Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation


Abstract
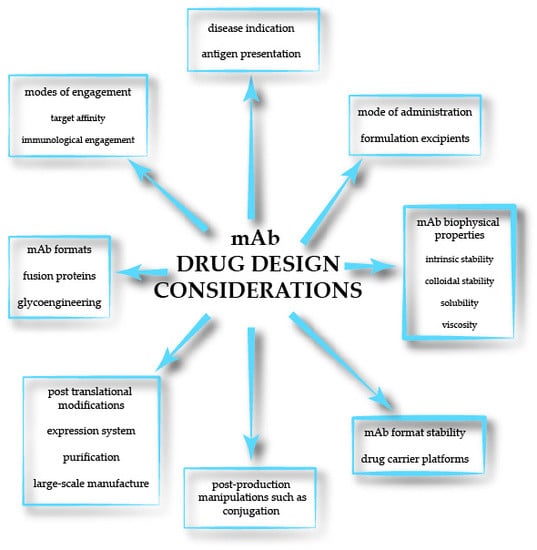
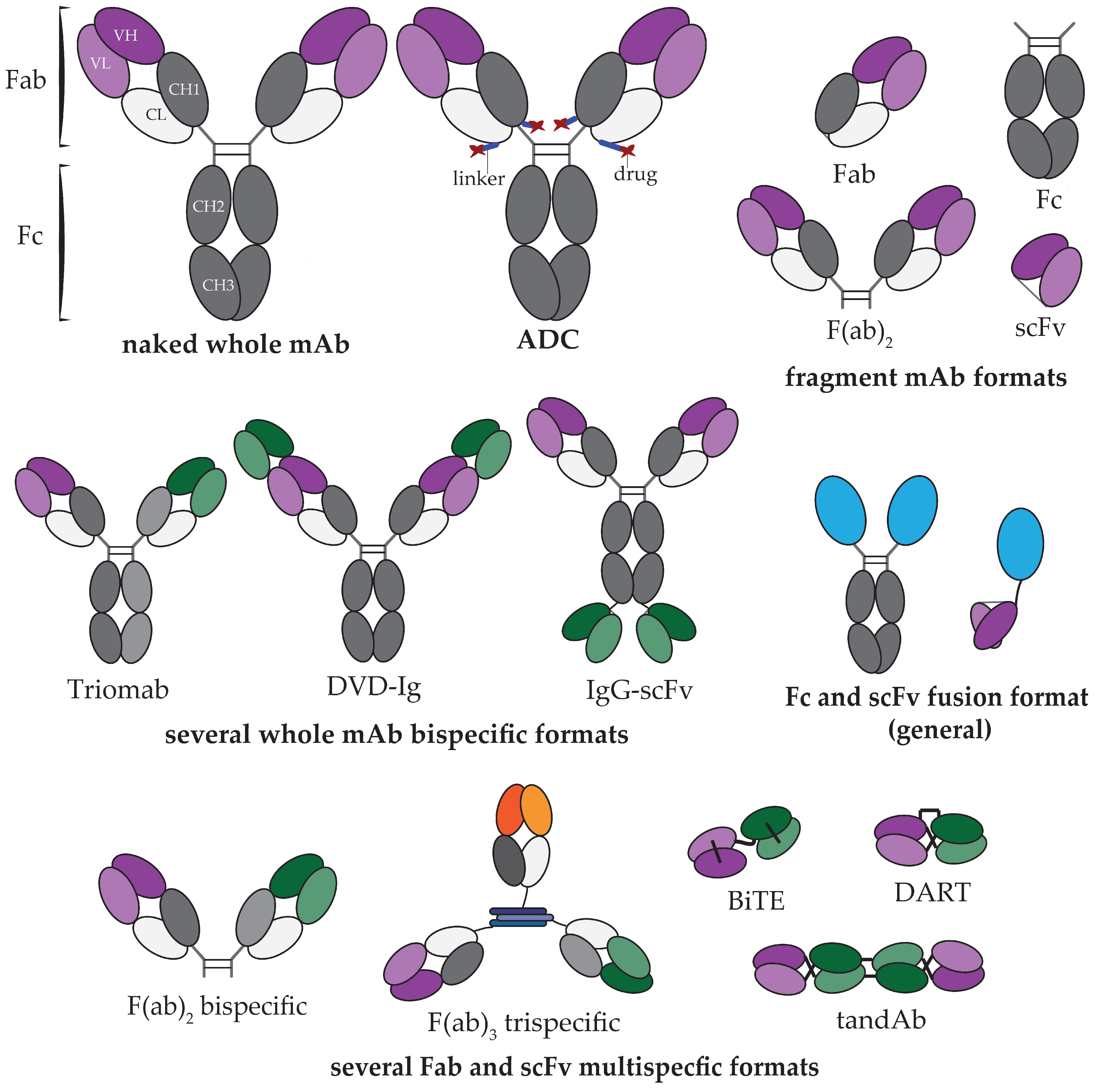
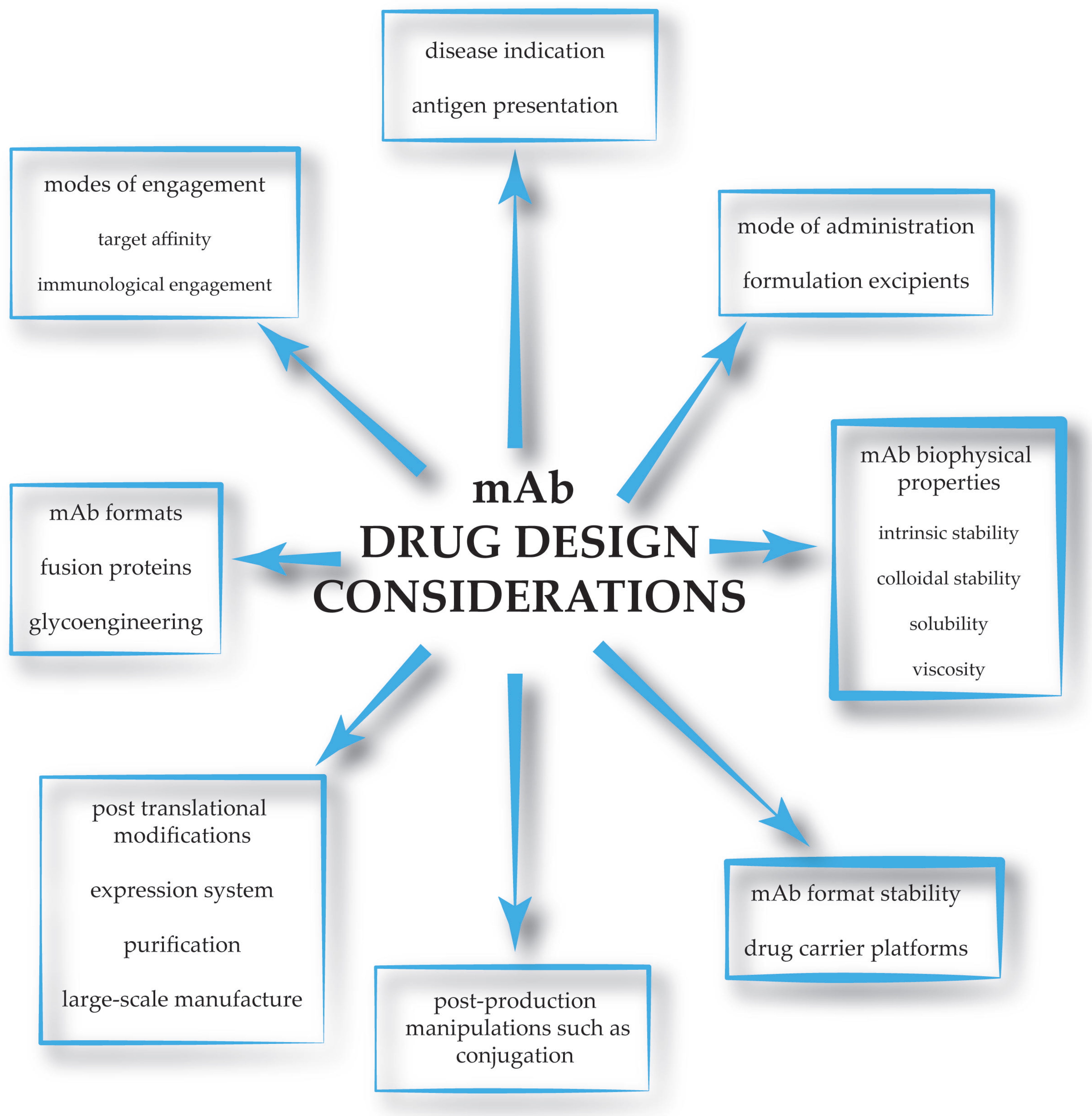
1. Introduction
2. Overview of mAb Production Challenges and Considerations
3. mAb Discovery and Manufacture Technologies
4. Formulation Strategies and Considerations
5. Improving mAb Tissue Penetration for Cancer Treatment
6. Strategic Modulation of mAb Immune Effector Functions
7. Computational Approaches for Aggregation Prediction and Rational Design of mAbs
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Urquhart, L. Top drugs and companies by sales in 2017. Nat. Rev. Drug Discov. 2018, 17, 232. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.; Dana Jones, S.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- An, Z. “Magic bullets” at the center stage of immune therapy: A special issue on therapeutic antibodies. Protein Cell 2018, 9, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.L.d.; Quintilio, W.; Manieri, T.M.; Tsuruta, L.R.; Moro, A.M. Advances and challenges in therapeutic monoclonal antibodies drug development. Bras. J. Pharm. Sci. 2018, 54. [Google Scholar] [CrossRef]
- Elgundi, Z.; Reslan, M.; Cruz, E.; Sifniotis, V.; Kayser, V. The state-of-play and future of antibody therapeutics. Adv. Drug Del. Rev. 2017, 122, 2–19. [Google Scholar] [CrossRef]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front. Immunol. 2018, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.N.; Luo, J.H. Research and development of innovative antibody-based drugs. Yaoxue Xuebao 2017, 52, 1811–1819. [Google Scholar]
- Awwad, S.; Angkawinitwong, U. Overview of antibody drug delivery. Pharmaceutics 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Klein, C. Special issue: Monoclonal antibodies. Antibodies 2018, 7, 17. [Google Scholar] [CrossRef]
- Wang, C.; Xu, P.; Zhang, L.; Huang, J.; Zhu, K.; Luo, C. Current strategies and applications for precision drug design. Front. Pharmacol. 2018, 9, 787. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Current progress in innovative engineered antibodies. Protein Cell 2018, 9, 86–120. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of antibody drug conjugates (adcs). OncoImmunology 2018, 7, e1395127. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mathieu, M.; Brezski, R.J. Igg fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.W.; Bajardi-Taccioli, A.; Houde, D.; Feschenko, M.; Carlage, T.; Kaltashov, I.A. Influence of glycan modification on igg1 biochemical and biophysical properties. J. Pharm. Biomed. Anal. 2018, 151, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.H.G.; Furtado, G.P.; Bezerra, M.R.L.; Pontes, L.Q.; Fernandes, C.F.C. Boosting half-life and effector functions of therapeutic antibodies by fc-engineering: An interaction-function review. Int. J. Biol. Macromol. 2018, 119, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, A.; Caron, A.L.; Picanço-Castro, V.; Swiech, K. Platforms for recombinant therapeutic glycoprotein production. In Recombinant Glycoprotein Production; Humana Press Inc.: New York, NY, USA, 2018; Volume 1674, pp. 1–14. [Google Scholar]
- Thomson, C.A. Igg structure and function. In Encyclopedia of Immunobiology; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 2, pp. 15–22. [Google Scholar]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Forrer, K.; Helk, B.; Trout, B.L. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol. J. 2011, 6, 38–44. [Google Scholar] [CrossRef]
- Pieracci, J.P.; Armando, J.W.; Westoby, M.; Thommes, J. Chapter 9—Industry review of cell separation and product harvesting methods. In Biopharmaceutical Processing; Jagschies, G., Lindskog, E., Łącki, K., Galliher, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 165–206. [Google Scholar]
- Huang, C.-J.; Lin, H.; Yang, X. Industrial production of recombinant therapeutics in escherichia coli and its recent advancements. J. Ind. Microbiol. Biotechnol. 2012, 39, 383–399. [Google Scholar] [CrossRef]
- Behme, S. Manufacturing of Pharmaceutical Proteins: From Technology to Economy: Second, Revised and Expanded Edition; Wiley: Hoboken, NJ, USA, 2015; pp. 1–427. [Google Scholar]
- Ulmer, N.; Vogg, S.; Müller-Späth, T.; Morbidelli, M. Chapter 7—Purification of human monoclonal antibodies and their fragments. In Human monoclonal Antibodies: Methods and Protocols; Steinitz, M., Ed.; Springer: New York, NY, USA, 2019; pp. 163–188. [Google Scholar]
- Arosio, P.; Barolo, G.; Müller-Späth, T.; Wu, H.; Morbidelli, M. Aggregation stability of a monoclonal antibody during downstream processing. Pharm. Res. 2011, 28, 1884–1894. [Google Scholar] [CrossRef]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Helk, B.; Trout, B.L. Conformational stability and aggregation of therapeutic monoclonal antibodies studied with ans and thioflavin t binding. mAbs 2011, 3, 408–411. [Google Scholar] [CrossRef]
- Bittner, B.; Richter, W.; Schmidt, J. Subcutaneous administration of biotherapeutics: An overview of current challenges and opportunities. Biodrugs 2018, 32, 425–440. [Google Scholar] [CrossRef]
- Viola, M.; Sequeira, J.; Seiça, R.; Veiga, F.; Serra, J.; Santos, A.C.; Ribeiro, A.J. Subcutaneous delivery of monoclonal antibodies: How do we get there? J. Control. Release 2018, 286, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L. 6.05—Basic principles of formulation for biotherapeutics: Approaches to alternative drug delivery. In Comprehensive Medicinal Chemistry III; Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 131–156. [Google Scholar]
- Morales, J.O.; Fathe, K.R.; Brunaugh, A.; Ferrati, S.; Li, S.; Montenegro-Nicolini, M.; Mousavikhamene, Z.; McConville, J.T.; Prausnitz, M.R.; Smyth, H.D.C. Challenges and future prospects for the delivery of biologics: Oral mucosal, pulmonary, and transdermal routes. AAPS J. 2017, 19, 652–668. [Google Scholar] [CrossRef] [PubMed]
- Muheem, A.; Shakeel, F.; Jahangir, M.A.; Anwar, M.; Mallick, N.; Jain, G.K.; Warsi, M.H.; Ahmad, F.J. A review on the strategies for oral delivery of proteins and peptides and their clinical perspectives. Saudi Pharm. J. 2016, 24, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.; Yuan, P.; Vavilala, D.; Fox, M. Optimization of protein expression in mammalian cells. Curr. Protoc. Protein Sci. 2019, 95, e77. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, E.K.; Fischer, S.; Wenger, T.; Schulz, P. Chapter 6—Host cells. In Biopharmaceutical Processing; Jagschies, G., Lindskog, E., Łącki, K., Galliher, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 111–130. [Google Scholar]
- Fliedl, L.; Grillari, J.; Grillari-Voglauer, R. Human cell lines for the production of recombinant proteins: On the horizon. New Biotechnol. 2015, 32, 673–679. [Google Scholar] [CrossRef]
- Matsuda, T.; Ito, T.; Takemoto, C.; Katsura, K.; Ikeda, M.; Wakiyama, M.; Kukimoto-Niino, M.; Yokoyama, S.; Kurosawa, Y.; Shirouzu, M. Cell-free synthesis of functional antibody fragments to provide a structural basis for antibody–antigen interaction. PLoS ONE 2018, 13, e0193158. [Google Scholar] [CrossRef]
- Stech, M.; Kubick, S. Cell-free synthesis meets antibody production: A review. Antibodies 2015, 4, 12. [Google Scholar] [CrossRef]
- Pujols, J.; Peña-Díaz, S.; Ventura, S. Aggrescan3d: Toward the prediction of the aggregation propensities of protein structures. In Methods Mol. Biol.; Humana Press Inc.: New York, NY, USA, 2018; Volume 1762, pp. 427–443. [Google Scholar]
- Van der Kant, R.; Karow-Zwick, A.R.; Van Durme, J.; Blech, M.; Gallardo, R.; Seeliger, D.; Aßfalg, K.; Baatsen, P.; Compernolle, G.; Gils, A.; et al. Prediction and reduction of the aggregation of monoclonal antibodies. J. Mol. Biol. 2017, 429, 1244–1261. [Google Scholar] [CrossRef]
- Lerch, T.F.; Sharpe, P.; Mayclin, S.J.; Edwards, T.E.; Lee, E.; Conlon, H.D.; Polleck, S.; Rouse, J.C.; Luo, Y.; Zou, Q. Infliximab crystal structures reveal insights into self-association. mAbs 2017, 9, 874–883. [Google Scholar] [CrossRef]
- Dobson, C.L.; Devine, P.W.A.; Phillips, J.J.; Higazi, D.R.; Lloyd, C.; Popovic, B.; Arnold, J.; Buchanan, A.; Lewis, A.; Goodman, J.; et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci. Rep. 2016, 6, 38644. [Google Scholar] [CrossRef]
- Courtois, F.; Agrawal, N.J.; Lauer, T.M.; Trout, B.L. Rational design of therapeutic mabs against aggregation through protein engineering and incorporation of glycosylation motifs applied to bevacizumab. mAbs 2016, 8, 99–112. [Google Scholar] [CrossRef]
- Courtois, F.; Schneider, C.P.; Agrawal, N.J.; Trout, B.L. Rational design of biobetters with enhanced stability. J. Pharm. Sci. 2015, 104, 2433–2440. [Google Scholar] [CrossRef]
- Yu, M.; Wu, J.; Shi, J.; Farokhzad, O.C. Nanotechnology for protein delivery: Overview and perspectives. J. Control. Release 2016, 240, 24–37. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2016, 17, 20. [Google Scholar] [CrossRef]
- Dalziel, M.; Beers, S.A.; Cragg, M.S.; Crispin, M. Through the barricades: Overcoming the barriers to effective antibody-based cancer therapeutics. Glycobiology 2018, 28, 697–712. [Google Scholar] [CrossRef]
- Lambert, J.M.; Berkenblit, A. Antibody-drug conjugates for cancer treatment. Annu. Rev. Med. 2018, 69, 191–207. [Google Scholar] [CrossRef]
- Lambert, J.M.; Morris, C.Q. Antibody–drug conjugates (adcs) for personalized treatment of solid tumors: A review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef]
- Levin, D.; Golding, B.; Strome, S.E.; Sauna, Z.E. Fc fusion as a platform technology: Potential for modulating immunogenicity. Trends Biotechnol. 2015, 33, 27–34. [Google Scholar] [CrossRef]
- Henry, K.A. Next-generation DNA sequencing of vh/vl repertoires: A primer and guide to applications in single-domain antibody discovery. In Phage Display: Methods and Protocols; Hust, M., Lim, T.S., Eds.; Springer: New York, NY, USA, 2018; pp. 425–446. [Google Scholar]
- Frenzel, A.; Roskos, L.; Klakamp, S.; Liang, M.; Arends, R.; Green, L. Antibody affinity. In Handbook of Therapeutic Antibodies, 2nd ed.; Wiley Blackwell: Hoboken, NJ, USA, 2014; Volume 1–4, pp. 115–140. [Google Scholar]
- Hu, J.; Han, J.; Li, H.; Zhang, X.; Liu, L.; Chen, F.; Zeng, B. Human embryonic kidney 293 cells: A vehicle for biopharmaceutical manufacturing, structural biology, and electrophysiology. Cells Tissues Organs 2018, 205, 1–8. [Google Scholar] [CrossRef]
- Jacobi, A.; Enenkel, B.; Garidel, P.; Eckermann, C.; Knappenberger, M.; Presser, I.; Kaufmann, H. Process development and manufacturing of therapeutic antibodies. In Handbook of Therapeutic Antibodies, 2nd ed.; Wiley Blackwell: Hoboken, NJ, USA, 2014; Volume 2–4, pp. 601–664. [Google Scholar]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Helk, B.; Forrer, K.; Trout, B.L. A screening tool for therapeutic monoclonal antibodies: Identifying the most stable protein and its best formulation based on thioflavin t binding. Biotechnol. J. 2012, 7, 127–132. [Google Scholar] [CrossRef]
- Tada, M.; Tatematsu, K.-I.; Ishii-Watabe, A.; Harazono, A.; Takakura, D.; Hashii, N.; Sezutsu, H.; Kawasaki, N. Characterization of anti-cd20 monoclonal antibody produced by transgenic silkworms (bombyx mori). mAbs 2015, 7, 1138–1150. [Google Scholar] [CrossRef]
- Maccani, A.; Landes, N.; Stadlmayr, G.; Maresch, D.; Leitner, C.; Maurer, M.; Gasser, B.; Ernst, W.; Kunert, R.; Mattanovich, D. Pichia pastoris secretes recombinant proteins less efficiently than chinese hamster ovary cells but allows higher space-time yields for less complex proteins. Biotechnol. J. 2014, 9, 526–537. [Google Scholar] [CrossRef]
- Frenzel, A.; Hust, M.; Schirrmann, T. Expression of recombinant antibodies. Front. Immunol. 2013, 4, 217. [Google Scholar] [CrossRef]
- Thoring, L.; Kubick, S. Versatile cell-free protein synthesis systems based on chinese hamster ovary cells. In Recombinant Protein Expression in Mammalian Cells: Methods and Protocols; Hacker, D.L., Ed.; Springer: New York, NY, USA, 2018; pp. 289–308. [Google Scholar]
- Tran, K.; Gurramkonda, C.; Cooper, M.A.; Pilli, M.; Taris, J.E.; Selock, N.; Han, T.-C.; Tolosa, M.; Zuber, A.; Peñalber-Johnstone, C.; et al. Cell-free production of a therapeutic protein: Expression, purification, and characterization of recombinant streptokinase using a cho lysate. Biotechnol. Bioeng. 2018, 115, 92–102. [Google Scholar] [CrossRef]
- Stech, M.; Nikolaeva, O.; Thoring, L.; Stöcklein, W.F.M.; Wüstenhagen, D.A.; Hust, M.; Dübel, S.; Kubick, S. Cell-free synthesis of functional antibodies using a coupled in vitro transcription-translation system based on cho cell lysates. Sci. Rep. 2017, 7, 12030. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Kwon, Y.-C.; Jewett, M.C. Establishing a high yielding streptomyces-based cell-free protein synthesis system. Biotechnol. Bioeng. 2017, 114, 1343–1353. [Google Scholar] [CrossRef]
- Jaroentomeechai, T.; Stark, J.C.; Natarajan, A.; Glasscock, C.J.; Yates, L.E.; Hsu, K.J.; Mrksich, M.; Jewett, M.C.; DeLisa, M.P. Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat. Commun. 2018, 9, 2686. [Google Scholar] [CrossRef]
- Gurramkonda, C.; Rao, A.; Borhani, S.; Pilli, M.; Deldari, S.; Ge, X.; Pezeshk, N.; Han, T.-C.; Tolosa, M.; Kostov, Y.; et al. Improving the recombinant human erythropoietin glycosylation using microsome supplementation in cho cell-free system. Biotechnol. Bioeng. 2018, 115, 1253–1264. [Google Scholar] [CrossRef]
- Nakamura, T.; Omasa, T. Optimization of cell line development in the gs-cho expression system using a high-throughput, single cell-based clone selection system. J. Biosci. Bioeng. 2015, 120, 323–329. [Google Scholar] [CrossRef]
- Zhou, Y.; Raju, R.; Alves, C.; Gilbert, A. Debottlenecking protein secretion and reducing protein aggregation in the cellular host. Curr. Opin. Biotechnol. 2018, 53, 151–157. [Google Scholar] [CrossRef]
- Inwood, S.; Betenbaugh, M.J.; Lal, M.; Shiloach, J. Genome-wide high-throughput rnai screening for identification of genes involved in protein production. In Recombinant Protein Expression in Mammalian Cells: Methods and Protocols; Hacker, D.L., Ed.; Springer: New York, NY, USA, 2018; pp. 209–219. [Google Scholar]
- Dangi, A.K.; Sinha, R.; Dwivedi, S.; Gupta, S.K.; Shukla, P. Cell line techniques and gene editing tools for antibody production: A review. Front. Pharmacol. 2018, 9, 630. [Google Scholar] [CrossRef]
- Delic, M.; Göngrich, R.; Mattanovich, D.; Gasser, B. Engineering of protein folding and secretion—Strategies to overcome bottlenecks for efficient production of recombinant proteins. Antioxid. Redox Signal. 2014, 21, 414–437. [Google Scholar] [CrossRef]
- Zhong, X.; Wright, J.F. Biological insights into therapeutic protein modifications throughout trafficking and their biopharmaceutical applications. Int. J. Cell Biol. 2013, 2013, 19. [Google Scholar] [CrossRef]
- Altamirano, C.; Berrios, J.; Vergara, M.; Becerra, S. Advances in improving mammalian cells metabolism for recombinant protein production. Electron. J. Biotechnol. 2013, 16. [Google Scholar] [CrossRef]
- Parola, C.; Mason, D.M.; Zingg, A.; Neumeier, D.; Reddy, S.T. Genome engineering of hybridomas to generate stable cell lines for antibody expression. In Recombinant Protein Expression in Mammalian Cells: Methods and Protocols; Hacker, D.L., Ed.; Springer: New York, NY, USA, 2018; pp. 79–111. [Google Scholar]
- You, M.; Yang, Y.; Zhong, C.; Chen, F.; Wang, X.; Jia, T.; Chen, Y.; Zhou, B.; Mi, Q.; Zhao, Q.; et al. Efficient mab production in cho cells with optimized signal peptide, codon, and utr. Appl. Microbiol. Biotechnol. 2018, 102, 5953–5964. [Google Scholar] [CrossRef]
- Mauro, V.P.; Chappell, S.A. Considerations in the use of codon optimization for recombinant protein expression. In Recombinant Protein Expression in Mammalian Cells: Methods and Protocols; Hacker, D.L., Ed.; Springer: New York, NY, USA, 2018; pp. 275–288. [Google Scholar]
- Michael, I.P.; Nagy, A. Inducible protein production in 293 cells using the piggybac transposon system. In Recombinant Protein Expression in Mammalian Cells: Methods and Protocols; Hacker, D.L., Ed.; Springer: New York, NY, USA, 2018; pp. 57–68. [Google Scholar]
- Balasubramanian, S. Recombinant cho cell pool generation using piggybac transposon system. In Recombinant Protein Expression in Mammalian Cells: Methods and Protocols; Hacker, D.L., Ed.; Springer: New York, NY, USA, 2018; pp. 69–78. [Google Scholar]
- Jäger, V.; Büssow, K.; Schirrmann, T. Transient recombinant protein expression in mammalian cells. In Animal Cell Culture; Al-Rubeai, M., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 27–64. [Google Scholar]
- Wijesuriya, S.D.; Pongo, E.; Tomic, M.; Zhang, F.; Garcia-Rodriquez, C.; Conrad, F.; Farr-Jones, S.; Marks, J.D.; Horwitz, A.H. Antibody engineering to improve manufacturability. Protein Expression Purif. 2018, 149, 75–83. [Google Scholar] [CrossRef]
- L’Abbé, D.; Bisson, L.; Gervais, C.; Grazzini, E.; Durocher, Y. Transient gene expression in suspension hek293-ebna1 cells. In Recombinant Protein Expression in Mammalian Cells: Methods and Protocols; Hacker, D.L., Ed.; Springer: New York, NY, USA, 2018; pp. 1–16. [Google Scholar]
- Arena, T.A.; Harms, P.D.; Wong, A.W. High throughput transfection of hek293 cells for transient protein production. In Recombinant Protein Expression in Mammalian Cells: Methods and Protocols; Hacker, D.L., Ed.; Springer: New York, NY, USA, 2018; pp. 179–187. [Google Scholar]
- Agrawal, V.; Yu, B.; Pagila, R.; Yang, B.; Simonsen, C.; Beske, O. A high-yielding, cho-k1-based transient transfection system. BioProcess Int. 2013, 11, 28–35. [Google Scholar]
- Hacker, D.L.; Kiseljak, D.; Rajendra, Y.; Thurnheer, S.; Baldi, L.; Wurm, F.M. Polyethyleneimine-based transient gene expression processes for suspension-adapted hek-293e and cho-dg44 cells. Protein Expression Purif. 2013, 92, 67–76. [Google Scholar] [CrossRef]
- Ritacco, F.V.; Wu, Y.; Khetan, A. Cell culture media for recombinant protein expression in chinese hamster ovary (cho) cells: History, key components, and optimization strategies. Biotechnol. Prog. 2018, 34, 1407–1426. [Google Scholar] [CrossRef]
- Whitford, W.G.; Lundgren, M.; Fairbank, A. Chapter 8—Cell culture media in bioprocessing. In Biopharmaceutical Processing; Jagschies, G., Lindskog, E., Łącki, K., Galliher, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 147–162. [Google Scholar]
- Davami, F.; Eghbalpour, F.; Barkhordari, F.; Mahboudi, F. Effect of peptone feeding on transient gene expression process in cho dg44. Avicenna J. Med. Biotechnol. 2014, 6, 147–155. [Google Scholar]
- Mahboudi, F.; Abolhassan, M.R.; Azarpanah, A.; Aghajani-Lazarjani, H.; Sadeghi-Haskoo, M.A.; Maleknia, S.; Vaziri, B. The role of different supplements in expression level of monoclonal antibody against human cd20. Avicenna J. Med. Biotechnol. 2013, 5, 140–147. [Google Scholar]
- You, M.; Liu, Y.; Chen, Y.; Guo, J.; Wu, J.; Fu, Y.; Shen, R.; Qi, R.; Luo, W.; Xia, N. Maximizing antibody production in suspension-cultured mammalian cells by the customized transient gene expression method. Biosci. Biotechnol. Biochem. 2013, 77, 1207–1213. [Google Scholar] [CrossRef]
- Backliwal, G.; Hildinger, M.; Kuettel, I.; Delegrange, F.; Hacker, D.L.; Wurm, F.M. Valproic acid: A viable alternative to sodium butyrate for enhancing protein expression in mammalian cell cultures. Biotechnol. Bioeng. 2008, 101, 182–189. [Google Scholar] [CrossRef]
- Elgundi, Z.; Sifniotis, V.; Reslan, M.; Cruz, E.; Kayser, V. Laboratory scale production and purification of a therapeutic antibody. JoVE 2017. [Google Scholar] [CrossRef]
- Kim, S.J.; Ha, G.S.; Lee, G.; Lim, S.I.; Lee, C.M.; Yang, Y.H.; Lee, J.; Kim, J.E.; Lee, J.H.; Shin, Y.; et al. Enhanced expression of soluble antibody fragments by low-temperature and overdosing with a nitrogen source. Enzyme Microb. Technol. 2018, 115, 9–15. [Google Scholar] [CrossRef]
- Lalonde, M.-E.; Durocher, Y. Therapeutic glycoprotein production in mammalian cells. J. Biotechnol. 2017, 251, 128–140. [Google Scholar] [CrossRef]
- Rajendra, Y.; Kiseljak, D.; Baldi, L.; Hacker, D.L.; Wurm, F.M. A simple high-yielding process for transient gene expression in cho cells. J. Biotechnol. 2011, 153, 22–26. [Google Scholar] [CrossRef]
- Codamo, J.; Munro, T.P.; Hughes, B.S.; Song, M.; Gray, P.P. Enhanced cho cell-based transient gene expression with the epi-cho expression system. Mol. Biotechnol. 2011, 48, 109–115. [Google Scholar] [CrossRef]
- Codamo, J.; Hou, J.J.C.; Hughes, B.S.; Gray, P.P.; Munro, T.P. Efficient mab production in cho cells incorporating pei-mediated transfection, mild hypothermia and the co-expression of xbp-1. J. Chem. Technol. Biotechnol. 2011, 86, 923–934. [Google Scholar] [CrossRef]
- Castan, A.; Schulz, P.; Wenger, T.; Fischer, S. Chapter 7—Cell line development. In Biopharmaceutical Processing; Jagschies, G., Lindskog, E., Łącki, K., Galliher, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 131–146. [Google Scholar]
- Lindskog, E.K. Chapter 31—The upstream process: Principal modes of operation. In Biopharmaceutical Processing; Jagschies, G., Lindskog, E., Łącki, K., Galliher, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 625–635. [Google Scholar]
- Fisher, A.C.; Kamga, M.-H.; Agarabi, C.; Brorson, K.; Lee, S.L.; Yoon, S. The current scientific and regulatory landscape in advancing integrated continuous biopharmaceutical manufacturing. Trends Biotechnol. 2018, 37, 253–267. [Google Scholar] [CrossRef]
- Rathore, A.S.; Agarwal, H.; Sharma, A.K.; Pathak, M.; Muthukumar, S. Continuous processing for production of biopharmaceuticals. Prep. Biochem. Biotechnol. 2015, 45, 836–849. [Google Scholar] [CrossRef]
- Shukla, A.A.; Suda, E. Chapter 3—Harvest and recovery of monoclonal antibodies: Cell removal and clarification. In Process Scale Purification of Antibodies; Gottschalk, U., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 55–79. [Google Scholar]
- Thömmes, J.; Twyman, R.M.; Gottschalk, U. Chapter 10—Alternatives to packed-bed chromatography for antibody extraction and purification. In Process Scale Purification of Antibodies; Gottschalk, U., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 215–231. [Google Scholar]
- Glynn, J. Chapter 11—Process-scale precipitation of impurities in mammalian cell culture broth. In Process Scale Purification of Antibodies; Gottschalk, U., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 233–246. [Google Scholar]
- Singh, N.; Arunkumar, A.; Chollangi, S.; Tan, Z.G.; Borys, M.; Li, Z.J. Clarification technologies for monoclonal antibody manufacturing processes: Current state and future perspectives. Biotechnol. Bioeng. 2016, 113, 698–716. [Google Scholar] [CrossRef]
- Singh, N.; Chollangi, S. Chapter 4—Next-generation clarification technologies for the downstream processing of antibodies. In Process scale Purification of Antibodies; Gottschalk, U., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 81–112. [Google Scholar]
- Kelley, B. Chapter 1—Downstream processing of monoclonal antibodies: Current practices and future opportunities. In Process Scale Purification of Antibodies; Gottschalk, U., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 1–21. [Google Scholar]
- Danielsson, Å. Chapter 17—Affinity chromatography. In Biopharmaceutical Processing; Jagschies, G., Lindskog, E., Łącki, K., Galliher, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 367–378. [Google Scholar]
- Kinna, A.; Tolner, B.; Rota, E.M.; Titchener-Hooker, N.; Nesbeth, D.; Chester, K. Imac capture of recombinant protein from unclarified mammalian cell feed streams. Biotechnol. Bioeng. 2016, 113, 130–140. [Google Scholar] [CrossRef]
- Ghose, S.; Jin, M.; Liu, J.; Hickey, J.; Lee, S. Chapter 14—Integrated polishing steps for monoclonal antibody purification. In Process Scale Purification of Antibodies; Gottschalk, U., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 303–323. [Google Scholar]
- Joshi, V.; Shivach, T.; Kumar, V.; Yadav, N.; Rathore, A. Avoiding antibody aggregation during processing: Establishing hold times. Biotechnol. J. 2014, 9, 1195–1205. [Google Scholar] [CrossRef]
- Liderfelt, J.; Royce, J. Chapter 23—Filtration methods for use in purification processes (concentration and buffer exchange). In Biopharmaceutical Processing; Jagschies, G., Lindskog, E., Łącki, K., Galliher, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 441–453. [Google Scholar]
- Anselmo, A.C.; Gokarn, Y.; Mitragotri, S. Non-invasive delivery strategies for biologics. Nat. Rev. Drug Discov. 2018, 18, 19. [Google Scholar] [CrossRef]
- Sousa, D.; Ferreira, D.; Rodrigues, J.L.; Rodrigues, L.R. Chapter 14—Nanotechnology in targeted drug delivery and therapeutics. In Applications of Targeted Nano Drugs and Delivery Systems; Mohapatra, S.S., Ranjan, S., Dasgupta, N., Mishra, R.K., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 357–409. [Google Scholar]
- Jani, R.; Krupa, G.; Rupal, J. Active targeting of nanoparticles: An innovative technology for drug delivery in cancer therapeutics. J. Drug Deliv. Ther. 2019. [Google Scholar] [CrossRef]
- Abdelaziz, H.M.; Gaber, M.; Abd-Elwakil, M.M.; Mabrouk, M.T.; Elgohary, M.M.; Kamel, N.M.; Kabary, D.M.; Freag, M.S.; Samaha, M.W.; Mortada, S.M.; et al. Inhalable particulate drug delivery systems for lung cancer therapy: Nanoparticles, microparticles, nanocomposites and nanoaggregates. J. Control. Release 2018, 269, 374–392. [Google Scholar] [CrossRef]
- Jackisch, C.; Kim, S.B.; Semiglazov, V.; Melichar, B.; Pivot, X.; Hillenbach, C.; Stroyakovskiy, D.; Lum, B.L.; Elliott, R.; Weber, H.A.; et al. Subcutaneous versus intravenous formulation of trastuzumab for her2-positive early breast cancer: Updated results from the phase iii hannah study. Ann. Oncol. 2015, 26, 320–325. [Google Scholar] [CrossRef]
- Lambertini, M.; Pondé, N.F.; Solinas, C.; de Azambuja, E. Adjuvant trastuzumab: A 10-year overview of its benefit. Expert Rev. Anticancer Ther. 2017, 17, 61–74. [Google Scholar] [CrossRef]
- Sanford, M. Subcutaneous trastuzumab: A review of its use in her2-positive breast cancer. Target. Oncol. 2014, 9, 85–94. [Google Scholar] [CrossRef]
- Reichert, J.M. Adalimumab (humira®). In Handbook of Therapeutic Antibodies, 2nd ed.; Wiley Blackwell: Hoboken, NJ, USA, 2014; Volume 3–4, pp. 1309–1322. [Google Scholar]
- Crommelin, D.J.A.; Hawe, A.; Jiskoot, W. Formulation of biologics including biopharmaceutical considerations. In Pharmaceutical Biotechnology: Fundamentals and Applications; Crommelin, D.J.A., Sindelar, R.D., Meibohm, B., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 83–103. [Google Scholar]
- Singh, S.K.; Mahler, H.-C.; Hartman, C.; Stark, C.A. Are injection site reactions in monoclonal antibody therapies caused by polysorbate excipient degradants? J. Pharm. Sci. 2018, 107, 2735–2741. [Google Scholar] [CrossRef]
- Rayaprolu, B.M.; Strawser, J.J.; Anyarambhatla, G. Excipients in parenteral formulations: Selection considerations and effective utilization with small molecules and biologics. Drug Dev. Ind. Pharm. 2018, 44, 1565–1571. [Google Scholar] [CrossRef]
- Pimentel, F.F.; Morgan, G.; Tiezzi, D.G.; de Andrade, J.M. Development of new formulations of biologics: Expectations, immunogenicity, and safety for subcutaneous trastuzumab. Pharmaceut. Med. 2018, 32, 319–325. [Google Scholar] [CrossRef]
- Garidel, P.; Kuhn, A.B.; Schäfer, L.V.; Karow-Zwick, A.R.; Blech, M. High-concentration protein formulations: How high is high? Eur. J. Pharm. Biopharm. 2017, 119, 353–360. [Google Scholar] [CrossRef]
- Kemter, K.; Altrichter, J.; Derwand, R.; Kriehuber, T.; Reinauer, E.; Scholz, M. Amino acid-based advanced liquid formulation development for highly concentrated therapeutic antibodies balances physical and chemical stability and low viscosity. Biotechnol. J. 2018, 13, e1700523. [Google Scholar] [CrossRef]
- Mandal, A.; Pal, D.; Agrahari, V.; Trinh, H.M.; Joseph, M.; Mitra, A.K. Ocular delivery of proteins and peptides: Challenges and novel formulation approaches. Adv. Drug Del. Rev. 2018, 126, 67–95. [Google Scholar] [CrossRef]
- Reslan, M.; Kayser, V. The effect of deuterium oxide on the conformational stability and aggregation of bovine serum albumin. Pharm. Dev. Technol. 2016, 1–7. [Google Scholar] [CrossRef]
- Reslan, M.; Ranganathan, V.; Macfarlane, D.R.; Kayser, V. Choline ionic liquid enhances the stability of herceptin(r) (trastuzumab). Chem. Commun. (Camb.) 2018, 54, 10622–10625. [Google Scholar] [CrossRef]
- Reslan, M.; Kayser, V. Ionic liquids as biocompatible stabilizers of proteins. Biophys. Rev. 2018, 10, 781–793. [Google Scholar] [CrossRef]
- Emami, F.; Vatanara, A.; Park, E.J.; Na, D.H. Drying technologies for the stability and bioavailability of biopharmaceuticals. Pharmaceutics 2018, 10, 131. [Google Scholar] [CrossRef]
- Izutsu, K.I. Applications of freezing and freeze-drying in pharmaceutical formulations. In Adv. Exp. Med. Biol.; Springer: New York, NY, USA, 2018; Volume 1081, pp. 371–383. [Google Scholar]
- Schermeyer, M.T.; Wöll, A.K.; Kokke, B.; Eppink, M.; Hubbuch, J. Characterization of highly concentrated antibody solution—A toolbox for the description of protein long-term solution stability. mAbs 2017, 9, 1169–1185. [Google Scholar] [CrossRef]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Helk, B.; Forrer, K.; Trout, B.L. Evaluation of a non-arrhenius model for therapeutic monoclonal antibody aggregation. J. Pharm. Sci. 2011, 100, 2526–2542. [Google Scholar] [CrossRef]
- Rabia, L.A.; Desai, A.A.; Jhajj, H.S.; Tessier, P.M. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem. Eng. J. 2018, 137, 365–374. [Google Scholar] [CrossRef]
- Van de Bovenkamp, F.S.; Derksen, N.I.L.; van Breemen, M.J.; de Taeye, S.W.; Ooijevaar-de Heer, P.; Sanders, R.W.; Rispens, T. Variable domain n-linked glycans acquired during antigen-specific immune responses can contribute to immunoglobulin g antibody stability. Front. Immunol. 2018, 9, 740. [Google Scholar] [CrossRef]
- Kuhn, A.B.; Kube, S.; Karow-Zwick, A.R.; Seeliger, D.; Garidel, P.; Blech, M.; Schäfer, L.V. Improved solution-state properties of monoclonal antibodies by targeted mutations. J. Phys. Chem. B 2017, 121, 10818–10827. [Google Scholar] [CrossRef]
- Singh, S.N.; Yadav, S.; Shire, S.J.; Kalonia, D.S. Dipole-dipole interaction in antibody solutions: Correlation with viscosity behavior at high concentration. Pharm. Res. 2014, 31, 2549–2558. [Google Scholar] [CrossRef]
- Ionescu, R.M.; Vlasak, J.; Price, C.; Kirchmeier, M. Contribution of variable domains to the stability of humanized igg1 monoclonal antibodies. J. Pharm. Sci. 2008, 97, 1414–1426. [Google Scholar] [CrossRef]
- Liu, L. Pharmacokinetics of monoclonal antibodies and fc-fusion proteins. Protein Cell 2018, 9, 15–32. [Google Scholar] [CrossRef]
- Schweizer, D.; Serno, T.; Goepferich, A. Controlled release of therapeutic antibody formats. Eur. J. Pharm. Biopharm. 2014, 88, 291–309. [Google Scholar] [CrossRef]
- Rudnick, S.I.; Adams, G.P. Affinity and avidity in antibody-based tumor targeting. Cancer Biother. Radiopharm. 2009, 24, 155–161. [Google Scholar] [CrossRef]
- Ickenstein, L.M.; Garidel, P. Hydrogel formulations for biologicals: Current spotlight from a commercial perspective. Ther. Deliv. 2018, 9, 221–230. [Google Scholar] [CrossRef]
- Saxena, A.; Wu, D. Advances in therapeutic fc engineering—Modulation of igg-associated effector functions and serum half-life. Front. Immunol. 2016, 7, 580. [Google Scholar] [CrossRef]
- Jefferis, R. Glycosylation of antibody molecules. In Handbook of Therapeutic Antibodies, 2nd ed.; Wiley Blackwell: Hoboken, NJ, USA, 2014; Volume 1–4, pp. 171–200. [Google Scholar]
- Zheng, K.; Bantog, C.; Bayer, R. The impact of glycosylation on monoclonal antibody conformation and stability. mAbs 2011, 3, 568–576. [Google Scholar] [CrossRef]
- Lei, C.; Gong, R.; Ying, T. Editorial: Antibody fc engineering: Towards better therapeutics. Front. Immunol. 2018, 9, 2450. [Google Scholar] [CrossRef]
- Booth, B.J.; Ramakrishnan, B.; Narayan, K.; Wollacott, A.M.; Babcock, G.J.; Shriver, Z.; Viswanathan, K. Extending human igg half-life using structure-guided design. mAbs 2018, 10, 1098–1110. [Google Scholar] [CrossRef]
- Kellner, C.; Otte, A.; Cappuzzello, E.; Klausz, K.; Peipp, M. Modulating cytotoxic effector functions by fc engineering to improve cancer therapy. Transfus. Med. Hemoth. 2017, 44, 327–336. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Pelletier, M.; Cvitkovic, R.; Bonnell, J.; Chang, C.Y.; Koksal, A.C.; O’Connor, E.; Gao, X.; Yu, X.Q.; et al. Enhancement of antibody functions through fc multiplications. mAbs 2017, 9, 393–403. [Google Scholar] [CrossRef][Green Version]
- Lefranc, M.P.; Ehrenmann, F.; Kossida, S.; Giudicelli, V.; Duroux, P. Use of imgt® databases and tools for antibody engineering and humanization. In Methods Mol. Biol.; Humana Press Inc.: New York, NY, USA, 2018; Volume 1827, pp. 35–69. [Google Scholar]
- Lefranc, M.-P.; Giudicelli, V.; Duroux, P.; Jabado-Michaloud, J.; Folch, G.; Aouinti, S.; Carillon, E.; Duvergey, H.; Houles, A.; Paysan-Lafosse, T.; et al. Imgt(®), the international immunogenetics information system(®) 25 years on. Nucleic Acids Res. 2015, 43, D413–D422. [Google Scholar] [CrossRef]
- Martin, A.C.R.; Allen, J. Bioinformatics tools for analysis of antibodies. In Handbook of Therapeutic Antibodies, 2nd ed.; Wiley Blackwell: Hoboken, NJ, USA, 2014; Volume 1–4, pp. 201–228. [Google Scholar]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Pandya, A.; Howard, M.J.; Zloh, M.; Dalby, P.A. An evaluation of the potential of nmr spectroscopy and computational modelling methods to inform biopharmaceutical formulations. Pharmaceutics 2018, 10, 1–24. [Google Scholar] [CrossRef]
- Rathore, D.; Faustino, A.; Schiel, J.; Pang, E.; Boyne, M.; Rogstad, S. The role of mass spectrometry in the characterization of biologic protein products. Expert Rev. Proteomic. 2018, 15, 431–449. [Google Scholar] [CrossRef]
- Wang, X.; An, Z.; Luo, W.; Xia, N.; Zhao, Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell 2018, 9, 74–85. [Google Scholar] [CrossRef]
- Baker Edward, N. Crystallography and the development of therapeutic medicines. IUCrJ 2018, 5, 118–119. [Google Scholar] [CrossRef]
- Vénien-Bryan, C.; Li, Z.; Vuillard, L.; Boutin, J.A. Cryo-electron microscopy and X-ray crystallography: Complementary approaches to structural biology and drug discovery. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 174–183. [Google Scholar] [CrossRef]
- Brader, M.L.; Baker, E.N.; Dunn, M.F.; Laue, T.M.; Carpenter, J.F. Using x-ray crystallography to simplify and accelerate biologics drug development. J. Pharm. Sci. 2017, 106, 477–494. [Google Scholar] [CrossRef]
- Nero, T.L.; Parker, M.W.; Morton, C.J. Protein structure and computational drug discovery. Biochem. Soc. Trans. 2018, 46, 1367–1379. [Google Scholar] [CrossRef]
- Blaffert, J.; Haeri, H.H.; Blech, M.; Hinderberger, D.; Garidel, P. Spectroscopic methods for assessing the molecular origins of macroscopic solution properties of highly concentrated liquid protein solutions. Anal. Biochem. 2018, 561–562, 70–88. [Google Scholar] [CrossRef]
- Brinson, R.G.; Marino, J.P.; Delaglio, F.; Arbogast, L.W.; Evans, R.M.; Kearsley, A.; Gingras, G.; Ghasriani, H.; Aubin, Y.; Pierens, G.K.; et al. Enabling adoption of 2d-nmr for the higher order structure assessment of monoclonal antibody therapeutics. mAbs 2019, 11, 94–105. [Google Scholar] [CrossRef]
- Young, J.A.; Gabrielson, J.P. Higher order structure methods for similarity assessment. In Biosimilars: Regulatory, Clinical, and Biopharmaceutical Development; Gutka, H.J., Yang, H., Kakar, S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 321–337. [Google Scholar]
- Kumar, S.; Plotnikov, N.V.; Rouse, J.C.; Singh, S.K. Biopharmaceutical informatics: Supporting biologic drug development via molecular modelling and informatics. J. Pharm. Pharmacol. 2018, 70, 595–608. [Google Scholar] [CrossRef]
- Westbrook, J.D.; Burley, S.K. How structural biologists and the protein data bank contributed to recent fda new drug approvals. Structure 2018, 27, 211–217. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Christie, C.; Duarte, J.M.; Feng, Z.; Westbrook, J.; Young, J.; Zardecki, C. Rcsb protein data bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018, 27, 316–330. [Google Scholar] [CrossRef]
- Kuroda, D.; Tsumoto, K. Antibody affinity maturation by computational design. In Methods Mol. Biol.; Humana Press Inc.: New York, NY, USA, 2018; Volume 1827, pp. 15–34. [Google Scholar]
- Fischman, S.; Ofran, Y. Computational design of antibodies. Curr. Opin. Struct. Biol. 2018, 51, 156–162. [Google Scholar] [CrossRef]
- Adolf-Bryfogle, J.; Kalyuzhniy, O.; Kubitz, M.; Weitzner, B.D.; Hu, X.; Adachi, Y.; Schief, W.R.; Dunbrack, R.L., Jr. Rosettaantibodydesign (rabd): A general framework for computational antibody design. PLoS Comp. Biol. 2018, 14, e1006112. [Google Scholar] [CrossRef]
- Branco, R.J.; Dias, A.M.; Roque, A.C. Understanding the molecular recognition between antibody fragments and protein a biomimetic ligand. J. Chromatogr. A 2012, 1244, 106–115. [Google Scholar] [CrossRef]
- Huang, B.; Liu, F.F.; Dong, X.Y.; Sun, Y. Molecular mechanism of the affinity interactions between protein a and human immunoglobulin g1 revealed by molecular simulations. J. Phys. Chem. B 2011, 115, 4168–4176. [Google Scholar] [CrossRef]
- Van der Kant, R.; van Durme, J.; Rousseau, F.; Schymkowitz, J. Solubis: Optimizing protein solubility by minimal point mutations. In Methods Mol. Biol.; Humana Press Inc.: New York, NY, USA, 2019; Volume 1873, pp. 317–333. [Google Scholar]
- Gil-Garcia, M.; Banó-Polo, M.; Varejao, N.; Jamroz, M.; Kuriata, A.; Díaz-Caballero, M.; Lascorz, J.; Morel, B.; Navarro, S.; Reverter, D.; et al. Combining structural aggregation propensity and stability predictions to redesign protein solubility. Mol. Pharm. 2018, 15, 3846–3859. [Google Scholar] [CrossRef]
- Seeliger, D.; Schulz, P.; Litzenburger, T.; Spitz, J.; Hoerer, S.; Blech, M.; Enenkel, B.; Studts, J.M.; Garidel, P.; Karow, A.R. Boosting antibody developability through rational sequence optimization. mAbs 2015, 7, 505–515. [Google Scholar] [CrossRef]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Prediction of aggregation prone regions of therapeutic proteins. J. Phys. Chem. B 2010, 114, 6614–6624. [Google Scholar] [CrossRef]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef]
- Majumder, S.; Jones, M.T.; Kimmel, M.; Alphonse Ignatius, A. Probing conformational diversity of fc domains in aggregation-prone monoclonal antibodies. Pharm. Res. 2018, 35, 220. [Google Scholar] [CrossRef]
- Nakamura, H.; Oda-Ueda, N.; Ueda, T.; Ohkuri, T. Introduction of a glycosylation site in the constant region decreases the aggregation of adalimumab fab. Biochem. Biophys. Res. Commun. 2018, 503, 752–756. [Google Scholar] [CrossRef]
- Pepinsky, R.B.; Silvian, L.; Berkowitz, S.A.; Farrington, G.; Lugovskoy, A.; Walus, L.; Eldredge, J.; Capili, A.; Mi, S.; Graff, C.; et al. Improving the solubility of anti-lingo-1 monoclonal antibody li33 by isotype switching and targeted mutagenesis. Protein Sci. 2010, 19, 954–966. [Google Scholar] [CrossRef]
- Wu, S.-J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.S.; et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Challenges | Advancements |
|---|---|
| Manufacture Considerations | |
| Hybridoma technology produces immunogenic mAbs | Humanization technologies [4,5,6] |
| Yield from hybridoma technology is variable | Commercial cell line development and recombinant technology [4,5,6]; lower organism systems for fragment mAbs, mammalian systems for whole mAbs, and Fc fusions [16,30,31] |
| Significance of post-translational modifications and higher-order structure in mAb product | |
| CHO expressed mAbs contain an immunogenic glycosylation profile | Human-based expression systems [16,30,31,32] |
| HEK 293 expressed mAbs are prone to aggregation | HKB-11 and PER.C6 cell lines [32] |
| Undesirable byproducts produced in the manufacture process | in vitro cell-free synthesis technology [33,34] |
| Stability of mAb affects manufacture yield due to product loss through aggregation in downstream processing steps | Enhancing mAb stability through framework mutations and hyperglycosylation [5,35,36,37,38,39,40] |
| Treatment Considerations for Drug Design and Formulation | |
| Susceptibility of mAb to degradation limits delivery to intravenous and subcutaneous only | Enhancing mAb stability through framework mutations, hyperglycosylation [5,35,36,37,38,39,40], and nanocarrier technologies [8,41] |
| Stability of mAb limits concentration of formulation | |
| Concentration of mAb affects viscosity and injection pressure for subcutaneous delivery | Excipients, fragment mAbs, PEGylation, and hyperglycosylation [25,26,27,41] |
| Poor tissue penetration and biodistribution | Fragment mAbs and nanocarrier technologies [8,41,42]; high affinity ADCs [43,44,45] |
| Reduced half-life in low MW species | PEGylation, hyper-glycosylation and Fc fusion proteins [8,41,46]; modulation of FcRn recycling through Fc mutation [11,13] |
| Modulation of immunological engagement | Isotype switching, glycoengineering, and Fc mutations [11,13,15,43,46] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sifniotis, V.; Cruz, E.; Eroglu, B.; Kayser, V. Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation. Antibodies 2019, 8, 36. https://doi.org/10.3390/antib8020036
Sifniotis V, Cruz E, Eroglu B, Kayser V. Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation. Antibodies. 2019; 8(2):36. https://doi.org/10.3390/antib8020036
Chicago/Turabian StyleSifniotis, Vicki, Esteban Cruz, Barbaros Eroglu, and Veysel Kayser. 2019. "Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation" Antibodies 8, no. 2: 36. https://doi.org/10.3390/antib8020036
APA StyleSifniotis, V., Cruz, E., Eroglu, B., & Kayser, V. (2019). Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation. Antibodies, 8(2), 36. https://doi.org/10.3390/antib8020036