Antibody Conjugates-Recent Advances and Future Innovations

 , , and
, , and
Abstract
1. Introduction
2. Critical Considerations for Antibody Conjugates
2.1. Target and Antibody Selection
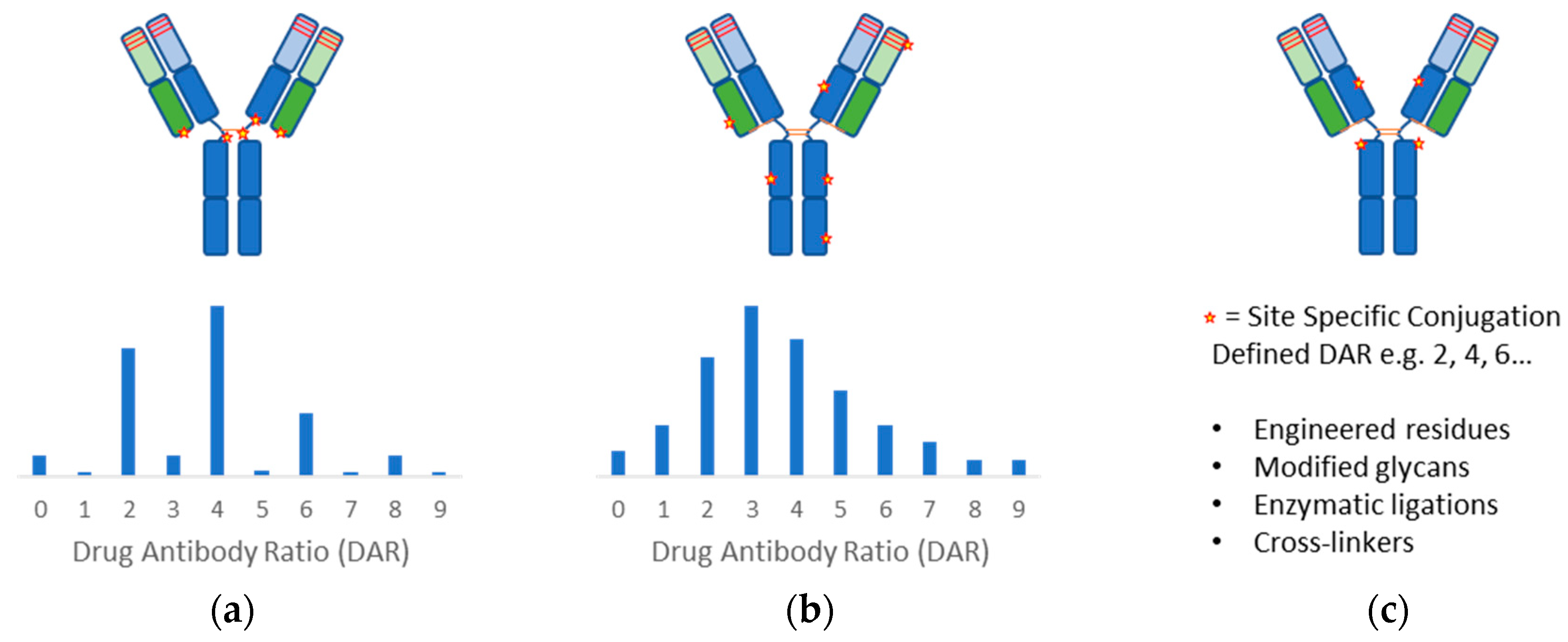
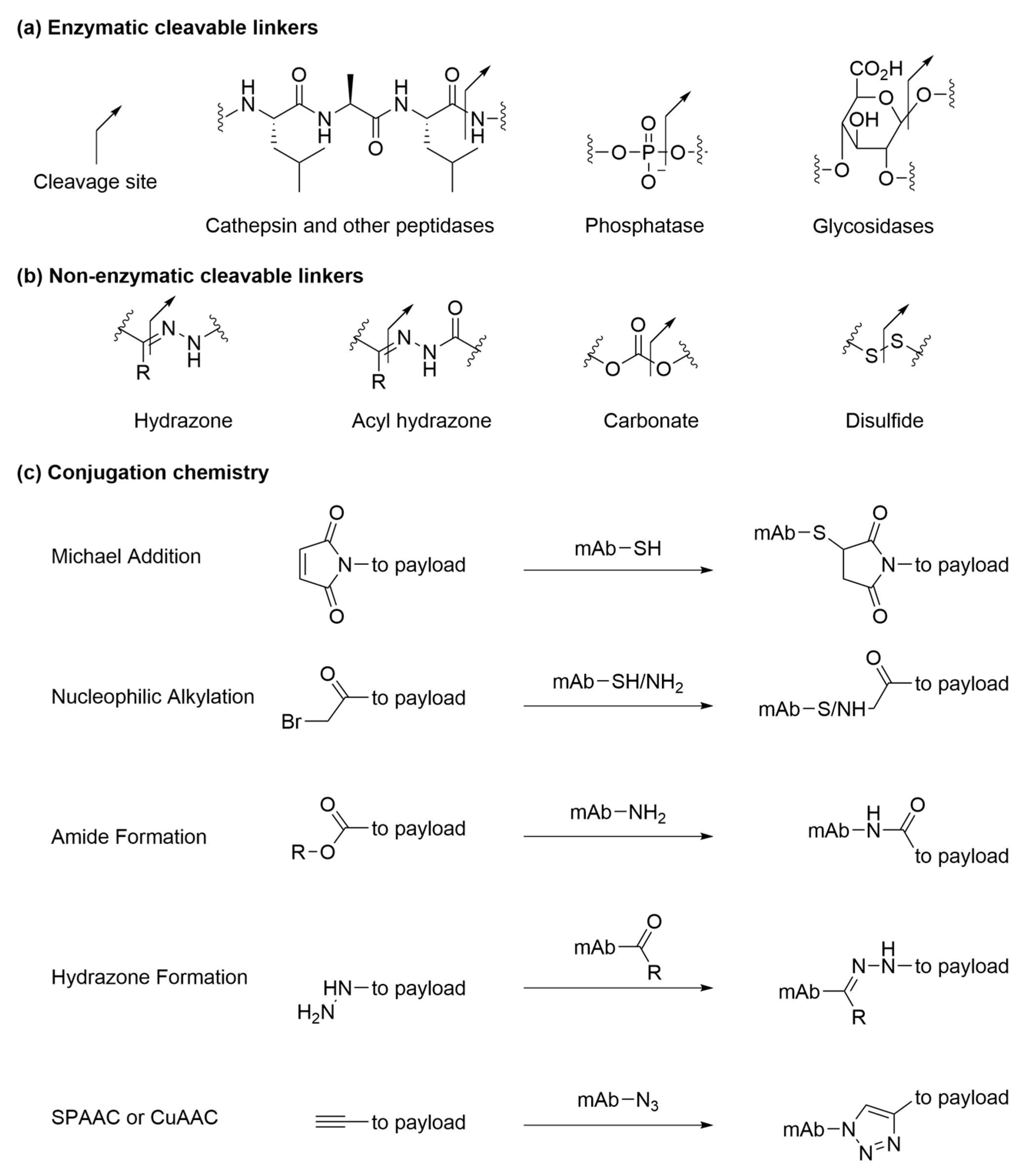
2.2. Conjugation Methods
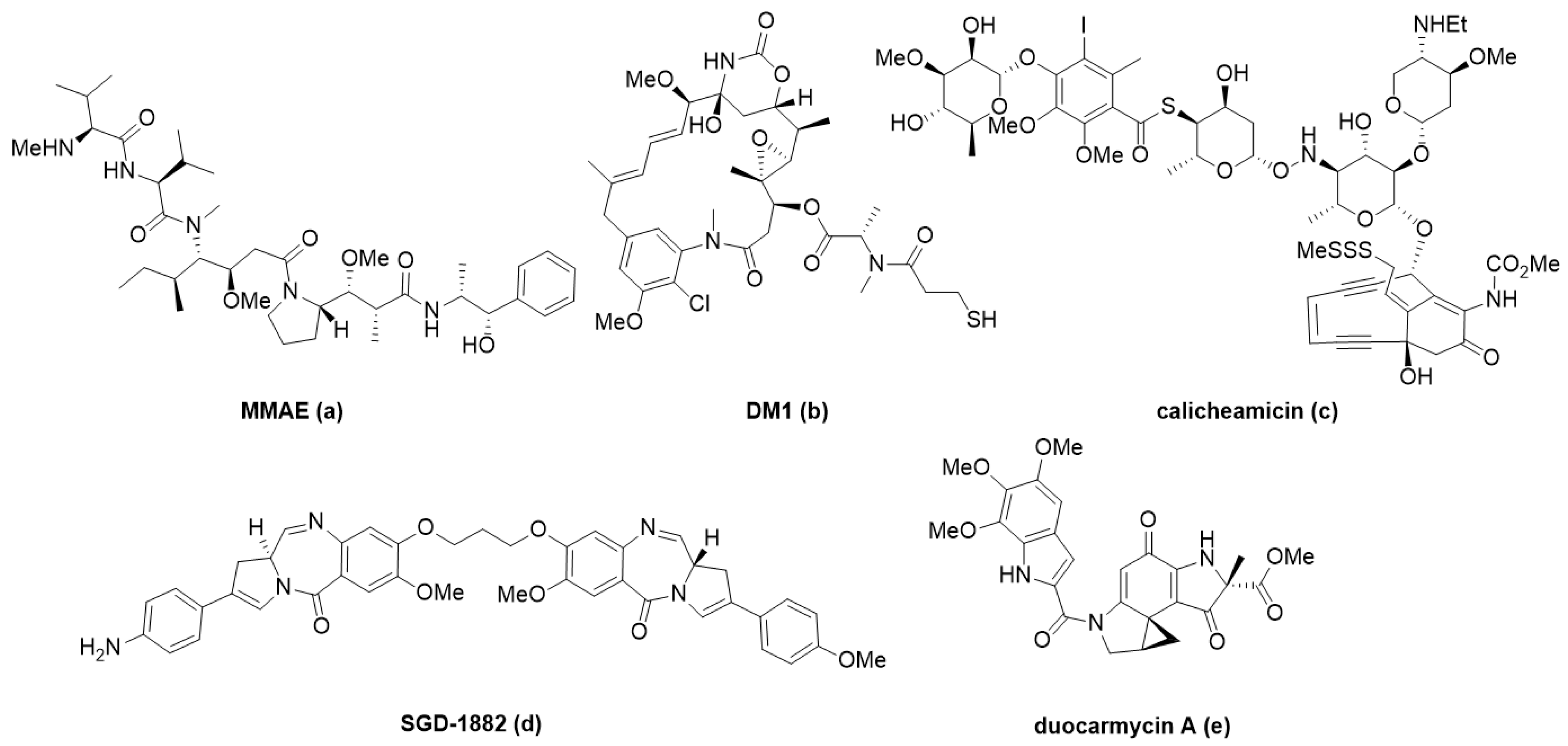
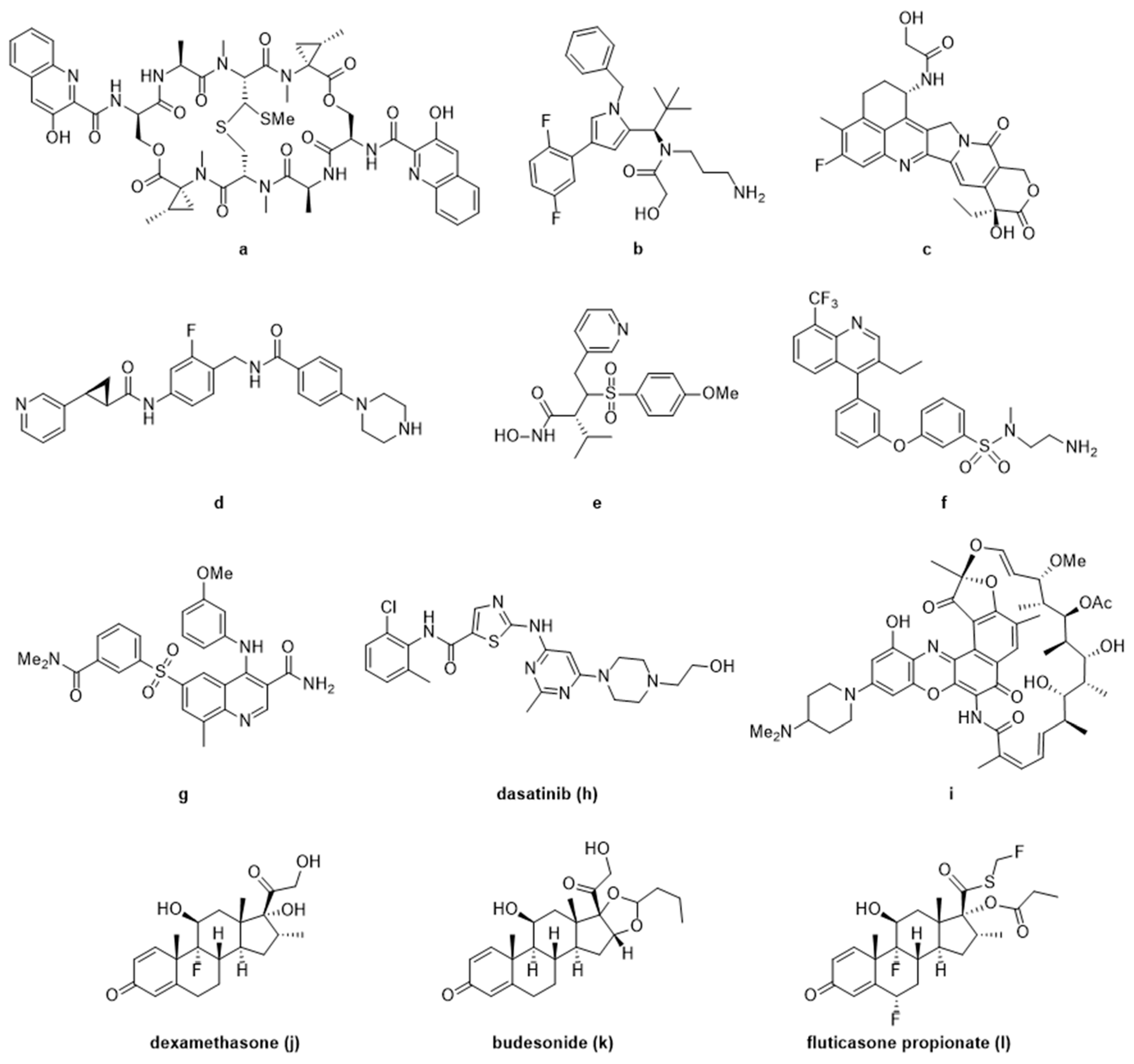
3. Current Small Molecule Payloads and Beyond
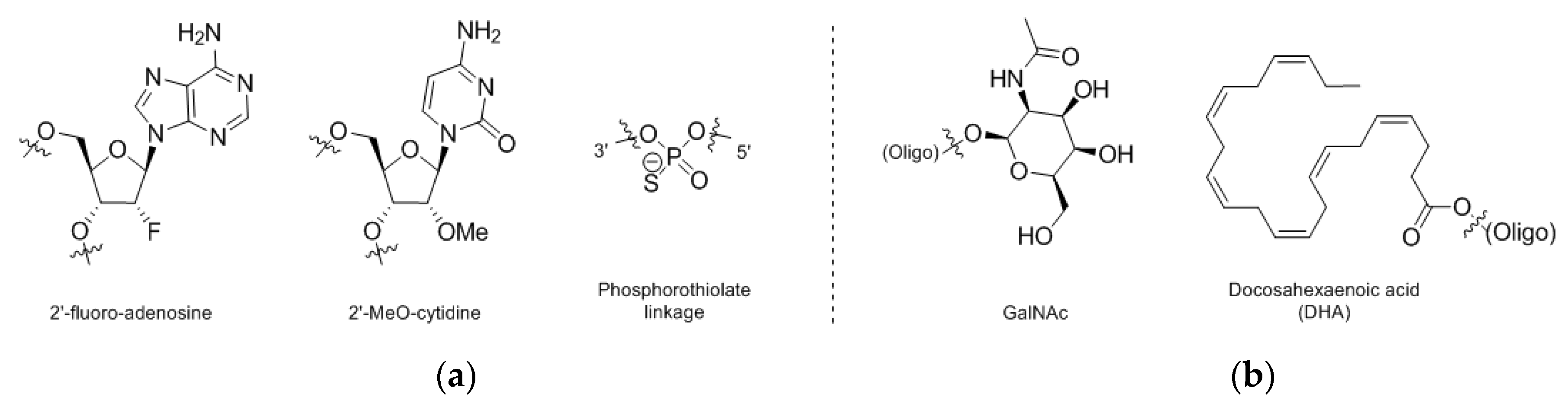
4. Nucleic Acid Conjugates
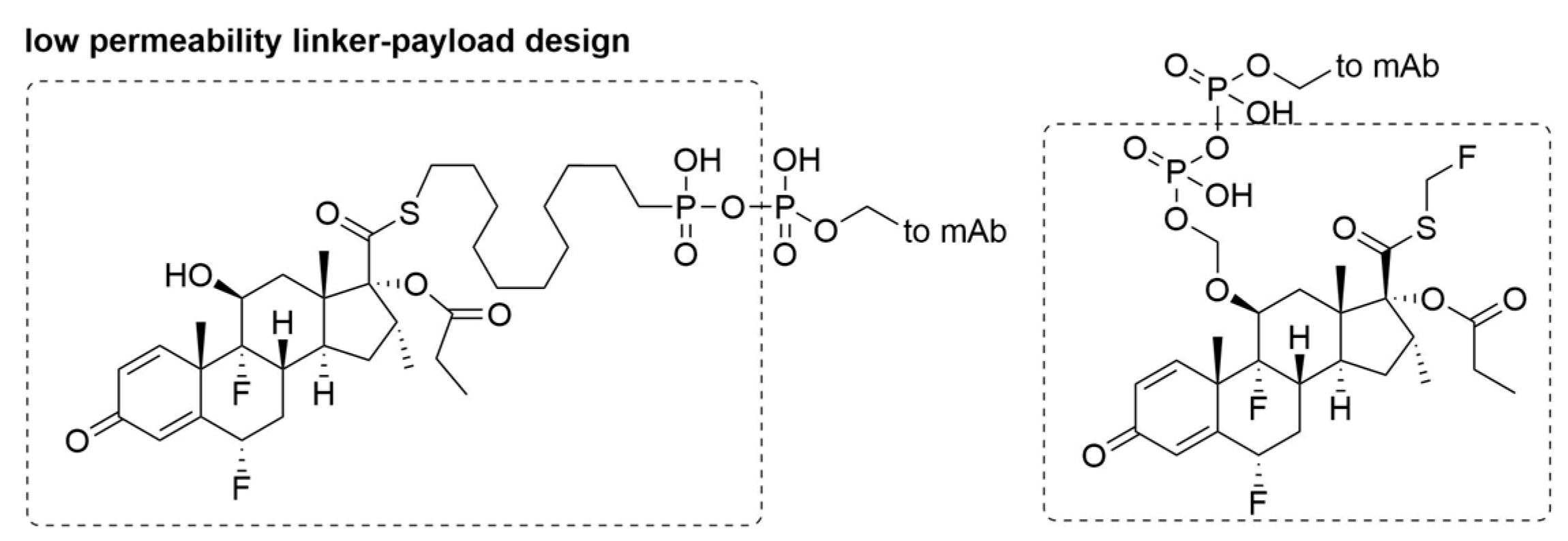
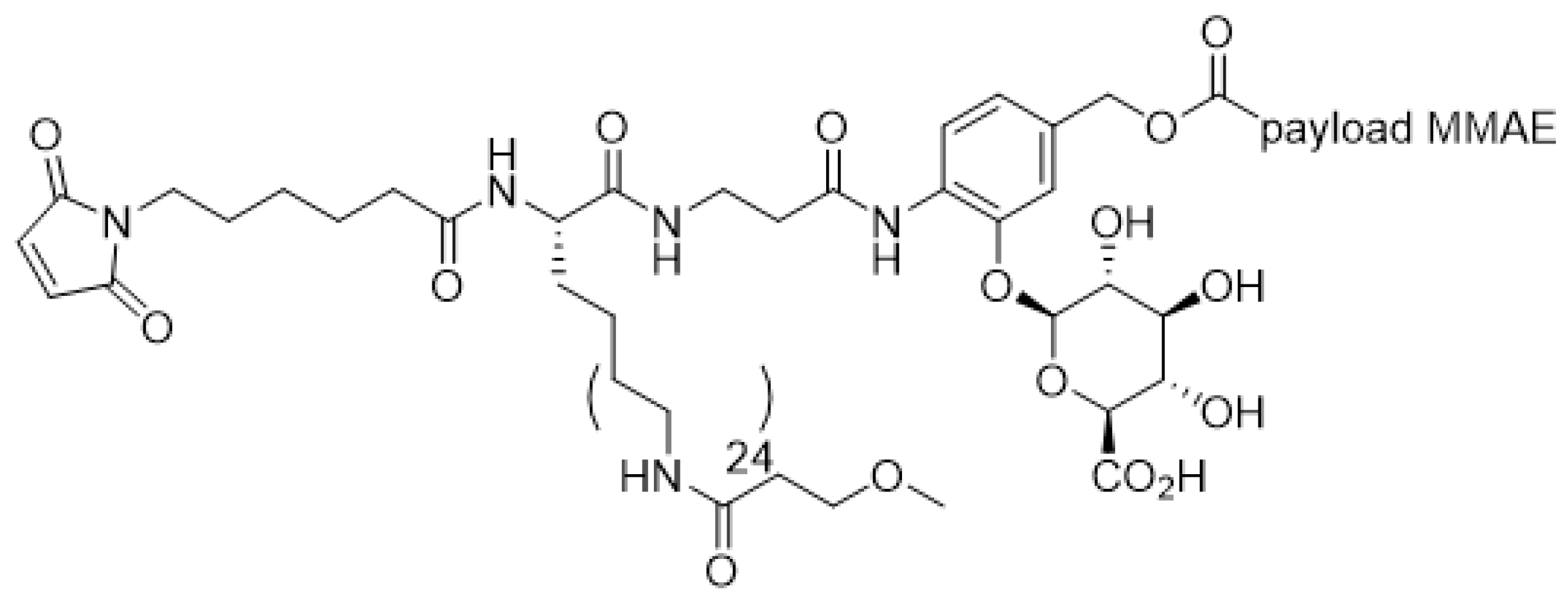
5. Linkers
6. Absorption, Distribution, Metabolism, and Excretion (ADME) of ADCs
7. Conjugate Developability, Formulations, and Characteristics
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Singh, S.; Kumar, N.K.; Dwiwedi, P.; Charan, J.; Kaur, R.; Sidhu, P.; Chugh, V.K. Monoclonal antibodies: A review. Curr. Clin. Pharmacol. 2018, 13, 85–99. [Google Scholar] [CrossRef]
- Grilo, A.L.; Mantalaris, A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. mAbs 2019, 11, 219–238. [Google Scholar] [CrossRef]
- Sifniotis, V.; Cruz, E.; Eroglu, B.; Kayser, V. Current advancements in addressing key challenges of therapeutic antibody design, manufacture, and formulation. Antibodies 2019, 8, 36. [Google Scholar] [CrossRef]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.F.U.H.; Wang, R.; Ling, S.; Wang, S. Antibody engineering for pursuing a healthier future. Front. Microbiol. 2017, 8, 495. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, L.; Clark, M.; Waldmann, H.; Winter, G. Reshaping human antibodies for therapy. Nature 1988, 332, 323–327. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs 2010, 2, 256–265. [Google Scholar] [CrossRef]
- Strohl, W.R. Human antibody discovery platforms. In Protein Therapeutics; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2017; pp. 113–159. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef]
- Mukherjee, A.; Waters, A.K.; Babic, I.; Nurmemmedov, E.; Glassy, M.C.; Kesari, S.; Yenugonda, V.M. Antibody drug conjugates: Progress, pitfalls, and promises. Hum. Antibodies 2019, 27, 53–62. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- FDA Approves First Chemoimmunotherapy Regimen for Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-chemoimmunotherapy-regimen-patients-relapsed-or-refractory-diffuse-large-b-cell (accessed on 25 June 2019).
- Petersen, B.H.; DeHerdt, S.V.; Schneck, D.W.; Bumol, T.F. The human immune response to KS1/4-desacetylvinblastine (LY256787) and KS1/4-desacetylvinblastine hydrazide (LY203728) in single and multiple dose clinical studies. Cancer Res. 1991, 51, 2286–2290. [Google Scholar] [PubMed]
- Abdollahpour-Alitappeh, M.; Lotfinia, M.; Gharibi, T.; Mardaneh, J.; Farhadihosseinabadi, B.; Larki, P.; Faghfourian, B.; Sepehr, K.S.; Abbaszadeh-Goudarzi, K.; Abbaszadeh-Goudarzi, G.; et al. Antibody-drug conjugates (ADCs) for cancer therapy: Strategies, challenges, and successes. J. Cell. Physiol. 2019, 234, 5628–5642. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.H.; Brembilla, N.C. Immunogenicity of biologic therapies: Causes and consequences. Expert Rev. Clin. Immunol. 2018, 14, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.; Castaneda Hernandez, G.; Jacobs, I.A.; Alvarez, D.F.; Carini, C. Assessing the immunogenicity of biopharmaceuticals. BioDrugs 2016, 30, 195–206. [Google Scholar] [CrossRef]
- Ghetie, V.; Vitetta, E. Immunotoxins in the therapy of cancer: From bench to clinic. Pharmacol. Ther. 1994, 63, 209–234. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The neonatal Fc Receptor (FcRn): A misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Choi, H.; Stokell, D.; Tang, J.; Murphy, A.; Wrobleski, A.; Feng, Y. The properties of cysteine-conjugated antibody-drug conjugates are impacted by the IgG subclass. AAPS J. 2018, 20, 103. [Google Scholar] [CrossRef]
- Mohammed, R.; Milne, A.; Kayani, K.; Ojha, U. How the discovery of rituximab impacted the treatment of B-cell non-Hodgkin’s lymphomas. J. Blood Med. 2019, 10, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2018, 7, e1395127. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Li, G.; Parsons, K.; Phillips, G.L.; Sliwkowski, M.X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 2011, 128, 347–356. [Google Scholar] [CrossRef] [PubMed]
- English, D.P.; Bellone, S.; Schwab, C.L.; Bortolomai, I.; Bonazzoli, E.; Cocco, E.; Buza, N.; Hui, P.; Lopez, S.; Ratner, E.; et al. T-DM1, a novel antibody-drug conjugate, is highly effective against primary HER2 overexpressing uterine serous carcinoma in vitro and in vivo. Cancer Med. 2014, 3, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, R.; Lopez, S.; Bellone, S.; Cocco, E.; Schwab, C.L.; Black, J.D.; Centritto, F.; Zhu, L.; Bonazzoli, E.; Buza, N.; et al. T-DM1, a novel antibody-drug conjugate, is highly effective against uterine and ovarian carcinosarcomas overexpressing HER2. Clin. Exp. Metastasis 2015, 32, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Black, J.; Menderes, G.; Bellone, S.; Schwab, C.L.; Bonazzoli, E.; Ferrari, F.; Predolini, F.; De Haydu, C.; Cocco, E.; Buza, N.; et al. SYD985, a novel duocarmycin-based HER2-targeting antibody-drug conjugate, shows antitumor activity in uterine serous carcinoma with HER2/Neu expression. Mol. Cancer Ther. 2016, 15, 1900–1909. [Google Scholar] [CrossRef]
- Menderes, G.; Bonazzoli, E.; Bellone, S.; Black, J.; Altwerger, G.; Masserdotti, A.; Pettinella, F.; Zammataro, L.; Buza, N.; Hui, P.; et al. SYD985, a novel duocarmycin-based HER2-targeting antibody-drug conjugate, shows promising antitumor activity in epithelial ovarian carcinoma with HER2/Neu expression. Gynecol. Oncol. 2017, 146, 179–186. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase i inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Arrojo, R.; Liu, D.; Rossi, E.A.; Chang, C.H.; Goldenberg, D.M. Sacituzumab govitecan (IMMU-132), an Anti-Trop-2/SN-38 antibody-drug conjugate: Characterization and efficacy in pancreatic, gastric, and other cancers. Bioconjug. Chem. 2015, 26, 919–931. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Guarino, M.J.; Masters, G.; Purcell, W.T.; Starodub, A.N.; Horn, L.; Scheff, R.J.; Bardia, A.; Messersmith, W.A.; Berlin, J.; et al. Therapy of advanced non-small-cell lung cancer with an SN-38-Anti-Trop-2 drug conjugate, sacituzumab govitecan. J. Clin. Oncol. 2017, 35, 2790–2797. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.A.; Chan, K.F.; Lin, P.C.; Song, Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. mAbs 2018, 10, 693–711. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef]
- Szot, C.; Saha, S.; Zhang, X.M.; Zhu, Z.; Hilton, M.B.; Morris, K.; Seaman, S.; Dunleavey, J.M.; Hsu, K.-S.; Yu, G.-J.; et al. Tumor stroma–targeted antibody-drug conjugate triggers localized anticancer drug release. J. Clin. Investig. 2018, 128, 2927–2943. [Google Scholar] [CrossRef]
- Liu, R.; Wang, R.E.; Wang, F. Antibody-drug conjugates for non-oncological indications. Expert Opin. Biol. Ther. 2016, 16, 591–593. [Google Scholar] [CrossRef]
- Yu, S.; Lim, A.; Tremblay, M.S. Next horizons: ADCs beyond oncology. In Innovations for Next-Generation Antibody-Drug Conjugates; Humana Press: Totowa, NJ, USA, 2018; pp. 321–347. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Mariathasan, S.; Tan, M.-W. Antibody–antibiotic conjugates: A novel therapeutic platform against bacterial infections. Trends Mol. Med. 2017, 23, 135–149. [Google Scholar] [CrossRef]
- Everts, M.; Kok, R.J.; Ásgeirsdóttir, S.A.; Melgert, B.N.; Moolenaar, T.J.M.; Koning, G.A.; van Luyn, M.J.A.; Meijer, D.K.F.; Molema, G. Selective intracellular delivery of dexamethasone into activated endothelial cells using an e-selectin-directed immunoconjugate. J. Immunol. 2002, 168, 883–889. [Google Scholar] [CrossRef]
- Lim, R.K.; Yu, S.; Cheng, B.; Li, S.; Kim, N.J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G.; et al. Targeted delivery of LXR agonist using a site-specific antibody-drug conjugate. Bioconjug. Chem. 2015, 26, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Pearson, A.D.; Lim, R.K.; Rodgers, D.T.; Li, S.; Parker, H.B.; Weglarz, M.; Hampton, E.N.; Bollong, M.J.; Shen, J.; et al. Targeted delivery of an anti-inflammatory PDE4 inhibitor to immune cells via an antibody-drug conjugate. Mol. Ther. 2016, 24, 2078–2089. [Google Scholar] [CrossRef]
- Beaumont, M.; Tomazela, D.; Hodges, D.; Ermakov, G.; Hsieh, E.; Figueroa, I.; So, O.-Y.; Song, Y.; Ma, H.; Antonenko, S.; et al. Antibody-drug conjugates: Integrated bioanalytical and biodisposition assessments in lead optimization and selection. AAPS Open 2018, 4, 6. [Google Scholar] [CrossRef]
- Kvirkvelia, N.; McMenamin, M.; Gutierrez, V.I.; Lasareishvili, B.; Madaio, M.P. Human anti-alpha3(IV)NC1 antibody drug conjugates target glomeruli to resolve nephritis. Am. J. Physiol. Ren. Physiol. 2015, 309, F680–F684. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.E.; Liu, T.; Wang, Y.; Cao, Y.; Du, J.; Luo, X.; Deshmukh, V.; Kim, C.H.; Lawson, B.R.; Tremblay, M.S.; et al. An immunosuppressive antibody-drug conjugate. J. Am. Chem. Soc. 2015, 137, 3229–3232. [Google Scholar] [CrossRef] [PubMed]
- Palchaudhuri, R.; Saez, B.; Hoggatt, J.; Schajnovitz, A.; Sykes, D.B.; Tate, T.A.; Czechowicz, A.; Kfoury, Y.; Ruchika, F.; Rossi, D.J.; et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat. Biotechnol. 2016, 34, 738–745. [Google Scholar] [CrossRef]
- Kern, J.C.; Dooney, D.; Zhang, R.; Liang, L.; Brandish, P.E.; Cheng, M.; Feng, G.; Beck, A.; Bresson, D.; Firdos, J.; et al. Novel phosphate modified cathepsin B linkers: Improving aqueous solubility and enhancing payload scope of ADCs. Bioconjug. Chem. 2016, 27, 2081–2088. [Google Scholar] [CrossRef]
- Brandish, P.E.; Palmieri, A.; Antonenko, S.; Beaumont, M.; Benso, L.; Cancilla, M.; Cheng, M.; Fayadat-Dilman, L.; Feng, G.; Figueroa, I.; et al. Development of Anti-CD74 antibody-drug conjugates to target glucocorticoids to immune cells. Bioconjug. Chem. 2018, 29, 2357–2369. [Google Scholar] [CrossRef]
- Graversen, J.H.; Svendsen, P.; Dagnaes-Hansen, F.; Dal, J.; Anton, G.; Etzerodt, A.; Petersen, M.D.; Christensen, P.A.; Moller, H.J.; Moestrup, S.K. Targeting the hemoglobin scavenger receptor CD163 in macrophages highly increases the anti-inflammatory potency of dexamethasone. Mol. Ther. 2012, 20, 1550–1558. [Google Scholar] [CrossRef]
- Thomsen, K.L.; Møller, H.J.; Graversen, J.H.; Magnusson, N.E.; Moestrup, S.K.; Vilstrup, H.; Grønbæk, H. Anti-CD163-dexamethasone conjugate inhibits the acute phase response to lipopolysaccharide in rats. World J. Hepatol. 2016, 8, 726–730. [Google Scholar] [CrossRef]
- Yarian, F.; Alibakhshi, A.; Eyvazi, S.; Arezumand, R.; Ahangarzadeh, S. Antibody-drug therapeutic conjugates: Potential of antibody-siRNAs in cancer therapy. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, D.; Kazane, S.; Javahishvili, T.; Tian, F.; Song, F.; Sellers, A.; Barnett, B.; Schultz, P.G. Site-specific antibody–polymer conjugates for siRNA delivery. J. Am. Chem. Soc. 2013, 135, 13885–13891. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Site-specific antibody conjugation for ADC and beyond. Biomedicines 2017, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.R.; Liu, B. Methods for site-specific drug conjugation to antibodies. mAbs 2014, 6, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Hackenberger, C.P.; Leonhardt, H.; Helma, J. Current status: Site-specific antibody drug conjugates. J. Clin. Immunol. 2016, 36 (Suppl. S1), 100–107. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Dimasi, N.; Fleming, R.; Zhong, H.; Bezabeh, B.; Kinneer, K.; Christie, R.J.; Fazenbaker, C.; Wu, H.; Gao, C. Efficient preparation of site-specific antibody-drug conjugates using cysteine insertion. Mol. Pharm. 2017, 14, 1501–1516. [Google Scholar] [CrossRef]
- Sussman, D.; Westendorf, L.; Meyer, D.W.; Leiske, C.I.; Anderson, M.; Okeley, N.M.; Alley, S.C.; Lyon, R.; Sanderson, R.J.; Carter, P.J.; et al. Engineered cysteine antibodies: An improved antibody-drug conjugate platform with a novel mechanism of drug-linker stability. Protein Eng. Des. Sel. 2018, 31, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Falck, G.; Müller, K.M. Enzyme-based labeling strategies for antibody–drug conjugates and antibody mimetics. Antibodies 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Bregeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grunberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 2010, 49, 9995–9997. [Google Scholar] [CrossRef] [PubMed]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Loo, C.; et al. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjug. Chem. 2015, 26, 650–659. [Google Scholar] [CrossRef]
- Ritzefeld, M. Sortagging: A robust and efficient chemoenzymatic ligation strategy. Chemistry 2014, 20, 8516–8529. [Google Scholar] [CrossRef]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase enzyme-mediated generation of site-specifically conjugated antibody drug conjugates with high in vitro and in vivo potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef]
- Pishesha, N.; Ingram, J.R.; Ploegh, H.L. Sortase A: A model for transpeptidation and its biological applications. Annu. Rev. Cell Dev. Biol. 2018, 34, 163–188. [Google Scholar] [CrossRef]
- Carrico, I.S.; Carlson, B.L.; Bertozzi, C.R. Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 2007, 3, 321. [Google Scholar] [CrossRef]
- Rabuka, D.; Rush, J.S.; deHart, G.W.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Barfield, R.M.; Rabuka, D. Site-specific bioconjugation using SMARTag((R)) Technology: A practical and effective chemoenzymatic approach to generate antibody-drug conjugates. Methods Mol. Biol. 2019, 2033, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; van der Weijden, J.; Sletten, E.M.; Rabuka, D.; Bertozzi, C.R. A Pictet-Spengler ligation for protein chemical modification. Proc. Natl. Acad. Sci. USA 2013, 110, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody–drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Hallam, T.J.; Smider, V.V. Unnatural amino acids in novel antibody conjugates. Future Med. Chem. 2014, 6, 1309–1324. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Axup, J.Y.; Schultz, P.G. Protein conjugation with genetically encoded unnatural amino acids. Curr. Opin. Chem. Biol. 2013, 17, 412–419. [Google Scholar] [CrossRef]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef]
- Tian, F.; Lu, Y.; Manibusan, A.; Sellers, A.; Tran, H.; Sun, Y.; Phuong, T.; Barnett, R.; Hehli, B.; Song, F.; et al. A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 1766–1771. [Google Scholar] [CrossRef]
- Hofer, T.; Skeffington, L.R.; Chapman, C.M.; Rader, C. Molecularly defined antibody conjugation through a selenocysteine interface. Biochemistry 2009, 48, 12047–12057. [Google Scholar] [CrossRef]
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tran, C.; Uter, N.T.; Yin, G.; Rivers, P.J.; et al. Production of site-specific antibody–drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug. Chem. 2014, 25, 351–361. [Google Scholar] [CrossRef]
- Yamada, K.; Ito, Y. Recent chemical approaches for site-specific conjugation of native antibodies: Technologies toward next generation antibody-drug conjugates. ChemBioChem 2019, 20, 2729–2737. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nagano, M.; Gavrilyuk, J.; Hakamata, W.; Inokuma, T.; Barbas, C.F., 3rd. Facile and stabile linkages through tyrosine: Bioconjugation strategies with the tyrosine-click reaction. Bioconjug. Chem. 2013, 24, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Zuberbuhler, K.; Casi, G.; Bernardes, G.J.; Neri, D. Fucose-specific conjugation of hydrazide derivatives to a vascular-targeting monoclonal antibody in IgG format. Chem. Commun. 2012, 48, 7100–7102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-specific antibody–drug conjugation through glycoengineering. Bioconjug. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef] [PubMed]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic conjugation of toxic payloads to the globally conserved N-Glycan of native mAbs provides homogeneous and highly efficacious antibody-drug conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, T.; Boons, G.J. Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew. Chem. Int. Ed. Engl. 2014, 53, 7179–7182. [Google Scholar] [CrossRef]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef]
- Behrens, C.R.; Ha, E.H.; Chinn, L.L.; Bowers, S.; Probst, G.; Fitch-Bruhns, M.; Monteon, J.; Valdiosera, A.; Bermudez, A.; Liao-Chan, S.; et al. Antibody-Drug Conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol. Pharm. 2015, 12, 3986–3998. [Google Scholar] [CrossRef]
- Bryant, P.; Pabst, M.; Badescu, G.; Bird, M.; McDowell, W.; Jamieson, E.; Swierkosz, J.; Jurlewicz, K.; Tommasi, R.; Henseleit, K.; et al. In vitro and in vivo evaluation of cysteine rebridged trastuzumab-MMAE antibody drug conjugates with defined drug-to-antibody ratios. Mol. Pharm. 2015, 12, 1872–1879. [Google Scholar] [CrossRef]
- Forte, N.; Chudasama, V.; Baker, J.R. Homogeneous antibody-drug conjugates via site-selective disulfide bridging. Drug Discov. Today Technol. 2018, 30, 11–20. [Google Scholar] [CrossRef]
- Schumacher, F.F.; Nunes, J.P.M.; Maruani, A.; Chudasama, V.; Smith, M.E.B.; Chester, K.A.; Baker, J.R.; Caddick, S. Next generation maleimides enable the controlled assembly of antibody-drug conjugates via native disulfide bond bridging. Org. Biomol. Chem. 2014, 12, 7261–7269. [Google Scholar] [CrossRef] [PubMed]
- Altwerger, G.; Bonazzoli, E.; Bellone, S.; Egawa-Takata, T.; Menderes, G.; Pettinella, F.; Bianchi, A.; Riccio, F.; Feinberg, J.; Zammataro, L.; et al. In vitro and in vivo activity of IMGN853, an antibody-drug conjugate targeting folate receptor alpha linked to DM4, in biologically aggressive endometrial cancers. Mol. Cancer Ther. 2018, 17, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Chen, C.; Chen, J.; Cruz-Chuh, J.D.; Delarosa, R.; Deng, Y.; Fourie-O’Donohue, A.; Figueroa, I.; Guo, J.; Jin, W.; et al. Exploration of pyrrolobenzodiazepine (PBD)-dimers containing disulfide-based prodrugs as payloads for antibody-drug conjugates. Mol. Pharm. 2018, 15, 3979–3996. [Google Scholar] [CrossRef] [PubMed]
- Mantaj, J.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From anthramycin to pyrrolobenzodiazepine (PBD)-containing antibody-drug conjugates (ADCs). Angew. Chem. Int. Ed. Engl. 2017, 56, 462–488. [Google Scholar] [CrossRef]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-drug conjugates for cancer therapy: Chemistry to clinical implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef]
- Ratnayake, A.S.; Chang, L.P.; Tumey, L.N.; Loganzo, F.; Chemler, J.A.; Wagenaar, M.; Musto, S.; Li, F.; Janso, J.E.; Ballard, T.E.; et al. Natural product bis-intercalator depsipeptides as a new class of payloads for antibody-drug conjugates. Bioconjug. Chem. 2019, 30, 200–209. [Google Scholar] [CrossRef]
- Lerchen, H.G.; Wittrock, S.; Stelte-Ludwig, B.; Sommer, A.; Berndt, S.; Griebenow, N.; Rebstock, A.S.; Johannes, S.; Cancho-Grande, Y.; Mahlert, C.; et al. Antibody-drug conjugates with pyrrole-based KSP inhibitors as the payload class. Angew. Chem. Int. Ed. Engl. 2018, 57, 15243–15247. [Google Scholar] [CrossRef]
- Karpov, A.S.; Abrams, T.; Clark, S.; Raikar, A.; D’Alessio, J.A.; Dillon, M.P.; Gesner, T.G.; Jones, D.; Lacaud, M.; Mallet, W.; et al. Nicotinamide phosphoribosyltransferase inhibitor as a novel payload for antibody-drug conjugates. ACS Med. Chem. Lett. 2018, 9, 838–842. [Google Scholar] [CrossRef]
- Love, E.A.; Sattikar, A.; Cook, H.; Gillen, K.; Large, J.M.; Patel, S.; Matthews, D.; Merritt, A. Developing an antibody–drug conjugate approach to selective inhibition of an extracellular protein. ChemBioChem 2019, 20, 754–758. [Google Scholar] [CrossRef]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- van der Goes, M.C.; Jacobs, J.W.; Bijlsma, J.W. The value of glucocorticoid co-therapy in different rheumatic diseases-positive and adverse effects. Arthritis Res. Ther. 2014, 16, S2. [Google Scholar] [CrossRef] [PubMed]
- Schäcke, H.; Döcke, W.-D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Kern, J.C.; Cancilla, M.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.; Liang, L.; Tomazela, D.; et al. Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody-drug conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Dovgan, I.; Koniev, O.; Kolodych, S.; Wagner, A. Antibody–oligonucleotide conjugates as therapeutic, imaging, and detection agents. Bioconjug. Chem. 2019, 30, 2483–2501. [Google Scholar] [CrossRef]
- Levin, A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef]
- Park, J.; Park, J.; Pei, Y.; Xu, J.; Yeo, Y. Pharmacokinetics and biodistribution of recently-developed siRNA nanomedicines. Adv. Drug Deliv. Rev. 2016, 104, 93–109. [Google Scholar] [CrossRef]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated oligonucleotides: Recent developments and therapeutic applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Sewing, S.; Gubler, M.; Gérard, R.; Avignon, B.; Mueller, Y.; Braendli-Baiocco, A.; Odin, M.; Moisan, A. GalNAc conjugation attenuates the cytotoxicity of antisense oligonucleotide drugs in renal tubular cells. Mol. Ther. Nucleic Acids 2019, 14, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Ämmälä, C.; Drury, W.J.; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.-M.; Valeur, E.; Jansson-Löfmark, R.; Janzén, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic β-cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef]
- Osborn, M.F.; Coles, A.H.; Biscans, A.; Haraszti, R.A.; Roux, L.; Davis, S.; Ly, S.; Echeverria, D.; Hassler, M.R.; Godinho, B.M.D.C.; et al. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 2018, 47, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kowolik, C.M.; Swiderski, P.M.; Kortylewski, M.; Yu, H.; Horne, D.A.; Jove, R.; Caballero, O.L.; Simpson, A.J.G.; Lee, F.-T.; et al. Humanized lewis-y specific antibody based delivery of STAT3 siRNA. ACS Chem. Biol. 2011, 6, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.-F.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB–siRNA conjugates. Nucleic Acids Res. 2014, 43, 1189–1203. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef]
- Satake, N.; Duong, C.; Yoshida, S.; Oestergaard, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. Novel targeted therapy for precursor B cell acute lymphoblastic leukemia: Anti-CD22 antibody-MXD3 antisense oligonucleotide conjugate. Mol. Med. 2016, 22, 632–642. [Google Scholar] [CrossRef]
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martinez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-antisense oligonucleotide conjugate downregulates a key gene in glioblastoma stem cells. Mol. Ther. Nucleic Acids 2018, 11, 518–527. [Google Scholar] [CrossRef]
- Humphreys, S.C.; Thayer, M.B.; Campuzano, I.D.G.; Netirojjanakul, C.; Rock, B.M. Quantification of siRNA-antibody conjugates in biological matrices by triplex-forming oligonucleotide ELISA. Nucleic Acid Ther. 2019, 29, 161–166. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Walker, M.A. Receptor-mediated and enzyme-dependent targeting of cytotoxic anticancer drugs. Pharmacol. Ther. 1999, 83, 67–123. [Google Scholar] [CrossRef]
- Kratz, F.; Müller, I.A.; Ryppa, C.; Warnecke, A. Prodrug strategies in anticancer chemotherapy. ChemMedChem 2008, 3, 20–53. [Google Scholar] [CrossRef] [PubMed]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid–valine–citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef]
- Wei, B.; Gunzner-Toste, J.; Yao, H.; Wang, T.; Wang, J.; Xu, Z.; Chen, J.; Wai, J.; Nonomiya, J.; Tsai, S.P.; et al. Discovery of peptidomimetic antibody–drug conjugate linkers with enhanced protease specificity. J. Med. Chem. 2018, 61, 989–1000. [Google Scholar] [CrossRef]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef]
- Kolodych, S.; Michel, C.; Delacroix, S.; Koniev, O.; Ehkirch, A.; Eberova, J.; Cianferani, S.; Renoux, B.; Krezel, W.; Poinot, P.; et al. Development and evaluation of beta-galactosidase-sensitive antibody-drug conjugates. Eur. J. Med. Chem. 2017, 142, 376–382. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor efficacy of a combination of CMC-544 (Inotuzumab Ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against Non-Hodgkin’s B-Cell lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [CrossRef]
- Zhou, D.; Casavant, J.; Graziani, E.I.; He, H.; Janso, J.; Loganzo, F.; Musto, S.; Tumey, N.; O’Donnell, C.J.; Dushin, R. Novel PIKK inhibitor antibody-drug conjugates: Synthesis and anti-tumor activity. Bioorg. Med. Chem. Lett. 2019, 29, 943–947. [Google Scholar] [CrossRef]
- Govindan, S.V.; Cardillo, T.M.; Sharkey, R.M.; Tat, F.; Gold, D.V.; Goldenberg, D.M. Milatuzumab-SN-38 conjugates for the treatment of CD74+ cancers. Mol. Cancer Ther. 2013, 12, 968–978. [Google Scholar] [CrossRef]
- Kellogg, B.A.; Garrett, L.; Kovtun, Y.; Lai, K.C.; Leece, B.; Miller, M.; Payne, G.; Steeves, R.; Whiteman, K.R.; Widdison, W.; et al. Disulfide-linked antibody−maytansinoid conjugates: Optimization of in vivo activity by varying the steric hindrance at carbon atoms adjacent to the disulfide linkage. Bioconjug. Chem. 2011, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, G.J.L.; Casi, G.; Trüssel, S.; Hartmann, I.; Schwager, K.; Scheuermann, J.; Neri, D. A traceless vascular-targeting antibody–drug conjugate for cancer therapy. Angew. Chem. Int. Ed. 2012, 51, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.D.; Kiick, K.L. Tunable degradation of maleimide—Thiol adducts in reducing environments. Bioconjug. Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M.; Manabe, S.; Matsumura, Y. Immunoregulation by IL-7R-targeting antibody-drug conjugates: Overcoming steroid-resistance in cancer and autoimmune disease. Sci. Rep. 2017, 7, 10735. [Google Scholar] [CrossRef]
- Hampe, C.S. Protective role of anti-idiotypic antibodies in autoimmunity—Lessons for type 1 diabetes. Autoimmunity 2012, 45, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef]
- Kraynov, E.; Kamath, A.V.; Walles, M.; Tarcsa, E.; Deslandes, A.; Iyer, R.A.; Datta-Mannan, A.; Sriraman, P.; Bairlein, M.; Yang, J.J.; et al. Current approaches for absorption, distribution, metabolism, and excretion characterization of antibody-drug conjugates: An industry white paper. Drug Metab. Dispos. 2016, 44, 617–623. [Google Scholar] [CrossRef]
- Drake, P.M.; Rabuka, D. Recent developments in ADC technology: Preclinical studies signal future clinical trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Betts, A.M. Antibody biodistribution coefficients. mAbs 2013, 5, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Yip, V.; Palma, E.; Tesar, D.B.; Mundo, E.E.; Bumbaca, D.; Torres, E.K.; Reyes, N.A.; Shen, B.Q.; Fielder, P.J.; Prabhu, S.; et al. Quantitative cumulative biodistribution of antibodies in mice. mAbs 2014, 6, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Herbertson, R.A.; Tebbutt, N.C.; Lee, F.T.; MacFarlane, D.J.; Chappell, B.; Micallef, N.; Lee, S.T.; Saunder, T.; Hopkins, W.; Smyth, F.E.; et al. Phase I biodistribution and pharmacokinetic study of Lewis Y-targeting immunoconjugate CMD-193 in patients with advanced epithelial cancers. Clin. Cancer Res. 2009, 15, 6709–6715. [Google Scholar] [CrossRef]
- Lu, D.; Joshi, A.; Wang, B.; Olsen, S.; Yi, J.H.; Krop, I.E.; Burris, H.A.; Girish, S. An integrated multiple-analyte pharmacokinetic model to characterize trastuzumab emtansine (T-DM1) clearance pathways and to evaluate reduced pharmacokinetic sampling in patients with HER2-positive metastatic breast cancer. Clin. Pharm. 2013, 52, 657–672. [Google Scholar] [CrossRef]
- Han, T.H.; Gopal, A.K.; Ramchandren, R.; Goy, A.; Chen, R.; Matous, J.V.; Cooper, M.; Grove, L.E.; Alley, S.C.; Lynch, C.M.; et al. CYP3A-mediated drug-drug interaction potential and excretion of brentuximab vedotin, an antibody-drug conjugate, in patients with CD30-positive hematologic malignancies. J. Clin. Pharmacol. 2013, 53, 866–877. [Google Scholar] [CrossRef]
- Collins, D.M.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired resistance to antibody-drug conjugates. Cancers 2019, 11, 394. [Google Scholar] [CrossRef]
- Bobaly, B.; Fleury-Souverain, S.; Beck, A.; Veuthey, J.L.; Guillarme, D.; Fekete, S. Current possibilities of liquid chromatography for the characterization of antibody-drug conjugates. J. Pharm. Biomed. Anal. 2018, 147, 493–505. [Google Scholar] [CrossRef]
- Lechner, A.; Giorgetti, J.; Gahoual, R.; Beck, A.; Leize-Wagner, E.; Francois, Y.N. Insights from capillary electrophoresis approaches for characterization of monoclonal antibodies and antibody drug conjugates in the period 2016–2018. J. Chromatogr. B 2019, 1122, 1–17. [Google Scholar] [CrossRef]
- Wagh, A.; Song, H.; Zeng, M.; Tao, L.; Das, T.K. Challenges and new frontiers in analytical characterization of antibody-drug conjugates. mAbs 2018, 10, 222–243. [Google Scholar] [CrossRef]
- Beck, A.; D’Atri, V.; Ehkirch, A.; Fekete, S.; Hernandez-Alba, O.; Gahoual, R.; Leize-Wagner, E.; Francois, Y.; Guillarme, D.; Cianferani, S. Cutting-edge multi-level analytical and structural characterization of antibody-drug conjugates: Present and future. Expert Rev. Proteom. 2019, 16, 337–362. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, A.V.; Yin, M.; Bodyak, N.; Stevenson, C.A.; Thomas, J.D.; Hammond, C.E.; Qin, L.; Zhu, B.; Gumerov, D.R.; Ter-Ovanesyan, E.; et al. A polymer-based antibody-vinca drug conjugate platform: Characterization and preclinical efficacy. Cancer Res. 2015, 75, 3365–3372. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef] [PubMed]
- Viricel, W.; Fournet, G.; Beaumel, S.; Perrial, E.; Papot, S.; Dumontet, C.; Joseph, B. Monodisperse polysarcosine-based highly-loaded antibody-drug conjugates. Chem. Sci. 2019, 10, 4048–4053. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Deweid, L.; Pirzer, T.; Yanakieva, D.; Englert, S.; Becker, B.; Avrutina, O.; Kolmar, H. Dextramabs: A novel format of antibody-drug conjugates featuring a multivalent polysaccharide scaffold. ChemistryOpen 2019, 8, 354–357. [Google Scholar] [CrossRef]
- Wang, L.; Amphlett, G.; Blattler, W.A.; Lambert, J.M.; Zhang, W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005, 14, 2436–2446. [Google Scholar] [CrossRef]
- Cao, M.; De Mel, N.; Jiao, Y.; Howard, J.; Parthemore, C.; Korman, S.; Thompson, C.; Wendeler, M.; Liu, D. Site-specific antibody-drug conjugate heterogeneity characterization and heterogeneity root cause analysis. mAbs 2019, 11, 1–13. [Google Scholar] [CrossRef]
- Mehta, G.; Scheinman, R.I.; Holers, V.M.; Banda, N.K. A new approach for the treatment of arthritis in mice with a novel conjugate of an Anti-C5aR1 antibody and C5 small interfering RNA. J. Immunol. 2015, 194, 5446–5454. [Google Scholar] [CrossRef]
- Ross, P.L.; Wolfe, J.L. Physical and chemical stability of antibody drug conjugates: Current status. J. Pharm. Sci. 2016, 105, 391–397. [Google Scholar] [CrossRef]
- Duerr, C.; Friess, W. Antibody-drug conjugates-stability and formulation. Eur. J. Pharm. Biopharm. 2019, 139, 168–176. [Google Scholar] [CrossRef]
- Wakankar, A.A.; Feeney, M.B.; Rivera, J.; Chen, Y.; Kim, M.; Sharma, V.K.; Wang, Y.J. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: Changes due to modification and conjugation processes. Bioconjug. Chem. 2010, 21, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.V.; Randolph, T.W.; Carpenter, J.F. Conjugation of emtansine onto trastuzumab promotes aggregation of the antibody-drug conjugate by reducing repulsive electrostatic interactions and increasing hydrophobic interactions. J. Pharm. Sci. 2019, 108, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Beckley, N.S.; Lazzareschi, K.P.; Chih, H.W.; Sharma, V.K.; Flores, H.L. Investigation into temperature-induced aggregation of an antibody drug conjugate. Bioconjug. Chem. 2013, 24, 1674–1683. [Google Scholar] [CrossRef]
- Adem, Y.T.; Schwarz, K.A.; Duenas, E.; Patapoff, T.W.; Galush, W.J.; Esue, O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug. Chem. 2014, 25, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Kumar, S.; Prashad, A.; Starkey, J.; Singh, S.K. Assessment of physical stability of an antibody drug conjugate by higher order structure analysis: Impact of thiol- maleimide chemistry. Pharm. Res. 2014, 31, 1710–1723. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Kumar, S.; Chipley, M.; Marcq, O.; Gupta, D.; Jin, Z.; Tomar, D.S.; Swabowski, C.; Smith, J.; Starkey, J.A.; et al. Characterization and higher-order structure assessment of an interchain cysteine-based ADC: Impact of drug loading and distribution on the mechanism of aggregation. Bioconjug. Chem. 2016, 27, 604–615. [Google Scholar] [CrossRef]
- Buecheler, J.W.; Winzer, M.; Tonillo, J.; Weber, C.; Gieseler, H. Impact of payload hydrophobicity on the stability of antibody-drug conjugates. Mol. Pharm. 2018, 15, 2656–2664. [Google Scholar] [CrossRef]
- Gandhi, A.V.; Arlotta, K.J.; Chen, H.N.; Owen, S.C.; Carpenter, J.F. Biophysical properties and heating-induced aggregation of lysine-conjugated antibody-drug conjugates. J. Pharm. Sci. 2018, 107, 1858–1869. [Google Scholar] [CrossRef]
- Ohri, R.; Bhakta, S.; Fourie-O’Donohue, A.; Dela Cruz-Chuh, J.; Tsai, S.P.; Cook, R.; Wei, B.; Ng, C.; Wong, A.W.; Bos, A.B.; et al. High-throughput cysteine scanning to identify stable antibody conjugation sites for maleimide- and disulfide-based linkers. Bioconjug. Chem. 2018, 29, 473–485. [Google Scholar] [CrossRef]
- Fathi, A.T.; Erba, H.P.; Lancet, J.E.; Stein, E.M.; Ravandi, F.; Faderl, S.; Walter, R.B.; Advani, A.S.; DeAngelo, D.J.; Kovacsovics, T.J.; et al. A phase 1 trial of vadastuximab talirine combined with hypomethylating agents in patients with CD33-positive AML. Blood 2018, 132, 1125–1133. [Google Scholar] [CrossRef]
- King, G.T.; Eaton, K.D.; Beagle, B.R.; Zopf, C.J.; Wong, G.Y.; Krupka, H.I.; Hua, S.Y.; Messersmith, W.A.; El-Khoueiry, A.B. A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Investig. New Drugs 2018, 36, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.; Barr, P.M.; Park, S.I.; Kolibaba, K.; Caimi, P.F.; Chhabra, S.; Kingsley, E.C.; Boyd, T.; Chen, R.; Carret, A.-S.; et al. A phase 1 trial of SGN-CD70A in patients with CD70-positive diffuse large B cell lymphoma and mantle cell lymphoma. Investig. New Drugs 2019, 37, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Saber, H.; Simpson, N.; Ricks, T.K.; Leighton, J.K. An FDA oncology analysis of toxicities associated with PBD-containing antibody-drug conjugates. Regul. Toxicol. Pharmacol. 2019, 107, 104429. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Carlson, A.; McFarland, J.M.; Banas, S.; Barfield, R.M.; Zmolek, W.; Kim, Y.C.; Huang, B.C.B.; Kudirka, R.; Rabuka, D. CAT-02-106, a site-specifically conjugated Anti-CD22 antibody bearing an MDR1-resistant maytansine payload yields excellent efficacy and safety in preclinical models. Mol. Cancer Ther. 2018, 17, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Buecheler, J.W.; Winzer, M.; Weber, C.; Gieseler, H. Oxidation-induced destabilization of model antibody-drug conjugates. J. Pharm. Sci. 2019, 108, 1236–1245. [Google Scholar] [CrossRef]
- Cockrell, G.M.; Wolfe, M.S.; Wolfe, J.L.; Schoneich, C. Photoinduced aggregation of a model antibody-drug conjugate. Mol. Pharm. 2015, 12, 1784–1797. [Google Scholar] [CrossRef]
- Liu, H.; Gaza-Bulseco, G.; Faldu, D.; Chumsae, C.; Sun, J. Heterogeneity of monoclonal antibodies. J. Pharm. Sci. 2008, 97, 2426–2447. [Google Scholar] [CrossRef]
- Chen, T.; Su, D.; Gruenhagen, J.; Gu, C.; Li, Y.; Yehl, P.; Chetwyn, N.P.; Medley, C.D. Chemical de-conjugation for investigating the stability of small molecule drugs in antibody-drug conjugates. J. Pharm. Biomed. Anal. 2016, 117, 304–310. [Google Scholar] [CrossRef]
- Wakankar, A.; Chen, Y.; Gokarn, Y.; Jacobson, F.S. Analytical methods for physicochemical characterization of antibody drug conjugates. mAbs 2011, 3, 161–172. [Google Scholar] [CrossRef]
- Su, D.; Kozak, K.R.; Sadowsky, J.; Yu, S.-F.; Fourie-O’Donohue, A.; Nelson, C.; Vandlen, R.; Ohri, R.; Liu, L.; Ng, C.; et al. Modulating antibody–drug conjugate payload metabolism by conjugation site and linker modification. Bioconjug. Chem. 2018, 29, 1155–1167. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC Product | Indications | Approval Date | Target Antigen | Antibody Conjugation | Average Drug Antibody Ratio (DAR) | Linker | Payload |
|---|---|---|---|---|---|---|---|
| Mylotarg® (Gemtuzumab ozogamicin) | Relapsed AML | 2001, withdrawn 2010; (reapproved 2017) | CD33 | Humanized IgG4—lysine | 2–3 | Hydrazone | Calicheamicin |
| Adcetris® (Brentuximab vedotin) | Relapsed HL and sALCL | 2011 | CD30 | Chimeric IgG1—cysteine | ~4 | Dipeptide cleavable (Val-Cit) | Monomethyl auristatin E (MMAE) |
| Kadcyla® (Trastuzumab emtansine) | HER2 + metastatic breast cancer | 2013 | HER2 | Humanized IgG1—lysine | 3.5 | Thioether Non-cleavable | Emtansine (DM1) |
| Besponsa® (Inotuzumab ozogamicin) | Relapsed or refractory CD22 + B-ALL | 2017 | CD22 | Humanized IgG4—lysine | ~4 | Hydrazone | Calicheamicin |
| Polivy® (Polatuzumab vedotin) | Relapsed or refractory DLBCL | 2019 | CD79b | Humanized IgG1—cysteine | 3.5 | Dipeptide cleavable (Val-Cit) | Monomethyl auristatin E (MMAE) |
| Enhertu® (fam-trastuzumab deruxtecan-nxki) | HER2 + unresectable metastic breast cancer | 2019 | HER2 | Humanized IgG1—cysteine | 7–8 | Tetrapeptide cleavable (Gly-gly-Phe-Gly) | Exatecan derivative (Dxd) |
| Species | ADME Information |
|---|---|
| Antibody |
|
| ADC |
|
| Payload |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, D.; Wurst, J.M.; Liu, T.; Martinez, R.M.; Datta-Mannan, A.; Feng, Y. Antibody Conjugates-Recent Advances and Future Innovations. Antibodies 2020, 9, 2. https://doi.org/10.3390/antib9010002
Leung D, Wurst JM, Liu T, Martinez RM, Datta-Mannan A, Feng Y. Antibody Conjugates-Recent Advances and Future Innovations. Antibodies. 2020; 9(1):2. https://doi.org/10.3390/antib9010002
Chicago/Turabian StyleLeung, Donmienne, Jacqueline M. Wurst, Tao Liu, Ruben M. Martinez, Amita Datta-Mannan, and Yiqing Feng. 2020. "Antibody Conjugates-Recent Advances and Future Innovations" Antibodies 9, no. 1: 2. https://doi.org/10.3390/antib9010002
APA StyleLeung, D., Wurst, J. M., Liu, T., Martinez, R. M., Datta-Mannan, A., & Feng, Y. (2020). Antibody Conjugates-Recent Advances and Future Innovations. Antibodies, 9(1), 2. https://doi.org/10.3390/antib9010002