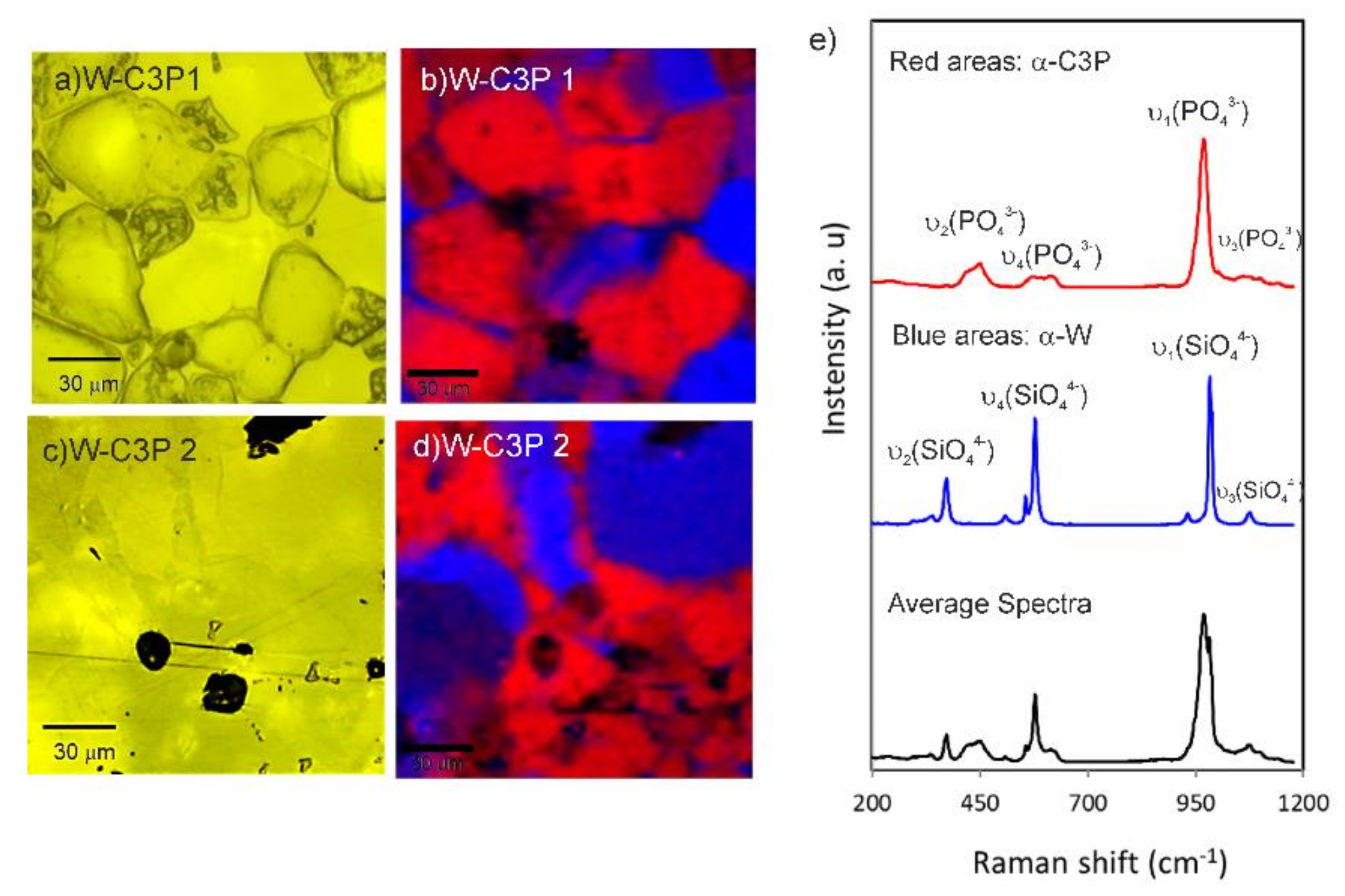
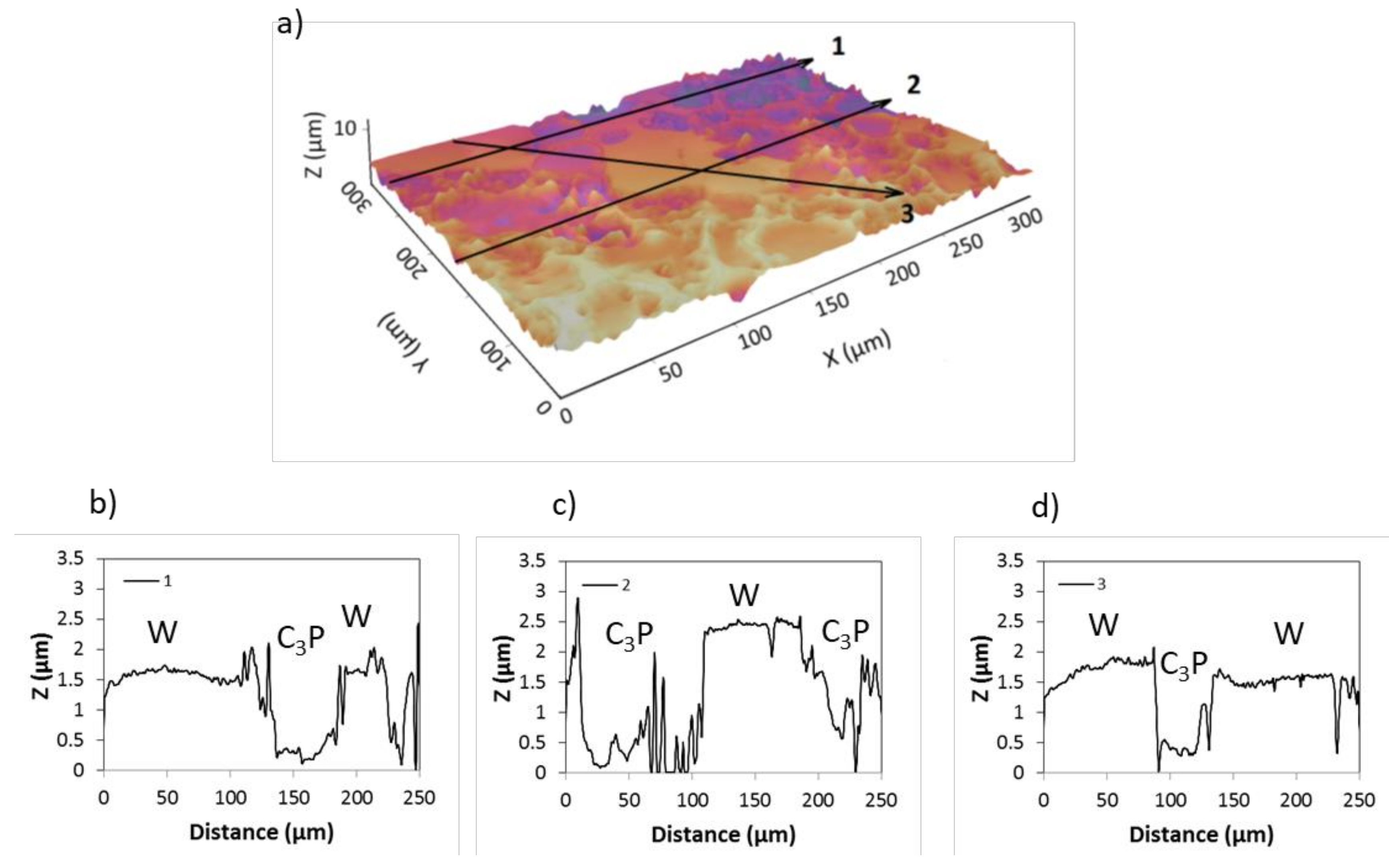
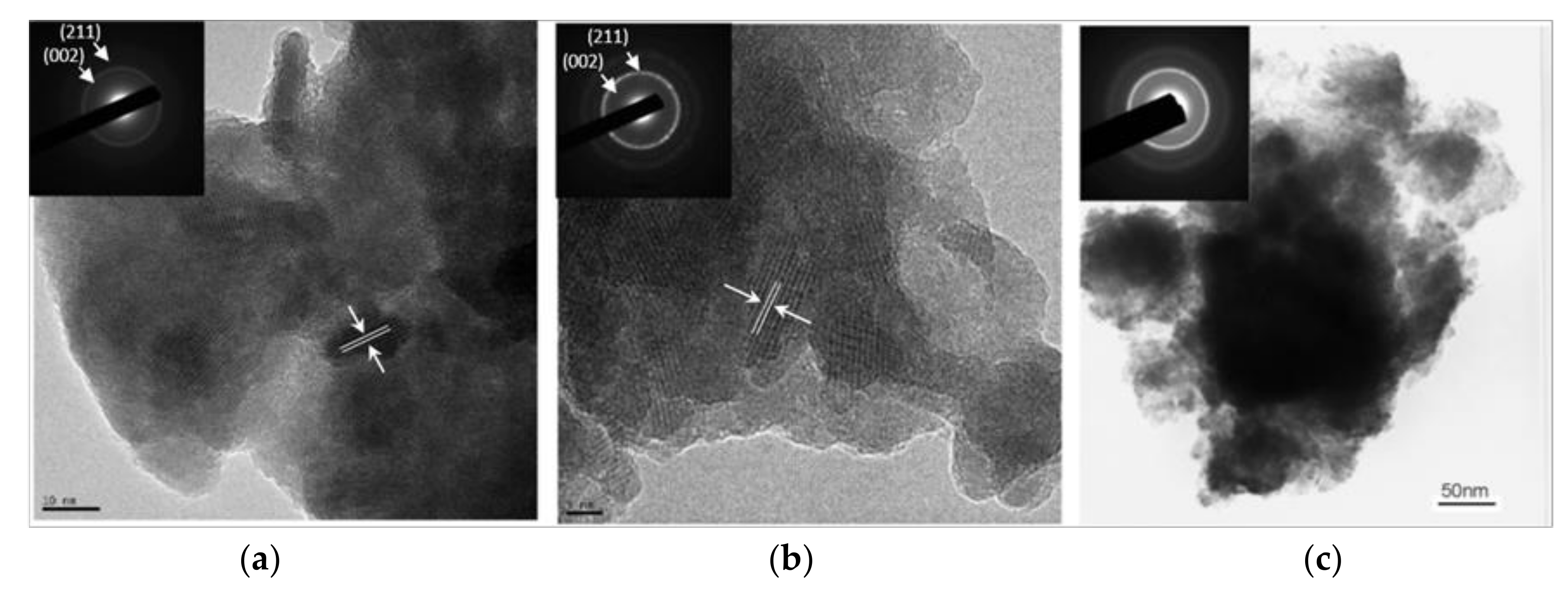
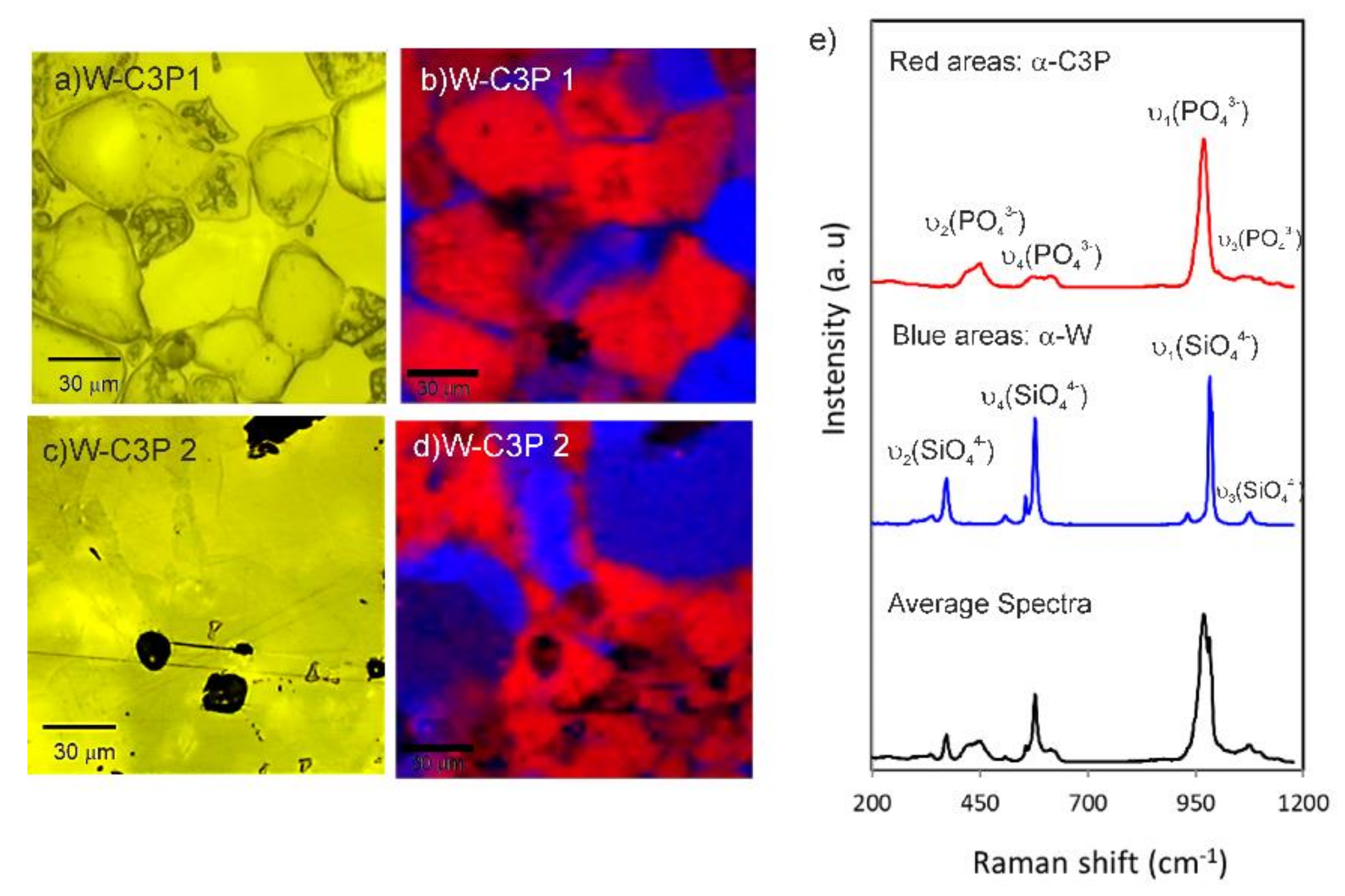
3.1. Biomaterial Characterization
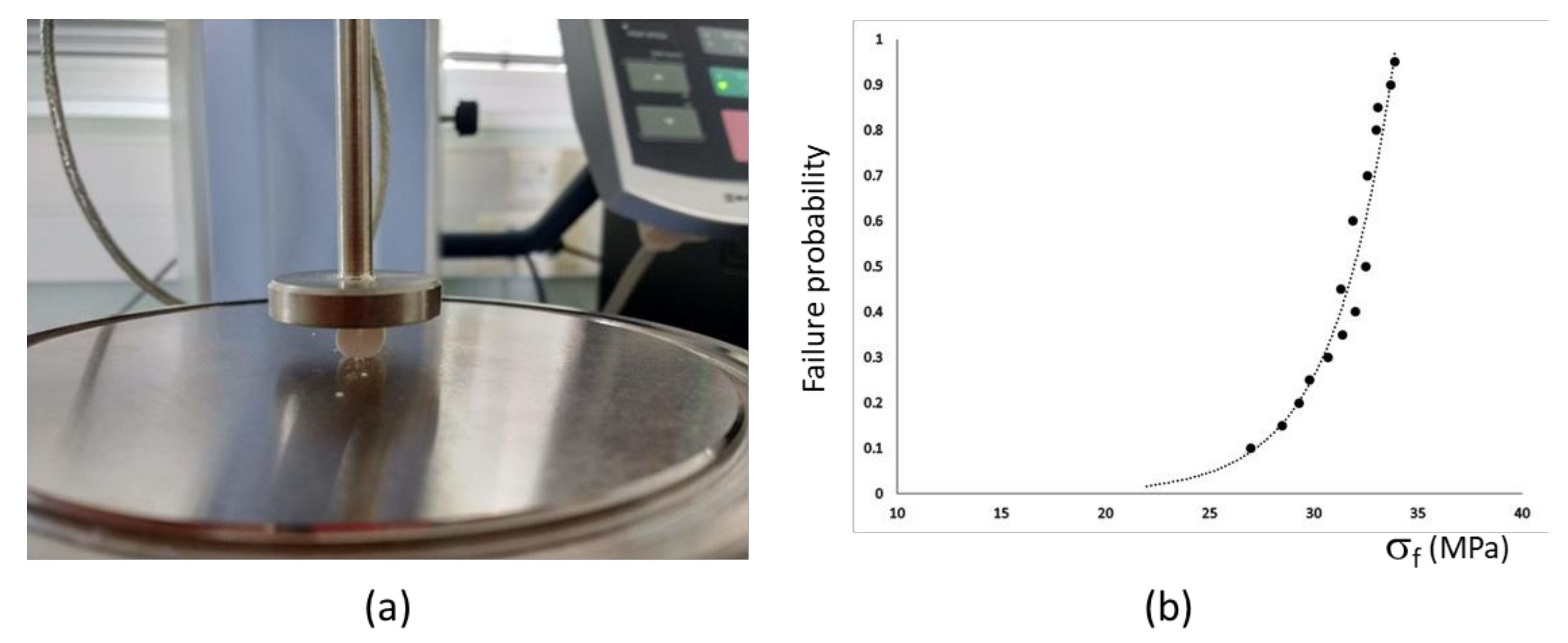
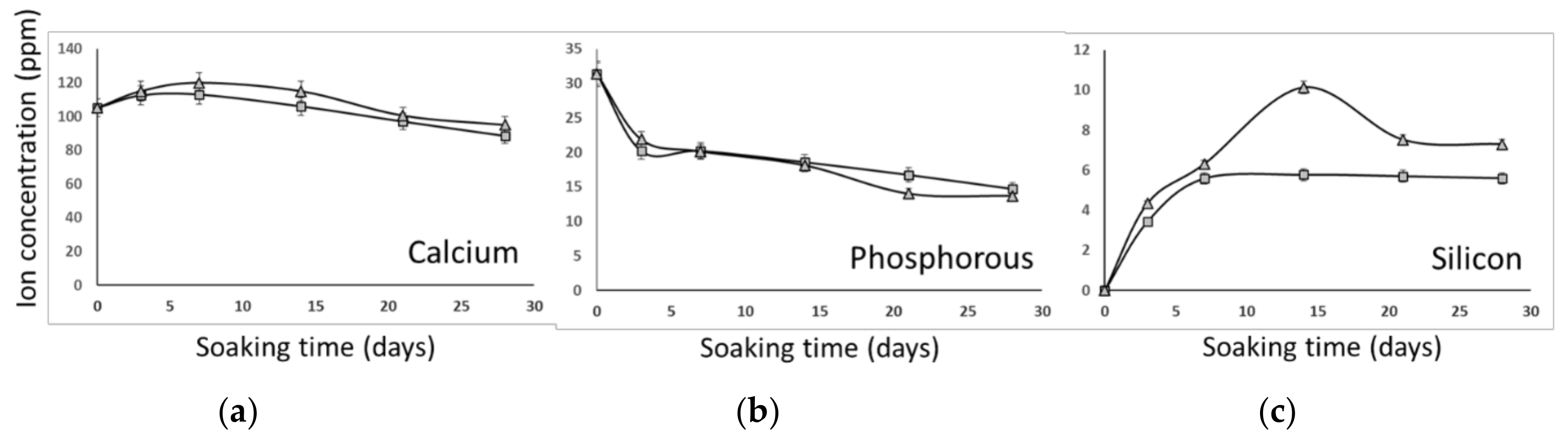
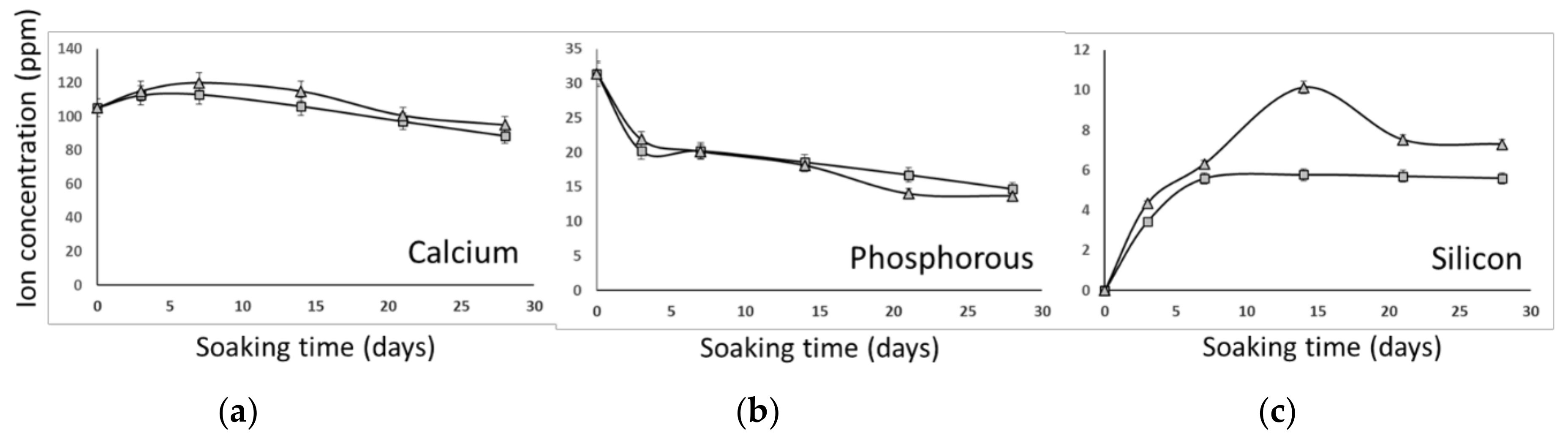
3.2. In Vitro Bioactivity
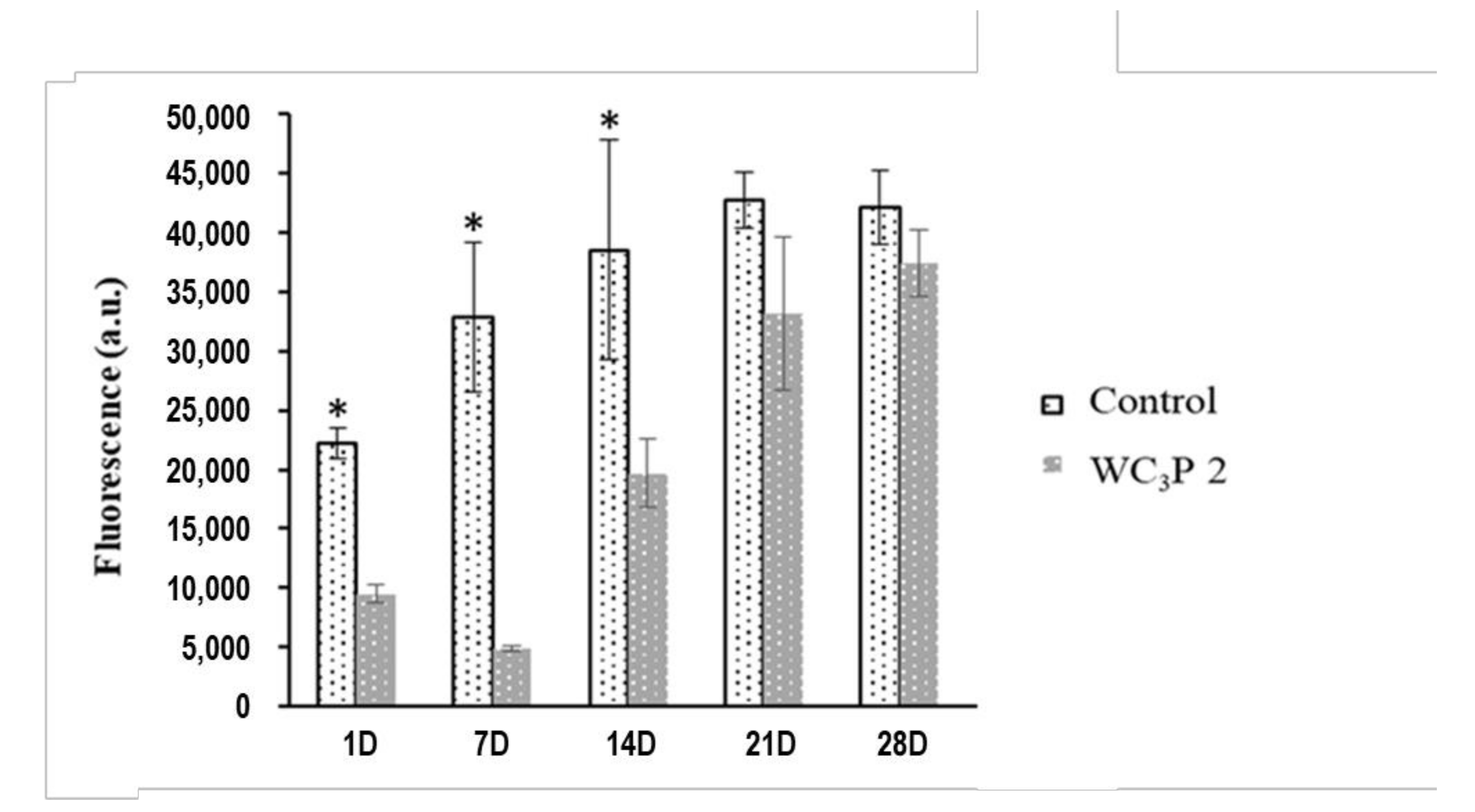
3.3. Cellular Adhesion, Morphology, and Proliferation Assays
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dorozhkin, S.V. Calcium orthophosphate bioceramics. Ceram. Int. 2015, 41, 13913–13966. [Google Scholar] [CrossRef]
- Islam, M.T.; Felfel, R.M.; Abou Neel, E.A.; Grant, D.M.; Ahmed, I.; Hossain, K.M.Z. Bioactive calcium phosphate–based glasses and ceramics and their biomedical applications: A review. J. Tissue Eng. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De la Casa-Lillo, M.A.; Velasquez, P.; De Aza, P.N. Influence of thermal treatment on the “in vitro” bioactivity of wollastonite materials. J. Mater. Sci: Mater. Med. 2011, 22, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Dashnyam, K.D.; Ahmed, E.-F.; Buitrago, J.O.; Pérez, R.A.; Knowles, J.C. ; Kim, H-W. A mini review focused on the proangiogenic role of silicate ions released from silicon-containing biomaterials. J. Tissue. Eng. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Eliaz, N.; Metoki, N. Calcium phosphate bioceramics: A review of their history, structure, properties, coating technologies and biomedical applications. Materials 2017, 3, 334. [Google Scholar] [CrossRef] [PubMed]
- Ros-Tárraga, P.; Mazón, P.; Rodríguez, M.A.; Meseguer-Olmo, L.; De Aza, P.N. Novel resorbable and osteoconductive calcium silicophosphate scaffold induced bone formation. Materials 2016, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Mazón, P.; De Aza, P.N. Porous scaffold prepared from α′L-dicalcium silicate doped with phosphorus for bone grafts. Ceram. Int. 2018, 44, 537–545. [Google Scholar] [CrossRef]
- Baino, F.; Caddeo, S.; Novajra, G.; Vitale-Brovarone, C. Using porous bioceramic scaffolds to model healthy and osteoporotic bone. J. Eur. Ceram. Soc. 2016, 36, 2175–2182. [Google Scholar] [CrossRef]
- Shao, H.F.; He, Y.; Fu, J.Z.; He, D.S.; Yang, X.Y.; Xie, J.J.; Ya, C.L.; Ye, J.; Xu, S.Z.; Gou, Z.R. 3D printing magnesium-doped wollastonite/beta-TCP bioceramics scaffolds with high strength and adjustable degradation. J. Eur. Ceram. Soc. 2016, 36, 1495–1503. [Google Scholar] [CrossRef]
- Itala, A.I.; Ylanen, H.O.; Ekholm, C.; Karlsson, K.H.; Aro, H.T. Pore diameter of more than 100 m is not requisite for bone ingrowth in rabbits. J. Biomed. Mat. Res. 2001, 58, 679–683. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 2005, 26, 5475–5491. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.A.; Mestres, G. Role of pore size and morphology in musculo-eskeletal tissue engine. Mat. Sci. Eng. C 2016, 61, 922–939. [Google Scholar] [CrossRef] [PubMed]
- De Aza, P.N.; Guitian, F.; De Aza, S. A new bioactive material, which transforms in situ into hydroxyapatite. Acta Mater. 1998, 46, 2541–2549. [Google Scholar] [CrossRef]
- De Aza, A.H.; Velásquez, P.; Alemany, M.I.; Pena, P.; De Aza, P.N. In situ bone-like apatite formation from a bioeutectic® ceramic in SBF dynamic flow. J. Am. Ceram. Soc. 2007, 90, 1200–1207. [Google Scholar] [CrossRef]
- De Aza, P.N.; Peña, J.I.; Luklinska, Z.B.; Meseguer-Olmo, L. Bioeutectic® ceramics for biomedical application obtained by laser floating zone method. In vivo evaluation. Materials 2014, 7, 2345–2410. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Chang, J.; Chou, L.; Zhai, W. Comparison of osteoblast-like cell responses to calcium silicate and tricalcium phosphate ceramics in vitro. J. Biomed. Mater. Res. B Appl. Biomater. 2007, 80, 174–183. [Google Scholar] [CrossRef] [PubMed]
- De Aza, P.N.; Guitian, F.; De Aza, S.; Valle, F.J. Analytical control of wollastonite for biomedical applications by use of atomic absorption spectrometry and inductively coupled plasma atomic emission spectrometry. Analyst 1998, 123, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Magallanes-Perdomo, M.; Pena, P.; De Aza, P.N.; Carrodeguas, R.G.; Rodríguez, M.A.; Turrillas, X.; De Aza, S.; De Aza, P.N. Devitrification studies of wollastonite-tricalcium phosphate eutectic glass. Acta Biomater. 2009, 5, 3057–3066. [Google Scholar] [CrossRef]
- Weibull, W. A statistical distribution function of wide applicability. J. Appl. Mech. 1951, 18, 292–297. [Google Scholar]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef]
- Meseguer-Olmo, L.; Aznar-Cervantes, S.; Mazón, P.; De Aza, P.N. In vitro behavior of adult mesenchymal stem cells of human bone marrow origin seeded on a novel bioactive ceramics in the Ca2SiO4-Ca3(PO4)2 system. J. Mater. Sci: Mater. Med. 2012, 23, 3003–30014. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Richet, P.; Mysen, B.O.; Ingrin, J. High-temperature X-ray diffraction and raman spectroscopy, of diopside and pseudowollastonite. Phys. Chem. Miner. 1998, 25, 401–414. [Google Scholar] [CrossRef]
- Pešková, Š.; Machovič, V.; Procházka, P. Raman spectroscopy structural study of fired concrete. Ceram. Silik. 2011, 55, 410–417. [Google Scholar]
- De Aza, P.N.; Santos, C.; Pazo, A.; De Aza, S.; Cusco, R.; Artus, L. Vibrational properties of calcium phosphate compounds 1. Raman spectrum of α-tricalcium phosphate. Chem. Mater. 1997, 9, 912–915. [Google Scholar] [CrossRef]
- Jillavenkatesa, A.; Condrate, R.A. The infrared and Raman spectra of β-and α-tricalcium phosphate (Ca3(PO4)2). Spectrosc. Lett. 1998, 31, 1619–1634. [Google Scholar] [CrossRef]
- De Aza, P.N.; Guitian, F.; De Aza, S. Phase-Diagram of wollastonite-tricalcium phosphate J. Am. Ceram. Soc. 1995, 78, 1653–1656. [Google Scholar] [CrossRef]
- Carrodeguas, R.G.; De Aza, A.H.; De Aza, P.N.; Baudín, C.; Jiménez, J.; López-Bravo, A.; Pena, P.; De Aza, S. Assesment of natural and synthetic wollasonite as source for bioceramics preparation. J. Biomed. Mat. Res. Part. A 2007, 83, 484–495. [Google Scholar] [CrossRef]
- Spector, M. High-resolution electron-microscope study of lattice images in biological apatites, J. Microsc. 1975, 103, 55–62. [Google Scholar] [CrossRef]
- De Aza, P.N.; Luklinska, Z.B.; Anseau, M.; Guitian, F.; De Aza, S. Electron microscopy of interfaces in a wollastonite-tricalcium phosphate Bioeutectic. J. Microsc. 1998, 189, 145–153. [Google Scholar] [CrossRef]
- Kerebel, B.; Daculsi, G.; Verbaere, A. High-resolution electron-microscopy and crystallographic study of some biological apatites. J. Ultrastruct. Res. 1976, 57, 266–275. [Google Scholar] [CrossRef]
- Minarelli-Gaspar, A.M.; Saska, S.; Da Cunha, L.R.; Bolini, P.D.A.; Carrodeguas, R.G.; De Aza, A.H.; Pena, P.; De Aza, P.N.; De Aza, S. Comparison of the biological behavior of wollastonite bioceramics prepared from synthetic and natural precursors. Key Eng. Mater. 2008, 361–363, 1083–1086. [Google Scholar]
- Martínez, I.; Velásquez, P.; Meseguer-Olmo, L.; De Aza, P.N. Production and study of in vitro behavior of monolithic α-tricalcium phosphate based ceramics in the system Ca3(PO4)2–Ca2SiO4. Ceram. Int. 2011, 37, 2527–2535. [Google Scholar] [CrossRef]
- Velásquez, P.; Luklinska, Z.B.; Meseguer-Olmo, L.; Mate-Sánchez de Val, J.E.; Delgado-Ruíz, R.A.; Calvo-Guirado, J.L.; Ramírez-Fernández, M.P.; De Aza, P.N. αTCP ceramic doped with dicalcium silicate for bone regeneration applications prepared by powder metallurgy method: In vitro and in vivo studies. J. Biomed. Mater. Res. Part A 2013, 101, 1943–1954. [Google Scholar] [CrossRef] [PubMed]
- Alemany, M.I.; Velásquez, P.; De la Casa-Lillo, M.A.; De Aza, P.N. Effect of materials’ processing methods on the in vitro bioactivity of wollastonite glass-ceramic materials J. Non-Cryst. Sol. 2005, 351, 1716–1726. [Google Scholar] [CrossRef]
- Carrodeguas, R.G.; De Aza, A.H.; Jimenez, J.; De Aza, P.N.; Pena, P.; López-Bravo, A.; De Aza, S. Preparation and in vitro characterization of wollastonite doped tricalcium phosphate bioceramics. Key Eng. Mater. 2008, 361–363, 237–240. [Google Scholar]
- Bauer, G.; Hohenberger, G. Causes of behavioral variation of bioactive calcium phosphate ceramics in living organism. Ber. Dtsch. Chem. Ges. 1989, 66, 23–27. [Google Scholar]
- Ibáñez, A. Síntesis de Wollastonita a Partir de Diatomitas Españolas y Estudio de su Aplicación en Revestimientos Cerámicos Porosos Procesados por Cocción Rápida. Ph.D. Thesis, Universidad Autónoma de Madrid, Madrid, Spain, 1993. [Google Scholar]
- Black, J.; Hastings, G. Handbook of Biomaterials Properties; Champan & Hall: Boca Raton, Fl, USA, 1998. [Google Scholar]
- Mazón, P.; García-Bernal, D.; Meseguer-Olmo, L.; Cragnolini, F.; De Aza, P.N. Human mesenchymal stem cell viability, proliferation and differentiation potential in response to ceramic chemistry and surface roughness. Ceram. Int. 2015, 44, 6631–6644. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | W-C3P 1 | W-C3P 2 | ||||
|---|---|---|---|---|---|---|
| Temperature (°C) | 1325 | 1350 | 1375 | 1325 | 1350 | 1375 |
| Shrinkage (%) | 15.3 ± 0.5 | 18.4 ± 0.5 | 22.1 ± 0.5 | 18.7 ± 0.5 | 19.2 ± 0.5 | 23.0 ± 0.5 |
| Apparent density (gr/cm3) | 2.55 ± 0.05 | 2.71 ± 0.05 | 3.10 ± 0.05 | 2.58 ± 0.05 | 2.83 ± 0.05 | 3.17 ± 0.05 |
| Porosity (%) | 3.7 ± 0.2 | 2.6 ± 0.2 | 1.5 ± 0.2 | 3.4 ± 0.2 | 2.8 ± 0.2 | 1.8 ± 0.2 |
| Atomic % | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| C3P | C3P | C3P | C3P | C3P | C3P | W | W | W | W | |
| Si | 0.95 | 1.15 | 0.80 | 1.01 | 1.19 | 0.99 | 48.84 | 49.07 | 49.24 | 49.16 |
| P | 38.86 | 38.96 | 38.17 | 37.95 | 37.96 | 38.64 | 0.04 | 0.08 | 0.23 | 0.12 |
| Ca | 60.19 | 59.89 | 61.03 | 61.04 | 60.85 | 60.37 | 51.12 | 50.85 | 50.53 | 50.72 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazón, P.; Ros-Tárraga, P.; Serena, S.; Meseguer-Olmo, L.; De Aza, P.N. In Vitro Bioactivity and Cell Biocompatibility of a Hypereutectic Bioceramic. Symmetry 2019, 11, 355. https://doi.org/10.3390/sym11030355
Mazón P, Ros-Tárraga P, Serena S, Meseguer-Olmo L, De Aza PN. In Vitro Bioactivity and Cell Biocompatibility of a Hypereutectic Bioceramic. Symmetry. 2019; 11(3):355. https://doi.org/10.3390/sym11030355
Chicago/Turabian StyleMazón, Patricia, Patricia Ros-Tárraga, Sara Serena, Luis Meseguer-Olmo, and Piedad N. De Aza. 2019. "In Vitro Bioactivity and Cell Biocompatibility of a Hypereutectic Bioceramic" Symmetry 11, no. 3: 355. https://doi.org/10.3390/sym11030355
APA StyleMazón, P., Ros-Tárraga, P., Serena, S., Meseguer-Olmo, L., & De Aza, P. N. (2019). In Vitro Bioactivity and Cell Biocompatibility of a Hypereutectic Bioceramic. Symmetry, 11(3), 355. https://doi.org/10.3390/sym11030355