Symmetry and Dissymmetry in Protein Structure—System-Coding Its Biological Specificity



Abstract
:1. Introduction
2. Materials and Methods
2.1. Proteins Subjected to Analysis
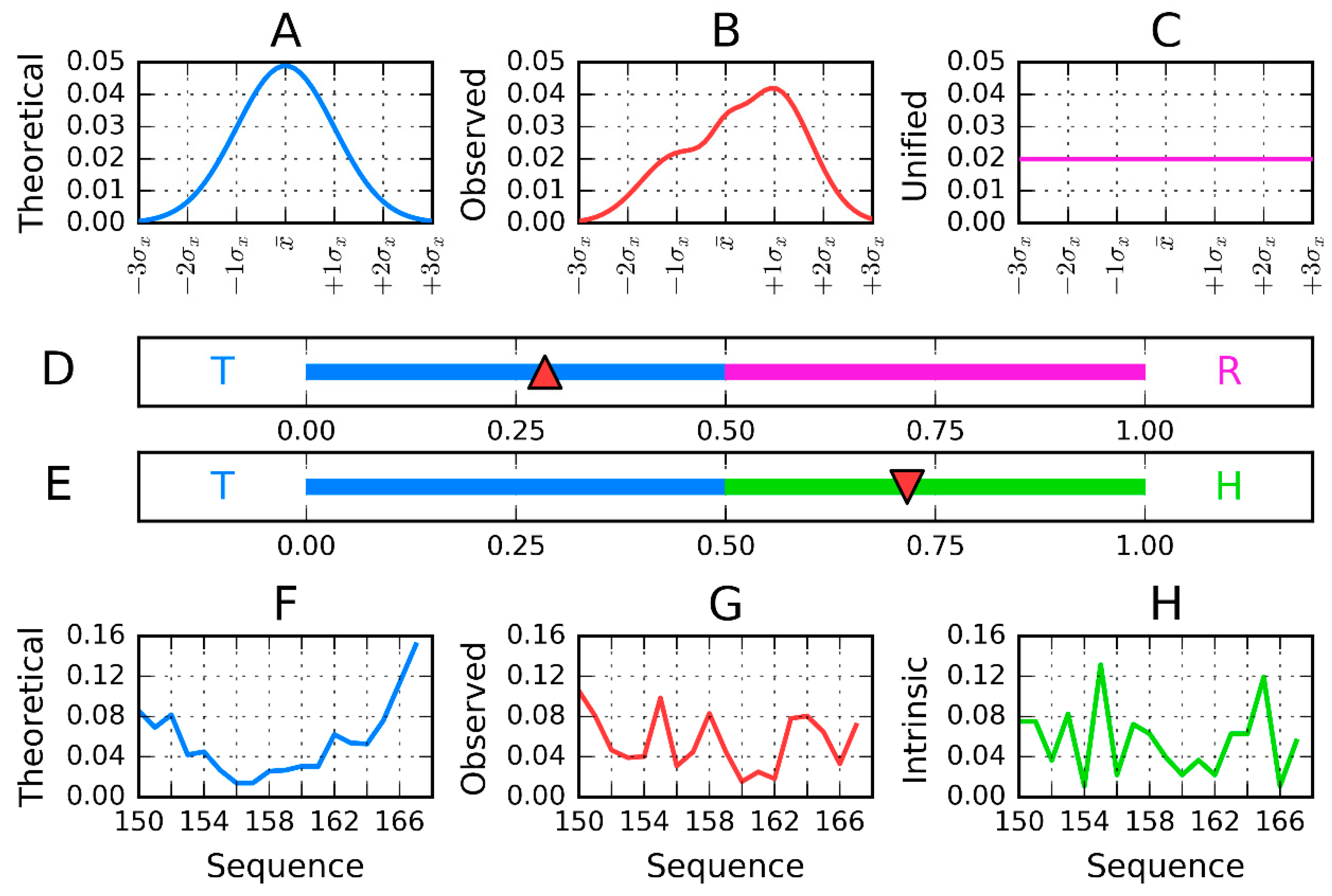
2.2. Model Description
2.3. Calculation Procedure
- the representation of the molecule with X, Y, Z coordinates (as available in PDB) given for each atom is changed to the effective atom representation. Effective atom—position of averaged coordinates of atoms belonging to certain amino acids;
- the molecule under consideration shall be oriented in coordinate system as follows:A—the geometric center localized in origin of coordinate system;B—the X-axis oriented according to two most distant effective atoms in molecule;C—the Y-axis oriented according to most distant effective atoms—distance limited to Y–Z-plane;
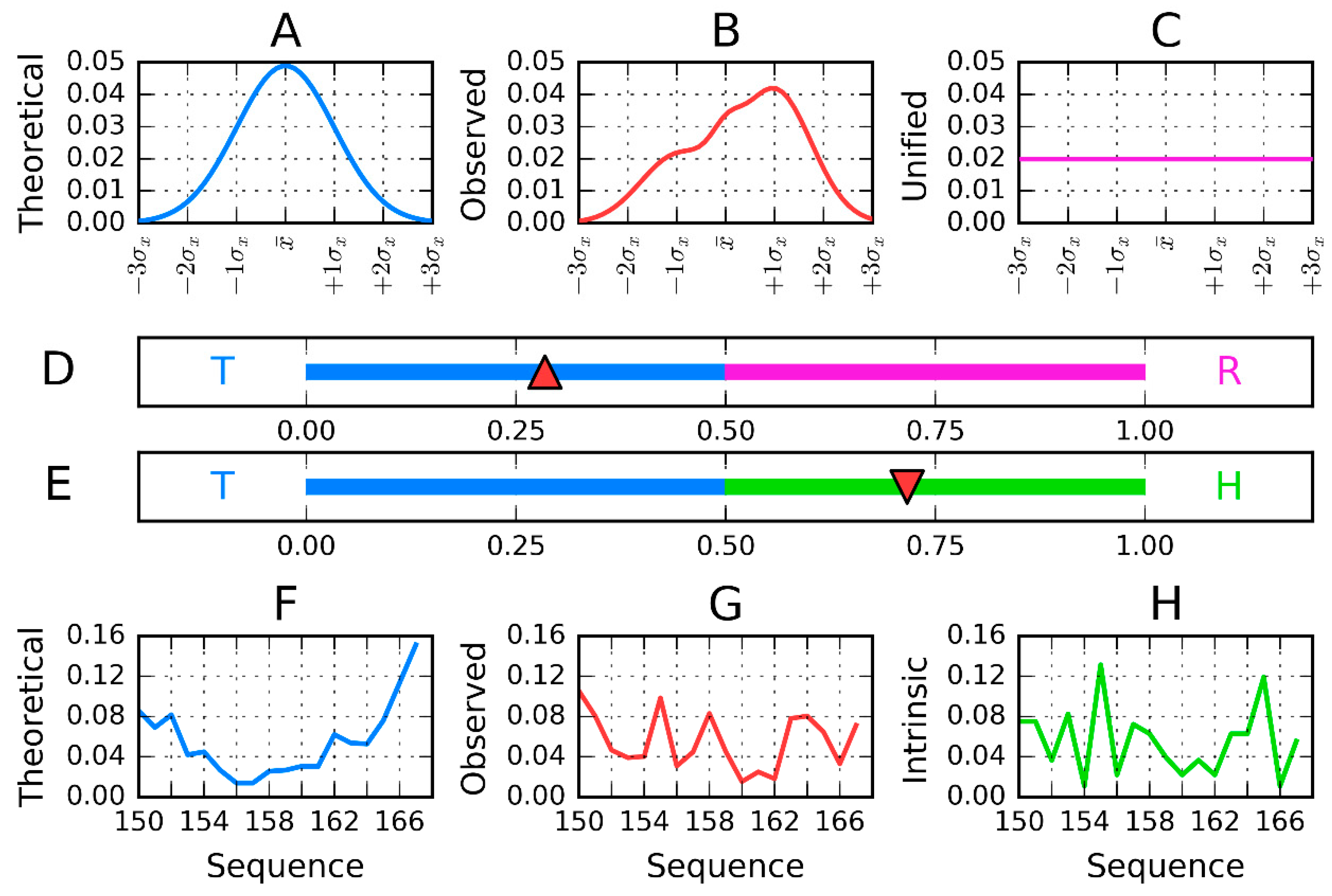
- the T-distribution–sigma parameters calculated as 1/6 of largest distance along each axis independently (according to 3-sigma rule for normal distribution);
- the second parameter (position of maximum) is calculated as a mean value of effective atoms positions;
- the Ti values are calculated as values of the 3D Gauss function in the position of each effective atom;
- the Oi values express the hydrophobic interaction between effective atoms—thus its value depends on the distance between two interacting residues and their intrinsic hydrophobicity taken according to the selected scale. The scale presented in [10] was taken in our calculations. The function expressing this interaction was taken according to Levitt function [32].
- Both T and O distribution shall be normalized—sum of all Ti and Oi values shall be equal to 1.0. This operation allows the comparison of T and O distributions;
- The Kullback Leibler divergence entropy (DKL) is applied to measure the difference between two profiles T and O [33]. Since the value of DKL entropy cannot be interpreted (entropy category) the second reference distribution is defined (Figure 1)—the unified one with Ri values for all residues equal to 1/N. The comparison of O distribution versus two reference distributions T and R may suggest the similarity to one of them. The closeness to one of them is expressed by RD parameter, the calculation of which is described above.
2.4. Antifreeze and Amyloids Versus Lyases
3. Results
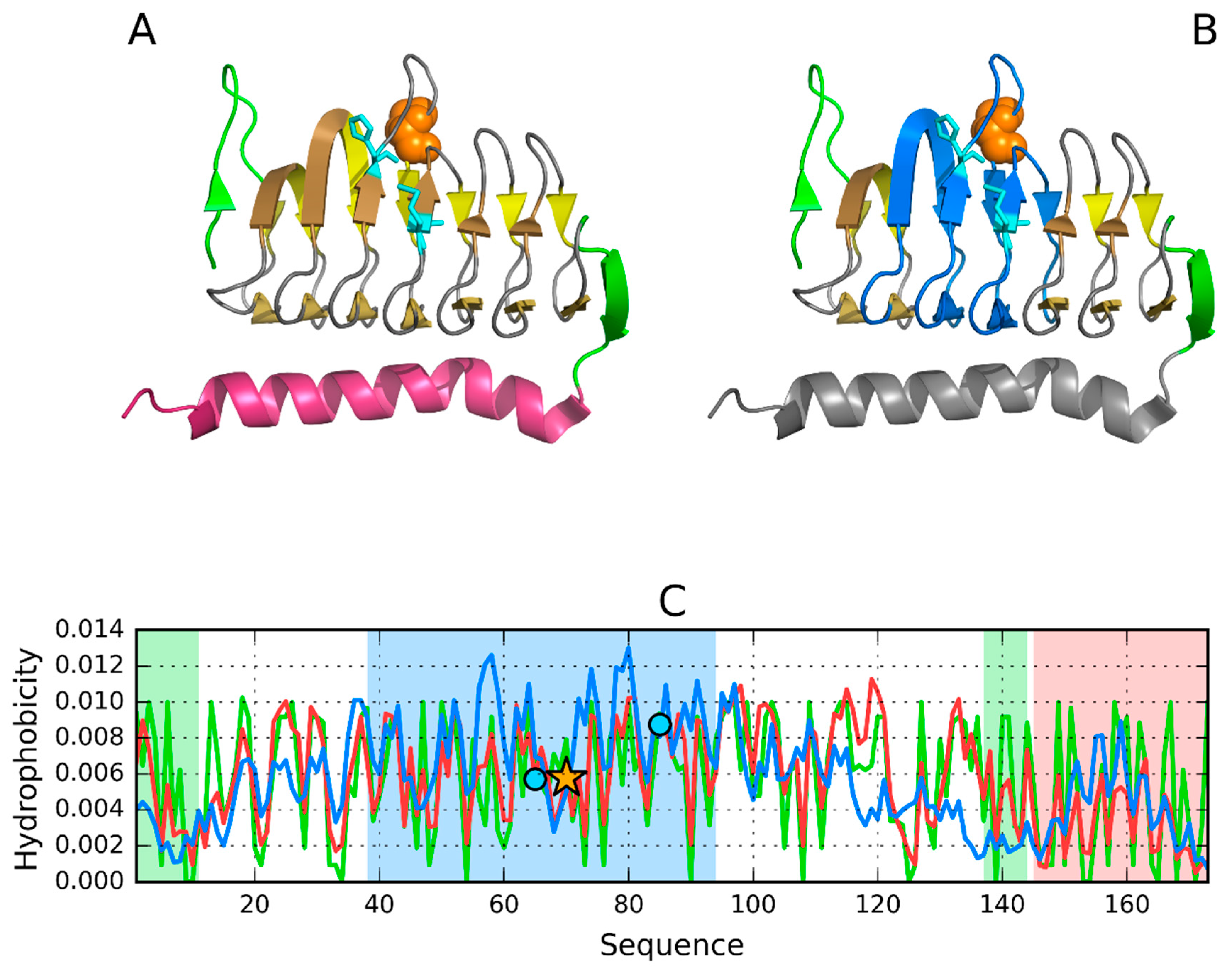
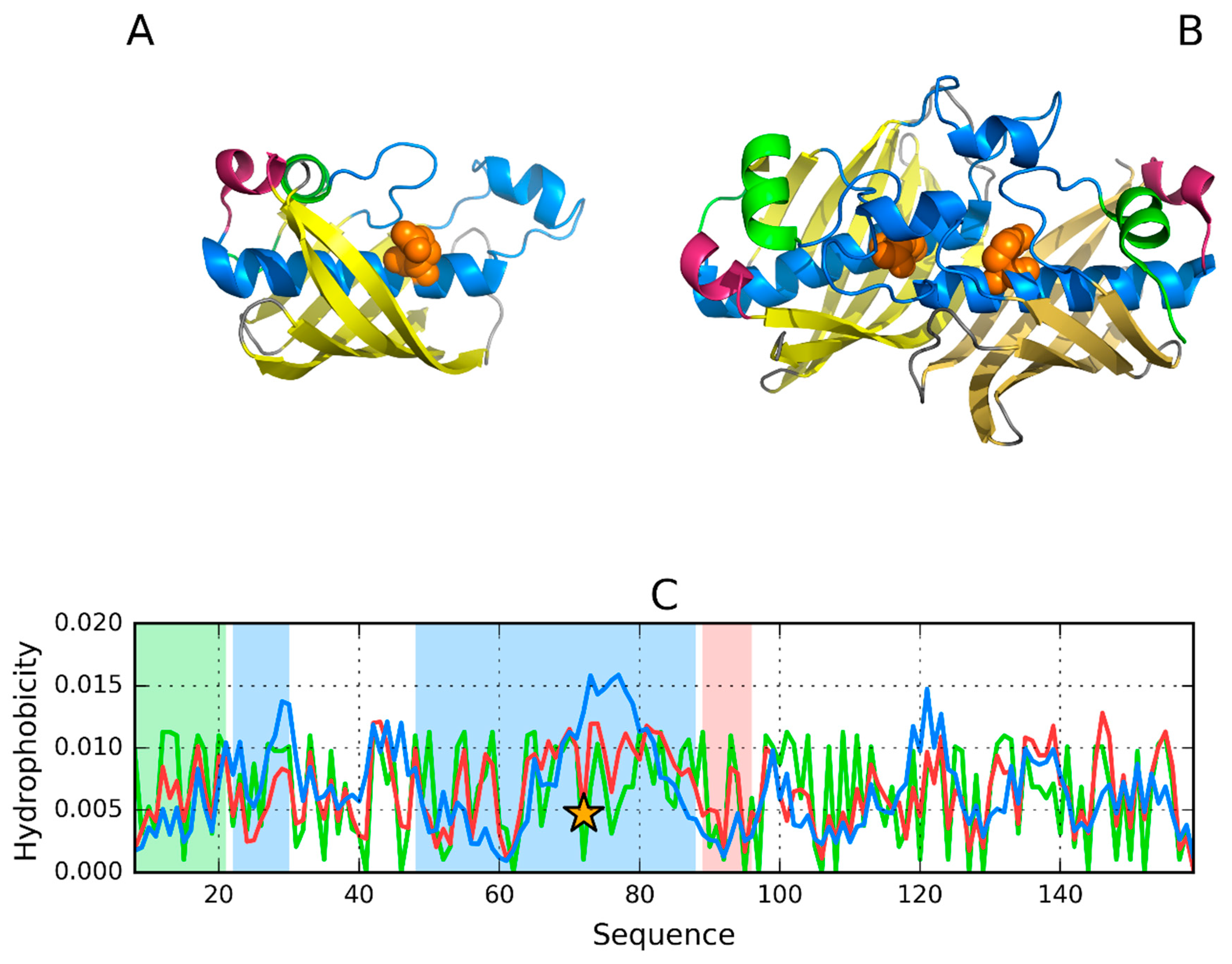
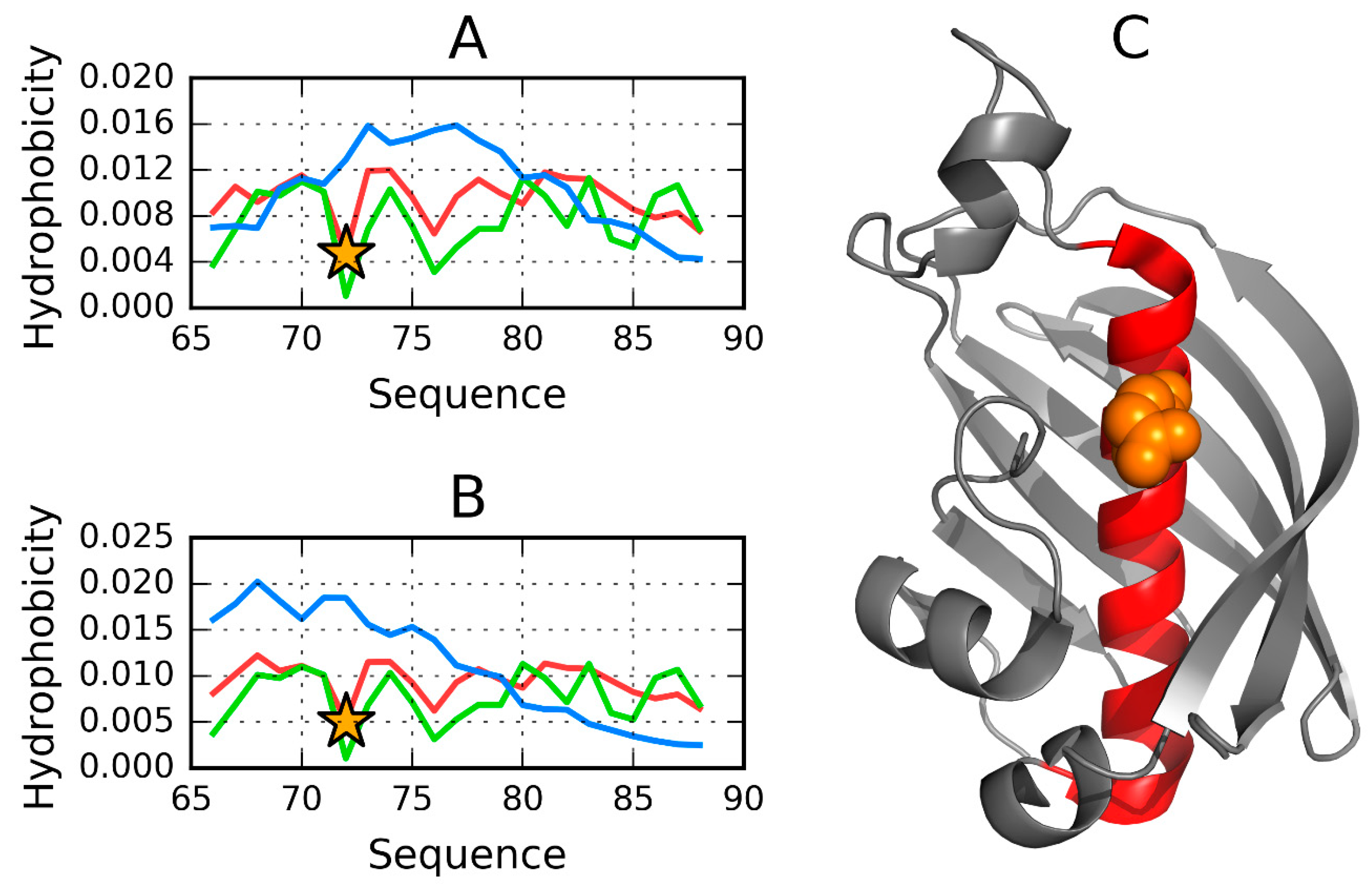
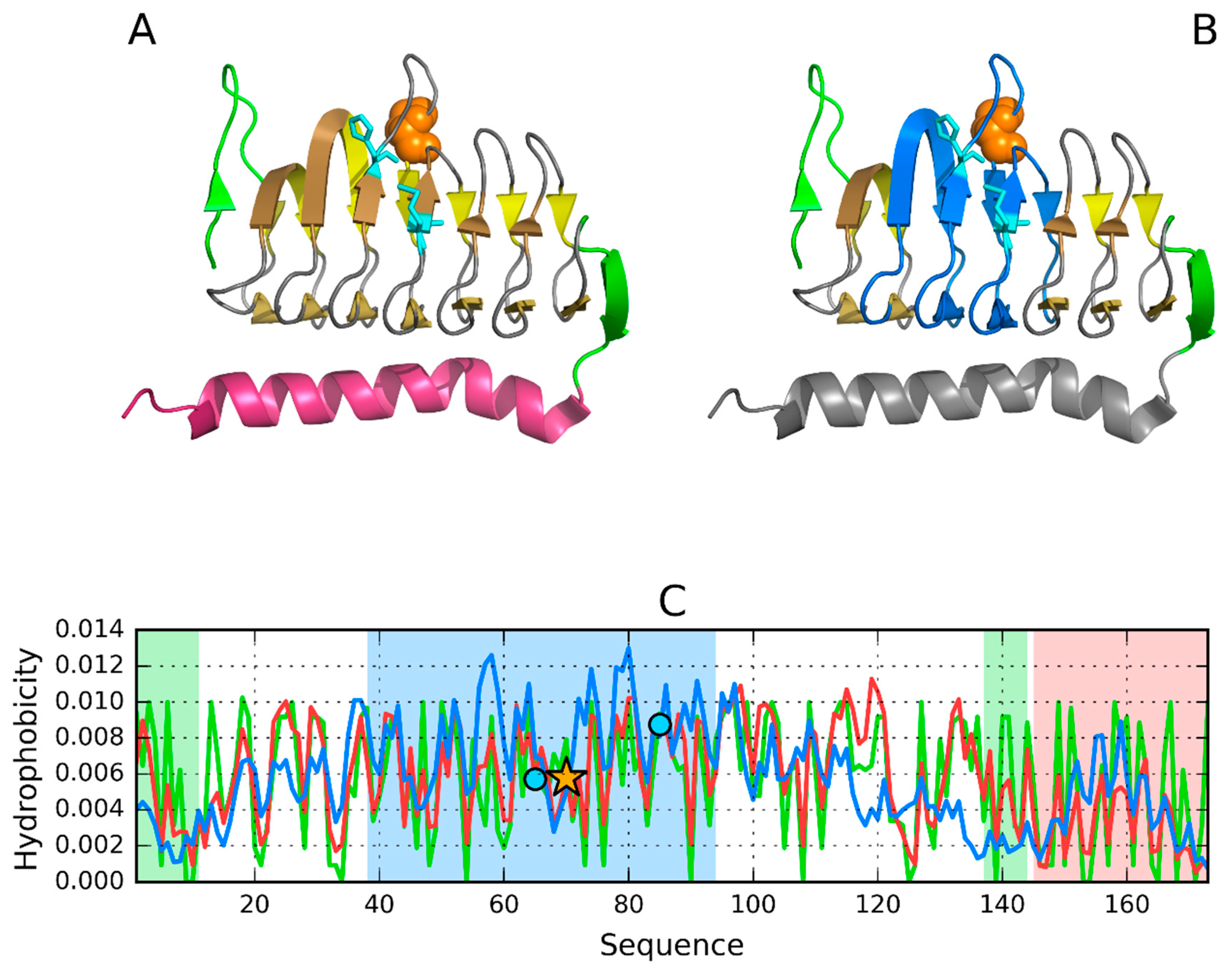
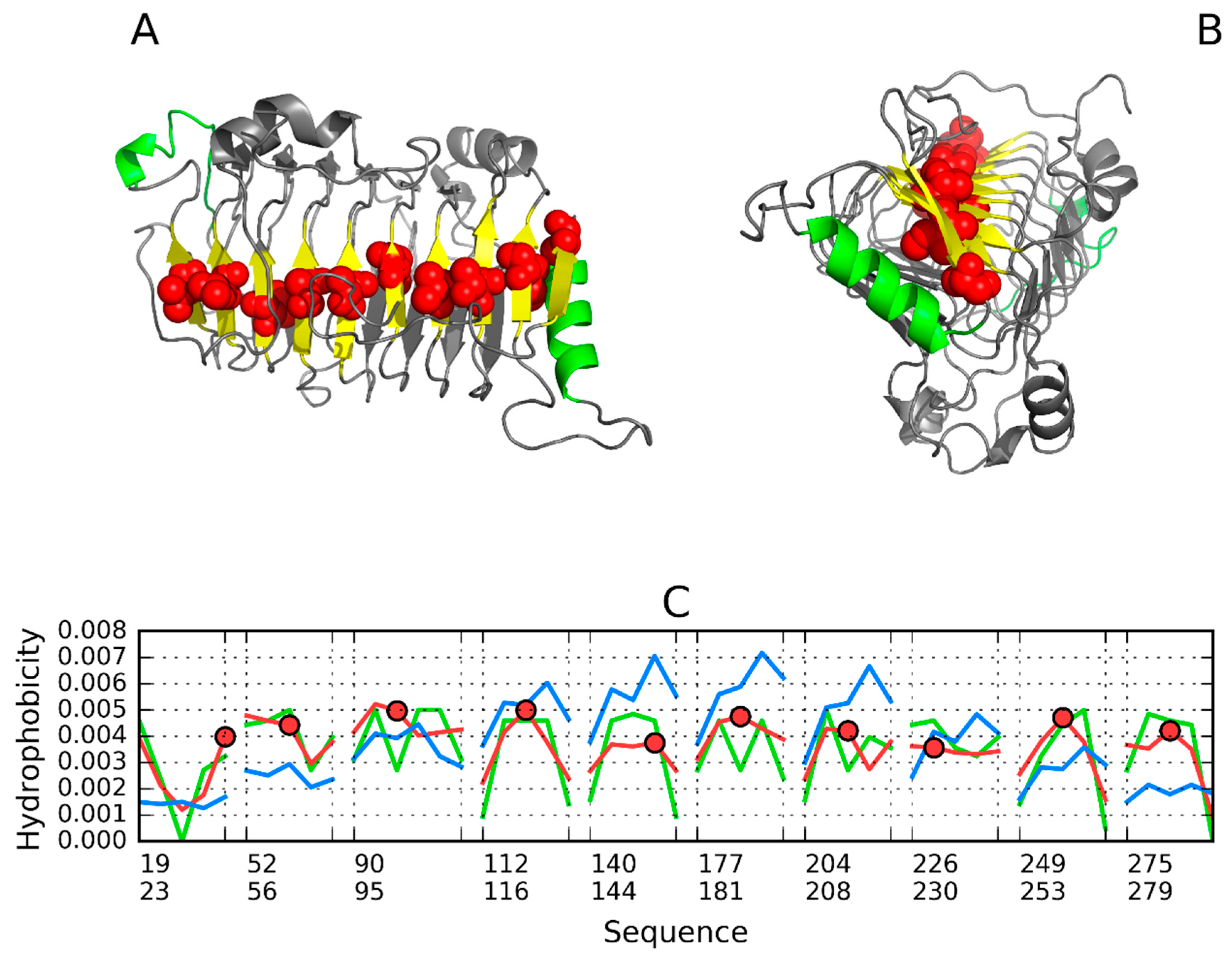
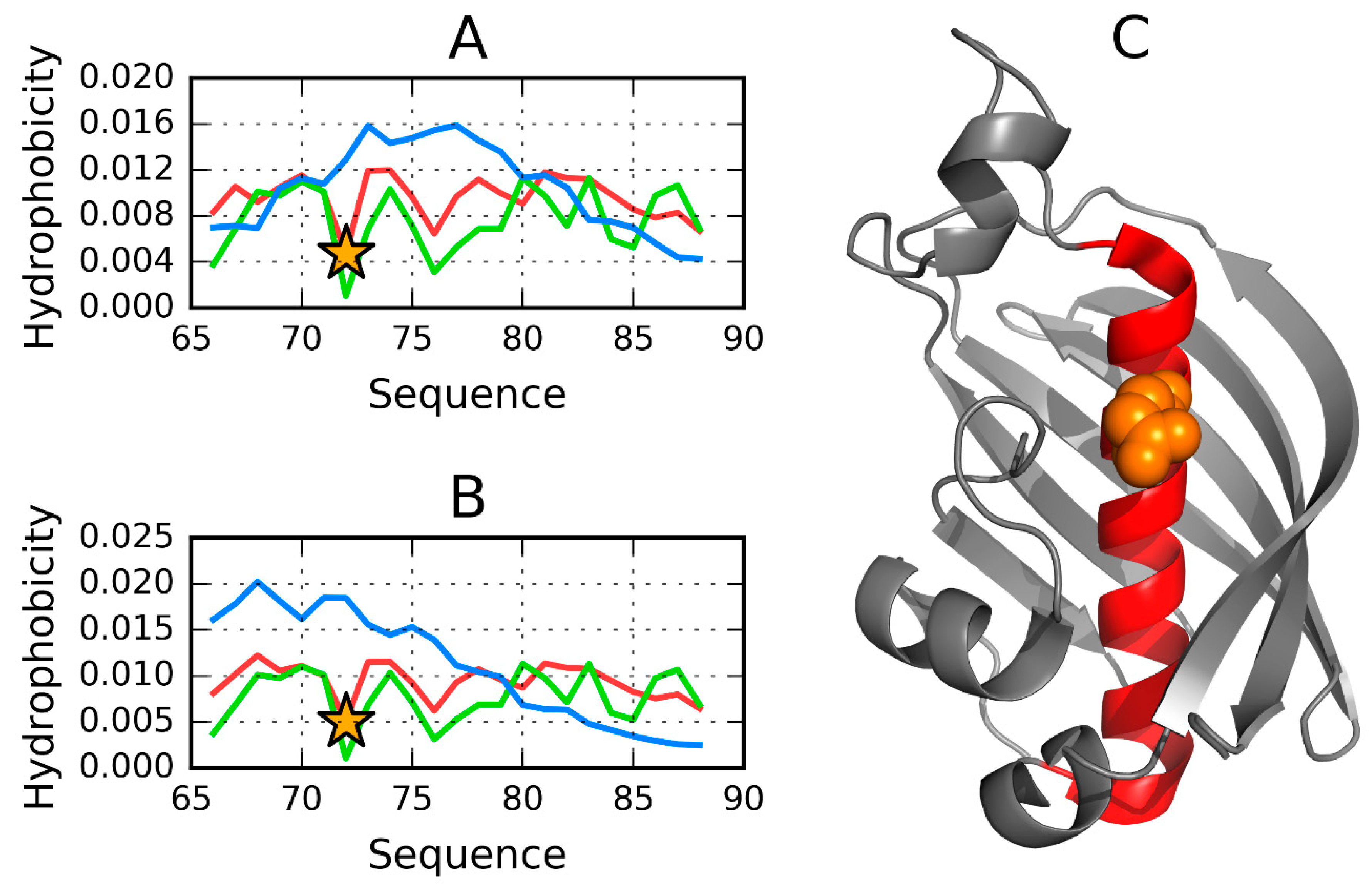
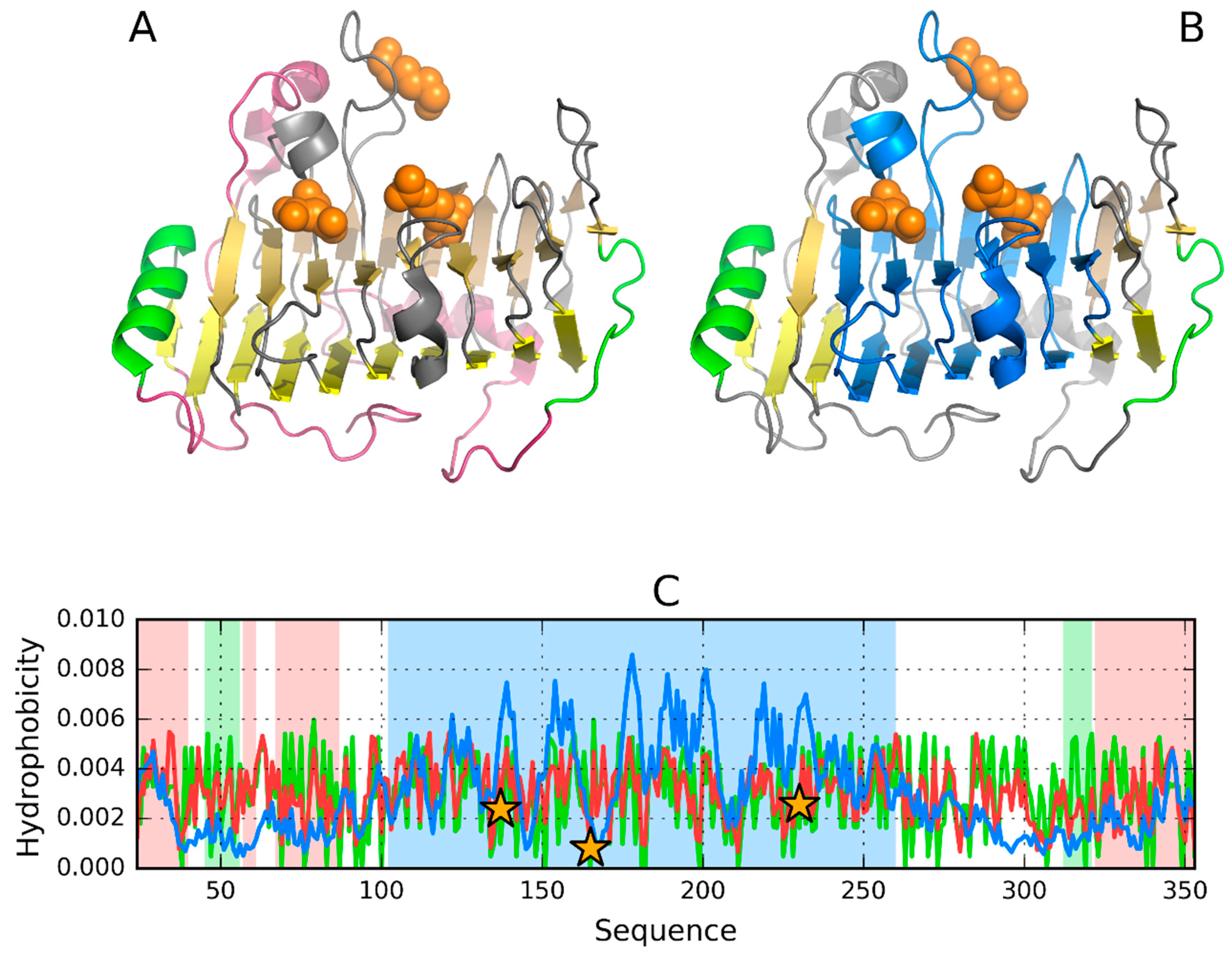
3.1. 2FKO—Representative of EC 4.2.1.1
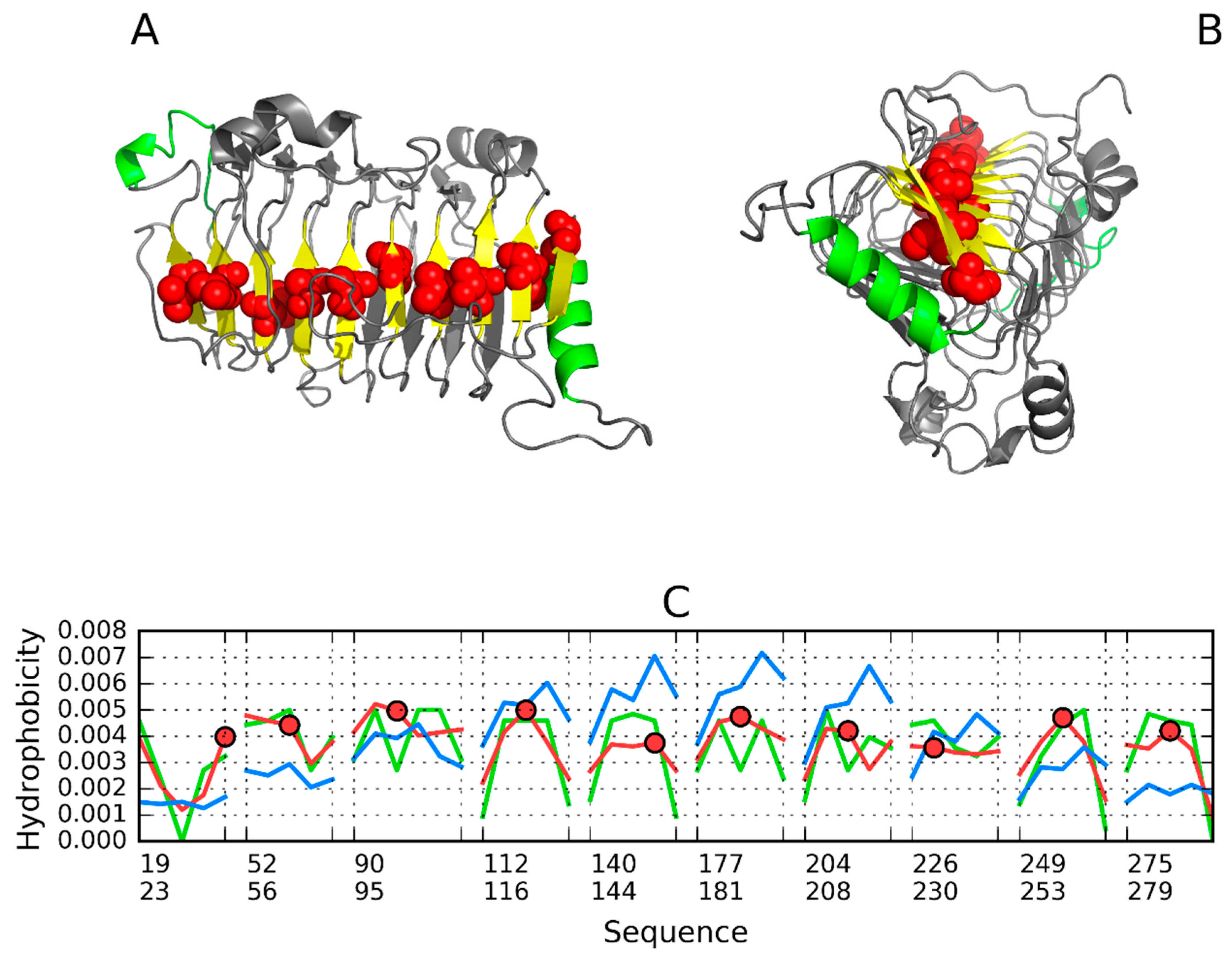
3.2. 1PLU—Representative of EC 4.2.2.2
3.3. Pectin Lyase (1IDJ)—Representative of EC 4.2.2.10
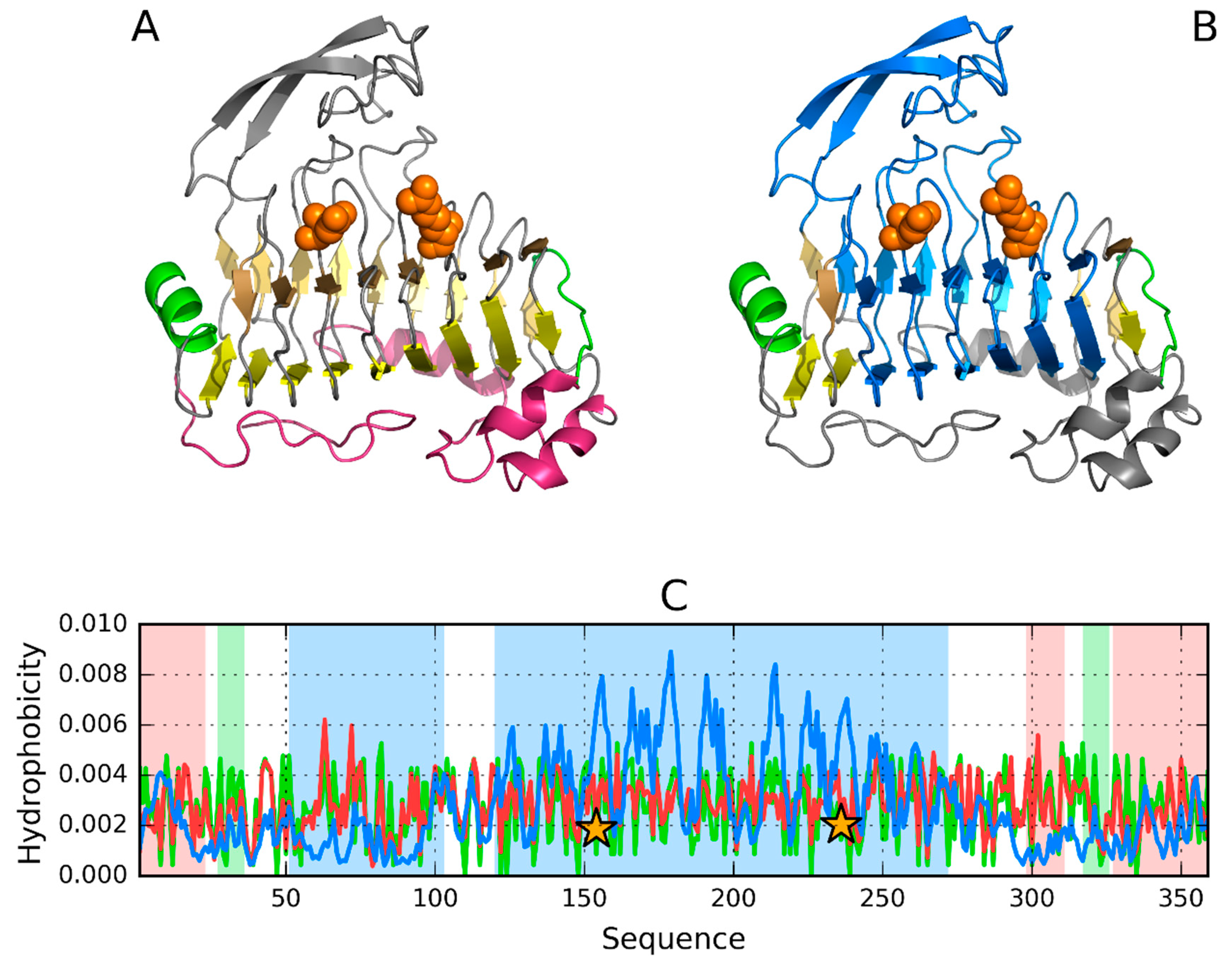
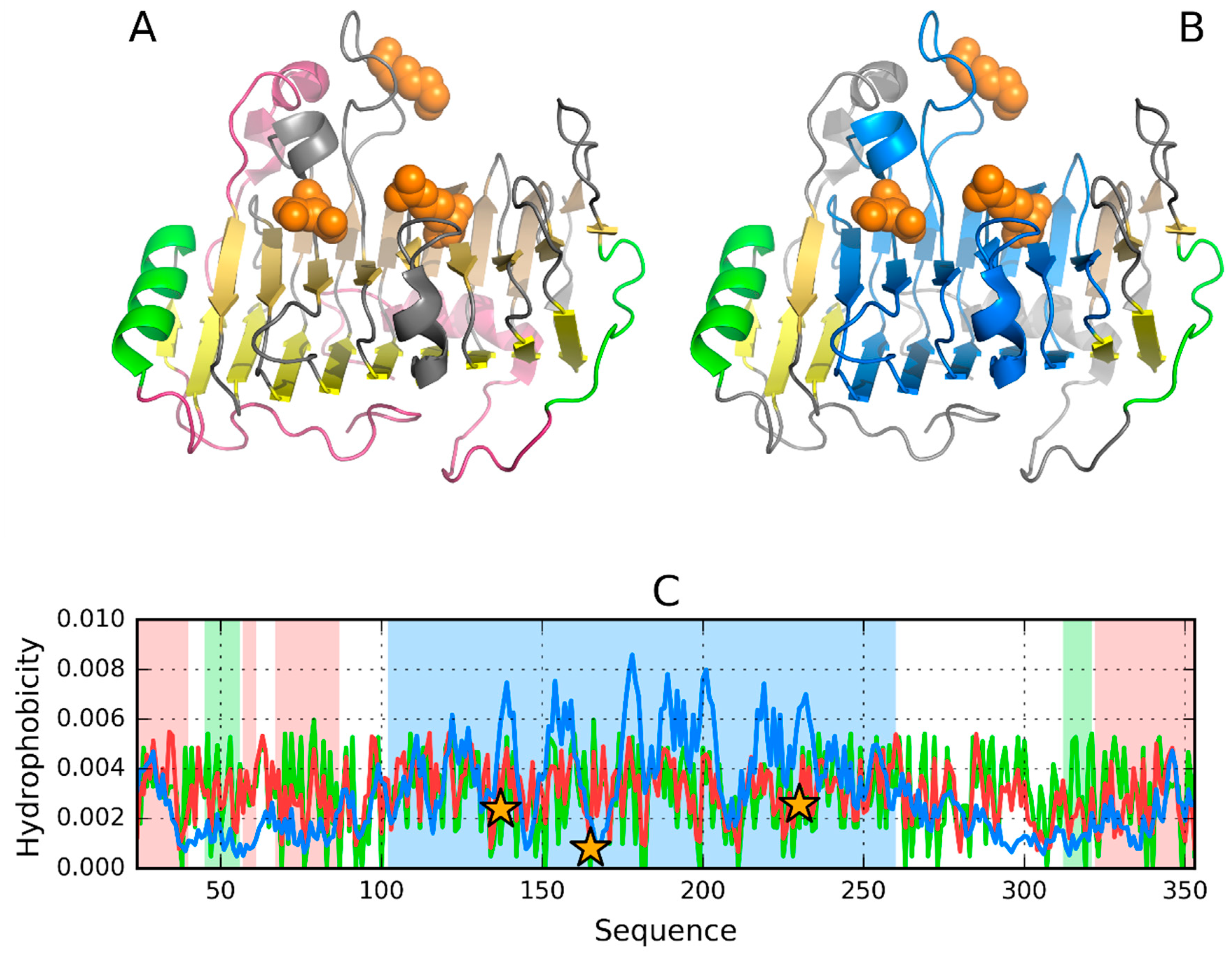
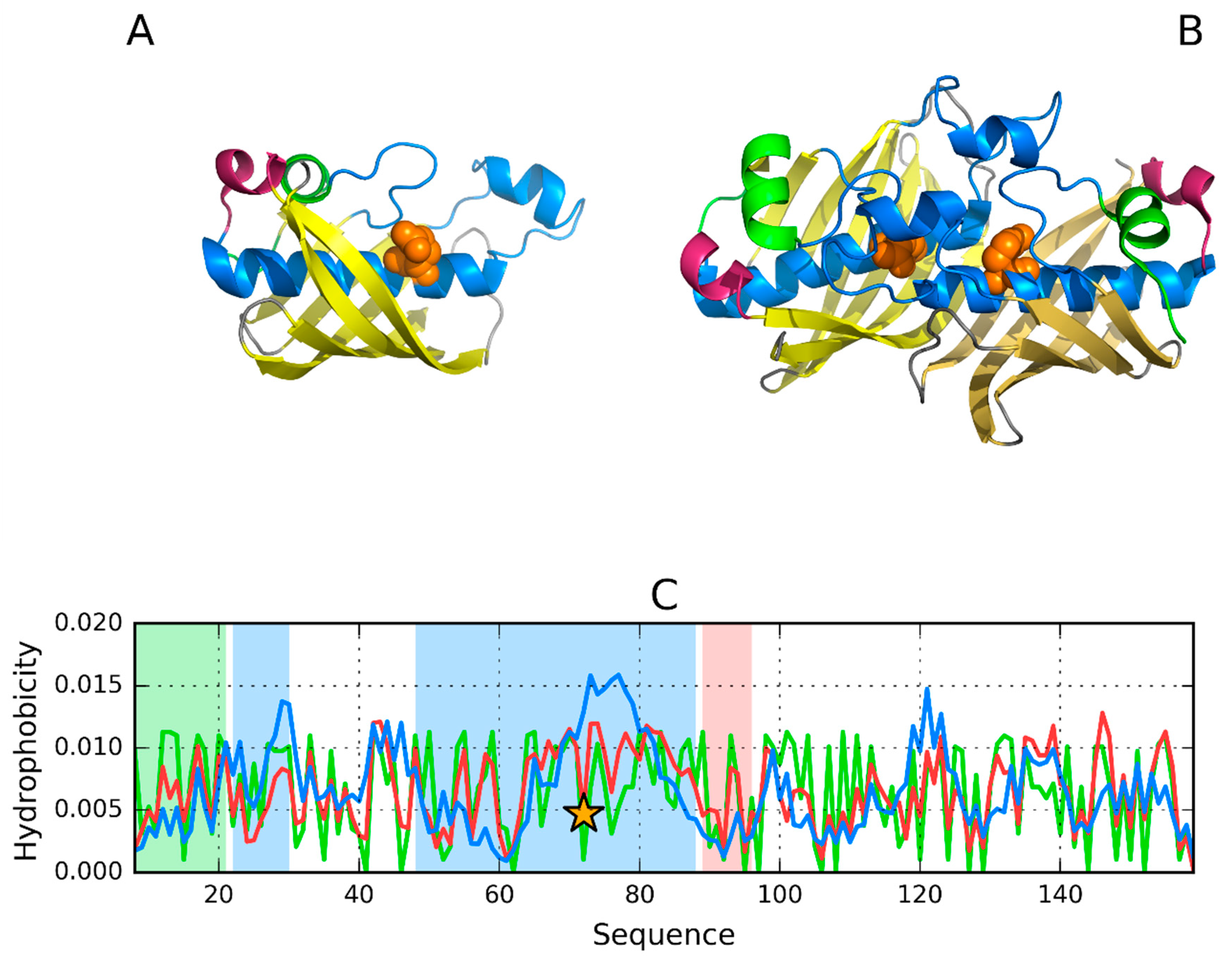
3.4. 3DP3—Representative of EC 4.2.2.59
3.5. Pectate Lyase—2QY1—Lyase II
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sternberg, M.J.E. Protein Structure Prediction—Principles and Approaches in Protein Structure Prediction; Ed Sternberg. IRL Press Oxford University Press: Oxford, UK; New York, NY, USA; Tokyo, Japan, 1996; p. 11. [Google Scholar]
- Andrade, M.A.; Perez-Iratxeta, C.; Ponting, C.P. Protein repeats: Structures, functions, and evolution. J. Struct. Biol. 2001, 134, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Kobe, B.; Kajava, A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001, 11, 725–732. [Google Scholar] [CrossRef]
- Kajava, A.V. Structural diversity of leucine-rich repeat proteins. J. Mol. Biol. 1998, 277, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.F.; Kajava, A.V. Design on a curve. Nat. Struct. Mol. Biol. 2015, 22, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Roterman, I.; Banach, M.; Konieczny, L. Application of the Fuzzy Oil Drop Model Describes Amyloid as a Ribbonlike Micelle. Entropy 2017, 19, 167. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. Why do antifreeze proteins require a solenoid? Biochimie 2018, 144, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Konieczny, L.; Bryliński, M.; Roterman, I. Gauss-function-Based model of hydrophobicity density in proteins. Silico Biol. 2006, 6, 15–22. [Google Scholar]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef] [Green Version]
- Kauzmann, W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 1959, 14, 1–63. [Google Scholar]
- Banach, M.; Konieczny, L.; Roterman, I. Can the Structure of Hydrophobic Core Determine the Complexation Site? In Identification of Ligand Binding Site and Protein-Protein Interaction Area; Roterman-Konieczna, I., Ed.; Springer: Dordrecht, The Netherlands, 2013; pp. 41–54. [Google Scholar]
- Banach, M.; Konieczny, L.; Roterman, I. Use of the “Fuzzy Oil Drop” Model to Identify the Complexation Area in Protein Homodimers. In Protein Folding; Roterman-Konieczna, I., Ed.; Silico, Woodhead Publishing (currently Elsevier): Oxford, UK; Cambridge, UK; Philadelphia, PA, USA; New Dehli, India, 2012; pp. 95–122. [Google Scholar]
- Banach, M.; Konieczny, L.; Roterman, I. Ligand-binding-site Recognition. In Protein Folding; Roterman-Konieczna, I., Ed.; Silico, Woodhead Publishing (currently Elsevier): Oxford, UK; Cambridge, UK; Philadelphia, PA, USA; New Dehli, India, 2012; pp. 78–94. [Google Scholar]
- Roterman, I.; Banach, M.; Kalinowska, B.; Konieczny, L. Influence of the Aqueous Environment on Protein Structure—A Plausible Hypothesis Concerning the Mechanism of Amyloidogenesis. Entropy 2016, 18, 351. [Google Scholar] [CrossRef]
- Fabian, P.; Stapor, K.; Banach, M.; Ptak-Kaczor, M.; Konieczny, L.; Roterman, I. Different synergy in amyloids and biologically active forms of proteins. Int. J. Mol. Sci. 2019, 20, 4436. [Google Scholar] [CrossRef] [PubMed]
- Roterman, I.; Banach, M.; Konieczny, L. Propagation of Fibrillar Structural Forms in Proteins Stopped by Naturally Occurring Short Polypeptide Chain Fragments. Pharmaceuticals 2017, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Iverson, T.M.; Alber, B.E.; Kisker, C.; Ferry, J.G.; Rees, D.C. A closer look at the active site of gamma-class carbonic anhydrases: High-resolution crystallographic studies of the carbonic anhydrase from Methanosarcina thermophila. Biochemistry 2000, 39, 9222–9231. [Google Scholar] [CrossRef] [PubMed]
- Jeyakanthan, J.; Rangarajan, S.; Mridula, P.; Kanaujia, S.P.; Shiro, Y.; Kuramitsu, S.; Yokoyama, S.; Sekar, K. Observation of a calcium-binding site in the gamma-class carbonic anhydrase from Pyrococcus horikoshii. Acta Crystallogr. D Biol. Crystallogr. 2008, 64 Pt 10, 1012–1019. [Google Scholar] [CrossRef]
- Yoder, M.D.; Jurnak, F. The Refined Three-Dimensional Structure of Pectate Lyase C from Erwinia chrysanthemi at 2.2 Angstrom Resolution (Implications for an Enzymatic Mechanism). Plant Physiol. 1995, 107, 349–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickersgill, R.; Jenkins, J.; Harris, G.; Nasser, W.; Robert-Baudouy, J. The structure of Bacillus subtilis pectate lyase in complex with calcium. Nat. Struct. Biol. 1994, 1, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Pickersgill, R.; Worboys, K.; Scott, M.; Cummings, N.; Cooper, A.; Jenkins, J.; Smith, D. The Conserved Arginine Proximal to the Essential Calcium of Bacillus Subtilis Pectate Lyase Stabilizes the Transition State. Available online: https://www.rcsb.org/structure/2BSP (accessed on 28 September 2019).
- Thomas, L.M.; Doan, C.N.; Oliver, R.L.; Yoder, M.D. Structure of pectate lyase A: Comparison to other isoforms. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1008–1015; [Google Scholar] [CrossRef]
- Herron, S.R.; Scavetta, R.D.; Garrett, M.; Legner, M.; Jurnak, F. Characterization and implications of Ca2+ binding to pectate lyase C. J. Biol. Chem. 2003, 278, 12271–12277; [Google Scholar] [CrossRef]
- Dehdashti, S.J.; Doan, C.N.; Chao, K.L.; Yoder, M.D. Effect of mutations in the T1.5 loop of pectate lyase A from Erwinia chrysanthemi EC16. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1339–1342. [Google Scholar] [CrossRef]
- Czerwinski, E.W.; Midoro-Horiuti, T.; White, M.A.; Brooks, E.G.; Goldblum, R.M. Crystal structure of Jun a 1, the major cedar pollen allergen from Juniperus ashei, reveals a parallel beta-helical core. J. Biol. Chem. 2005, 280, 3740–3746. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.; Shevchik, V.E.; Hugouvieux-Cotte-Pattat, N.; Pickersgill, R.W. The crystal structure of pectate lyase Pel9A from Erwinia chrysanthemi. J. Biol. Chem. 2004, 279, 9139–9145. [Google Scholar] [CrossRef]
- Mayans, O.; Scott, M.; Connerton, I.; Gravesen, T.; Benen, J.; Visser, J.; Pickersgill, R.; Jenkins, J. Two crystal structures of pectin lyase A from Aspergillus reveal a pH driven conformational change and striking divergence in the substrate-binding clefts of pectin and pectate lyases. Structure 1997, 5, 677–689. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Zhang, L.; Liu, X.; Li, X.; Zheng, M.; Li, H.; Yu, K.; Chen, K.; Shen, X.; Jiang, H.; et al. Discovering potent inhibitors against the beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ) of Helicobacter pylori: Structure-based design, synthesis, bioassay, and crystal structure determination. J. Med. Chem. 2009, 52, 2465–2481. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, L.; Zhang, Y.; Zhang, H.; Du, J.; Ding, J.; Guo, Y.; Jiang, H.; Shen, X. Emodin targets the beta-hydroxyacyl-acyl carrier protein dehydratase from Helicobacter pylori: Enzymatic inhibition assay with crystal structural and thermodynamic characterization. BMC Microbiol. 2009, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Bergeron, H.; Grosse, S.; Beauchemin, M.; Garron, M.L.; Shaya, D.; Sulea, T.; Cygler, M.; Lau, P.C. Improvement of the thermostability and activity of a pectate lyase by single amino acid substitutions, using a strategy based on melting-temperature-guided sequence alignment. Appl. Environ. Microbiol. 2008, 74, 1183–1189. [Google Scholar] [CrossRef]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- de Beer, T.A.P.; Berka, K.; Thornton, J.M.; Laskowski, R.A. PDBsum additions. Nucleic Acids Res. 2014, 42, D292–D296. [Google Scholar] [CrossRef]
- Schutzius, T.M.; Jung, S.; Maitra, T.; Graeber, G.; Köhme, M.; Poulikakos, D. Spontaneous droplet trampolining on rigid superhydrophobic surfaces. Nature 2015, 527, 82–85. [Google Scholar] [CrossRef]
- Prymula, K.; Jadczyk, T.; Roterman, I. Catalytic residues in hydrolases: Analysis of methods designed for ligand-binding site prediction. J. Comput. Aided. Mol. Des. 2011, 25, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Riek, R. On the possible amyloid origin of protein folds. J. Mol. Biol. 2012, 421, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Rufo, C.M.; Moroz, Y.S.; Stöhr, J.; Smith, T.A.; Hu, X.; DeGrado, W.F.; Korendovych, I.V. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 2014, 6, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelig, B.; Szostak, J.W. Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature 2007, 448, 828–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.A.; Hecht, M.H. Novel proteins: From fold to function. Curr. Opin. Chem. Biol. 2011, 15, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.S.; Greisman, J.B.; Hecht, M.H. De Novo proteins with life-sustaining functions are structurally dynamic. J. Mol. Biol. 2016, 428, 399–411. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Organism | Ref |
|---|---|---|
| EC 4.2.1.1. Carbonic anhydrase H2CO3 = CO2 + H2O | ||
| 1QRG Zn2+ | Methanosarcina thermophila | [18] |
| 1QRM Zn2+ | Methanosarcina thermophila | [18] |
| 2FKO Zn2+ | Pyrococcus horikoshi | [19] |
| EC 4.2.2.2. Pectate lyaseEliminative cleavage of pectate to give oligosaccharides with 4-deoxy-alpha-D-gluc-4-enuronosyl groups at their non-reducing ends | ||
| 1PLU Lu | Erwinia chrysanthemiplant | [20] |
| 1BN8 Ca2+ | Bacillus subtilis | [21] |
| 2BSP Ca2+ | Bacillus subtilis | [22] |
| 1JTA | Erwinia chrysanthemi | [23] |
| 1JRG | Erwinia chrysanthemi | [23] |
| 1O88 Ca2+ | Erwinia chrysanthemi | [24] |
| 1OOC | Erwinia chrysanthemi | [25] |
| 1PXZ | Juniperus ashei | [26] |
| 1RU4 2Ca2+ | Erwinia chrysanthemi | [27] |
| EC 4.2.2.10. Pectin lyase Eliminative cleavage of pectin to give oligosaccharides with terminal 4-deoxy-6-methyl-alpha-D-galact-4-enuronosyl groups | ||
| 1IDJ | Aspergillus niger | [28] |
| EC 4.2.1.59 3-hydroxyacyl-[acyl-carrier-protein] dehydratase (3R)-3-hydroxyacyl-[acyl-carrier protein] = a trans-2-enoyl-[acyl-carrier protein] + H2O | ||
| 3DP3 | Helicobacter pylori | [29] |
| 3ED0 | Helicobacter pylori | [30] |
| EC 3.4.11.18 Methionyl aminopeptidase Pectate II | ||
| 2QXZ | xanthomonas campestris | [31] |
| 2QY1 | xanthomonas campestris | [31] |
| PDB ID IONS | CHAIN | SOLENOID | ENVELOPE | STOP |
|---|---|---|---|---|
| EC 4.2.1.1 | ||||
| 1QRG | 0.418 | 0.506 | Helix—0.373 | 0.298 |
| 1QRM | 0.476 | 0.601 | Helix—0.426 | 0.313 |
| 2FKO | 0.457 | 0.526 | Helix—0.348 | 0.399 |
| EC 4.2.2.2 | ||||
| 1PLU | 0.654 | 0.627 | 0.594 | 0.362 |
| 1BN8 | 0.684 | 0.416 | 0.469 0.607 | 0.314 |
| 2BSP | 0.688 | 0.729 | 0.667 | 0.304 |
| 1JTA | 0.632 | 0.603 | 0.457 | 0.376 |
| 1JRG | 0.649 | 0.310 | ||
| 1O88 | 0.650 | 0.608 | 0.667 | 0.304 |
| 1OOC | 0.643 | 0.719 | 0.437 | 0.369 |
| 1PXZ | 0.591 | 0.620 | 0.509 | 0.503 |
| 1RU4 | 0.685 | 0.705 | 0.724 * | 0.752 |
| EC 4.2.2.10 | ||||
| 1IDJ | 0.722 | 0.769 | 0.634 | 0.255 |
| EC 4.2.2.59 | ||||
| 3DP3 | 0.463 | 0. | 0.770 | 0.343 |
| 3ED0 | 0.457 | 0.431 | 0.778 | 0.333 |
| Lyase II | ||||
| 2QXZ | 0.675 | 0.693 | 0.442 | 0.532 |
| 2QY1 | 0.674 | 0.665 | 0.444 | 0.494 |
| Fragment | Structural Properties | RD | Correlation Coefficient | |||
|---|---|---|---|---|---|---|
| T–O–R | T–O–H | HvT | TvO | HvO | ||
| 2–144 | SOLENOID | 0.526 | 0.468 | 0.295 | 0.516 | 0.775 |
| 2–4 | Beta-sheet | 0.542 | 0.703 | 0.408 | 0.462 | 0.804 |
| 12–14 * | Beta-sheet | 0.386 | 0.297 | 0.374 | 0.639 | 0.848 |
| 18–20 | Beta-sheet | 0.643 | 0.544 | 0.145 | 0.516 | 0.658 |
| STOP | ||||||
| 1–11 | STOP N-term | 0.305 | 0.143 | 0.427 | 0.790 | 0.767 |
| 137–144 | STOP signal | 0.399 | 0.163 | 0.531 | 0.652 | 0.794 |
| ENVELOPE | ||||||
| 144–173 | Helix C-term | 0.348 | 0.160 | 0.423 | 0.694 | 0.785 |
| CATAL. Center | ||||||
| 60–80 Y70 | Catalytic residue | 0.361 | 0.423 | 0.392 | 0.733 | 0.766 |
| 38–94 | surrounding | 0.376 | 0.299 | 0.363 | 0.668 | 0.750 |
| ION BINDING | ||||||
| 41–51 | 0.372 | 0.441 | 0.588 | 0.682 | 0.885 | |
| 60–70 | Zn2+ (65) | 0.367 | 0.380 | 0.289 | 0.728 | 0.609 |
| 82–87 | Zn2+ (87) | 0.419 | 0.246 | 0.544 | 0.552 | 0.885 |
| 100–110 | 0.502 | 0.283 | 0.425 | 0.308 | 0.825 | |
| Fragment | Structural Properties | RD | Correlation Coefficient | |||
|---|---|---|---|---|---|---|
| T–O–R | T–O–H | H/T | T/O | H/O | ||
| SOLENOID | 0.627 | 0.543 | 0.250 | 0.214 | 0.800 | |
| 19–23 * | Beta-sheet | 0.756 | 0.590 | 0.210 | 0.278 | 0.700 |
| 80–86 * | Beta-sheet E | 0.356 | 0.335 | 0.648 | 0.720 | 0.853 |
| 118–122 * | Beta-sheet | 0.521 | 0.523 | 0.431 | 0.278 | 0.772 |
| STOP | ||||||
| 25–37 | Helix | 0.371 | 0.314 | 0.775 | 0.664 | 0.861 |
| 286–297 | 0.348 | 0.306 | 0.583 | 0.710 | 0.782 | |
| ENVELOPE | ||||||
| 1–17 | Random Coil | 0.455 | 0.515 | 0.031 | 0.590 | 0.456 |
| 37–50 | Random Coil | 0.432 | 0.251 | 0.252 | 0.642 | 0.754 |
| 61–70 | Helix | 0.480 | 0.604 | 0.564 | 0.623 | 0.774 |
| 282–358 | Random Coil | 0.511 | 0.280 | 0.326 | 0.523 | 0.763 |
| 286–297 | C-term stop | 0.348 | 0.306 | 0.583 | 0.710 | 0.782 |
| 327–336 | helix | 0.428 | 0.273 | 0.498 | 0.489 | 0.965 |
| All above | 0.571 | 0.388 | 0.265 | 0.517 | 0.706 | |
| 1–26 | 0.536 | 0.413 | 0.105 | 0.529 | 0.543 | |
| 6–26 ** | 0.495 | 0.330 | 0.165 | 0.646 | 0.513 | |
| 38–50 | 0.393 | 0.225 | 0.351 | 0.717 | 0.761 | |
| 57–73 | 0.600 | 0.614 | 0.096 | 0.315 | 0.808 | |
| 57–73 ** | 0.441 | 0.501 | 0.536 | 0.724 | 0.654 | |
| 313–353 | 0.587 | 0.434 | 0.243 | 0.414 | 0.795 | |
| 313–345 * | 0.467 | 0.285 | 0.266 | 0.523 | 0.791 | |
| All above | 0.626 | 0.439 | 0.186 | 0.426 | 0.726 | |
| All **above | 0.467 | 0.285 | 0.656 | 0.844 | 0.852 | |
| CATAL. C. | ||||||
| 96–248 | 0.617 | 0.556 | 0.330 | 0.380 | 0.780 | |
| 58–80 | loop | 0.679 | 0.660 | −0.179 | −0.185 | 0.848 |
| 123–132 | loop | 0.596 | 0.430 | 0.239 | 0.223 | 0.871 |
| 150–167 | loop | 0.671 | 0.716 | −0.047 | 0.205 | 0.696 |
| 195–202 | loop | 0.111 | 0.407 | 0.029 | 0.725 | 0.487 |
| 126–136 | 131D—Beta fr. | 0.572 | 0.436 | 0.321 | 0.491 | 0.897 |
| 213–223 | 218R—Beta fr. | 0.471 | 0.384 | 0.498 | 0.549 | 0.930 |
| 117–224 | 0.621 | 0.525 | 0.314 | 0.402 | 0.774 | |
| 117–224 58–80 | 0.720 | 0.650 | 0.239 | 0.372 | 0.782 | |
| Fragment | Structural Properties | RD | Correlation Coefficient | |||
|---|---|---|---|---|---|---|
| T–R | T–H | H/T | T/O | H/O | ||
| SOLENOID | 0.769 | 0.635 | 0.167 | 0.205 | 0.788 | |
| 56…97 | Beta-sheet | 0.584 | 0.766 | 0.464 | 0.124 | 0.770 |
| 74…150 | Beta-sheet | 0.567 | 0.162 | 0.365 | 0.144 | 0.729 |
| 264…272 | Beta-sheet | 0.591 | 0.614 | 0.238 | 0.271 | 0.784 |
| STOP | ||||||
| 27–36 | Helix | 0.255 | 0.258 | 0.727 | 0.868 | 0.937 |
| 317–326 | 0.682 | 0.705 | 0.094 | −0.096 | 0.676 | |
| ENVELOPE | ||||||
| 1–23 | 0.659 | 0.683 | 0.016 | 0.214 | 0.572 | |
| 298–311 | loop | 0.647 | 0.632 | 0.159 | 0.250 | 0.728 |
| 327–359 | 0.553 | 0.484 | 0.148 | 0.494 | 0.632 | |
| 295–307 | helix | 0.345 | 0.445 | 0.640 | 0.668 | 0.742 |
| 293–312 | 0.489 | 0.611 | 0.509 | 0.553 | 0.810 | |
| 1–26 295–359 | Together | 0.662 | 0.656 | 0.082 | 0.258 | 0.664 |
| CATAL. C. | ||||||
| 51–103 | 0.609 | 0.766 | 0.321 | 0.321 | 0.761 | |
| 120–272 | 0.715 | 0.476 | 0.264 | 0.226 | 0.762 | |
| 144–164 | D154 | 0.601 | 0.363 | 0.334 | 0.375 | 0.858 |
| 166–186 | R176 | 0.806 | 0.661 | 0.354 | 0.167 | 0.771 |
| 226–246 | R236 | 0.672 | 0.319 | −0.103 | 0.043 | 0.760 |
| 51–103 | Loop | 0.609 | 0.766 | 0.321 | 0.321 | 0.761 |
| 145–152 | Loop D154 | 0.282 | 0.073 | 0.648 | 0.808 | 0.775 |
| 211–235 | Loop R236 | 0.760 | 0.424 | 0.344 | 0.172 | 0.606 |
| 146–154 | Loop | 0.544 | 0.176 | −0.003 | 0.288 | 0.691 |
| 199–210 | Loop | 0.701 | 0.731 | 0.132 | −0.066 | 0.742 |
| 103–242 | All Eres includ. | 0.732 | 0.461 | 0.289 | 0.239 | 0.781 |
| Fragment | Structural Properties | RD | Correlation Coefficient | |||
|---|---|---|---|---|---|---|
| T–R | T–H | H/T | T/O | H/O | ||
| MONOMER | ||||||
| Chain A | 0.463 | |||||
| SOLENOID * | 0.433 | 0.208 | 0.277 | 0.547 | 0.728 | |
| 32–37 | Beta-sheet | 0.414 | 0.185 | 0.333 | 0.566 | 0.725 |
| 66–88 | Helix 72E | 0.770 | 0.555 | −0.245 | 0.259 | 0.557 |
| Beta-sheet | 0.433 | 0.208 | 0.276 | 0.547 | 0.728 | |
| No P-P | 0.429 | 0.295 | 0.350 | 0.660 | 0.736 | |
| P-P | 0.625 | 0.351 | 0.006 | 0.278 | 0.634 | |
| STOP | ||||||
| 90–95 | Helix | 0.333 | 0.202 | 0.529 | 0.709 | 0.931 |
| 8–21 | STOP | 0.244 | 0.212 | 0.549 | 0.880 | 0.664 |
| 14–21 | helix | 0.346 | 0.281 | 0.705 | 0.876 | 0.872 |
| 53–58 | Helix-interface | 0.450 | 0.849 | 0.633 | 0.481 | 0.906 |
| CATAL. Center | ||||||
| 22–30 | 0.095 | 0.127 | 0.669 | 0.966 | 0.647 | |
| 48–88 | 0.533 | 0.521 | 0.128 | 0.635 | 0.674 | |
| Envelope | 0.534 | 0.527 | 0.188 | 0.586 | 0.634 | |
| DIMER | ||||||
| Dimer | 0.588 | |||||
| 66–88 | Helix 72E | 0.906 | 0.783 | −0.159 | 0.243 | 0.582 |
| Beta-sheet | 0.609 | 0.318 | 0.152 | 0.224 | 0.780 | |
| No P-P | 0.580 | 0.432 | 0.303 | 0.589 | 0.739 | |
| P-P | 0.512 | 0.291 | 0.263 | 0.562 | 0.729 | |
| 90–95 | Stop-helix | 0.421 | 0.277 | 0.385 | 0.529 | 0.940 |
| 8–21 | Stop-helix | 0.473 | 0.449 | 0.331 | 0.651 | 0.683 |
| 53–58 | Helix-interface | 0.251 | 0.600 | 0.601 | 0.836 | 0.858 |
| COMPLEX | ||||||
| Six units | 0.745 | |||||
| 66–88 | Helix 72E | 0.908 | 0.786 | −0.237 | 0.047 | 0.610 |
| Beta-sheet | 0.637 | 0.366 | 0.112 | 0.185 | 0.779 | |
| No P-P | 0.659 | 0.516 | 0.257 | 0.305 | 0.745 | |
| P-P | 0.712 | 0.461 | −0.058 | −0.042 | 0.774 | |
| 90–95 | Stop-helix | 0.598 | 0.395 | −0.284 | −0.225 | 0.897 |
| 14–21 | Stop-helix | 0.538 | 0.493 | 0.333 | 0.216 | 0.906 |
| 53–58 | Helix-interface | 0.620 | 0.844 | −0.605 | −0.880 | −0.856 |
| SOLENOID | 0.634 | 0.363 | 0.111 | 0.185 | 0.785 | |
| Fragment | Structural Properties | RD | Correlation Coefficient | |||
|---|---|---|---|---|---|---|
| T–R | T–H | H/T | T/O | H/O | ||
| SOLENOID | 0.665 | 0.477 | 0.205 | 0.321 | 0.761 | |
| 41–43 | Beta-sheet | 0.762 | 0.453 | 0.077 | 0.153 | 0.735 |
| 88–92 | Beta-sheet | 0.694 | 0.550 | 0.282 | 0.381 | 0.829 |
| 125–129 | Beta-sheet | 0.672 | 0.477 | 0.184 | 0.232 | 0.840 |
| 201–203 | Beta-sheet | 0.824 | 0.756 | 0.022 | 0.143 | 0.874 |
| STOP | ||||||
| 45–55 | Helix | 0.444 | 0.391 | 0.654 | 0.669 | 0.838 |
| ENVELOPE | ||||||
| 24–40 | N-terminal loop | 0.475 | 0.314 | 0.374 | 0.574 | 0.316 |
| 57–61 | 0.244 | 0.329 | 0.786 | 0.866 | 0.580 | |
| 67–87 | 0.525 | 0.178 | 0.295 | 0.272 | 0.878 | |
| 322–353 | 0.497 | 0.240 | 0.227 | 0.556 | 0.589 | |
| 39–61 | 0.488 | 0.459 | 0.474 | 0.536 | 0.654 | |
| 303–343 | 0.191 | 0.437 | 0.362 | 0.611 | 0.574 | |
| 344–353 | C-terminal loop | 0.133 | 0.288 | 0.471 | 0.857 | 0.622 |
| Loops complete | 0.541 | 0.329 | 0.167 | 0.542 | 0.504 | |
| CATAL. Center | ||||||
| 102–260 | 0.639 | 0.518 | 0.246 | 0.317 | 0.747 | |
| 127–240 | 0.652 | 0.472 | 0.268 | 0.392 | 0.764 | |
| 132–142 | Cat. Res D137 | 0.624 | 0.510 | 0.477 | 0.456 | 0.901 |
| 160–170 | Cat. Res. K165 | 0.697 | 0.598 | 0.251 | 0.273 | 0.734 |
| 225–235 | Cat. Res R230 | 0.246 | 0.294 | 0.469 | 0.745 | 0.858 |
| 67–87 | loop | 0.525 | 0.178 | 0.295 | 0.272 | 0.878 |
| 129–137 | loop | 0.553 | 0.721 | 0.044 | 0.115 | 0.877 |
| 161–177 | loop | 0.716 | 0.650 | −0.005 | 0.349 | 0.654 |
| 204–215 | 0.230 | 0.153 | 0.413 | 0.852 | 0.626 | |
| 129–237 | 0.629 | 0.468 | 0.271 | 0.400 | 0.764 | |
| 67–222 | total | 0.652 | 0.441 | 0.262 | 0.404 | 0.753 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banach, M.; Konieczny, L.; Roterman, I. Symmetry and Dissymmetry in Protein Structure—System-Coding Its Biological Specificity. Symmetry 2019, 11, 1215. https://doi.org/10.3390/sym11101215
Banach M, Konieczny L, Roterman I. Symmetry and Dissymmetry in Protein Structure—System-Coding Its Biological Specificity. Symmetry. 2019; 11(10):1215. https://doi.org/10.3390/sym11101215
Chicago/Turabian StyleBanach, Mateusz, Leszek Konieczny, and Irena Roterman. 2019. "Symmetry and Dissymmetry in Protein Structure—System-Coding Its Biological Specificity" Symmetry 11, no. 10: 1215. https://doi.org/10.3390/sym11101215
APA StyleBanach, M., Konieczny, L., & Roterman, I. (2019). Symmetry and Dissymmetry in Protein Structure—System-Coding Its Biological Specificity. Symmetry, 11(10), 1215. https://doi.org/10.3390/sym11101215