David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Antibody Fragment Formats
2.1. Fragment Variable (Fv)-Based Formats
2.1.1. Single Chain Fragment Variable (scFv)
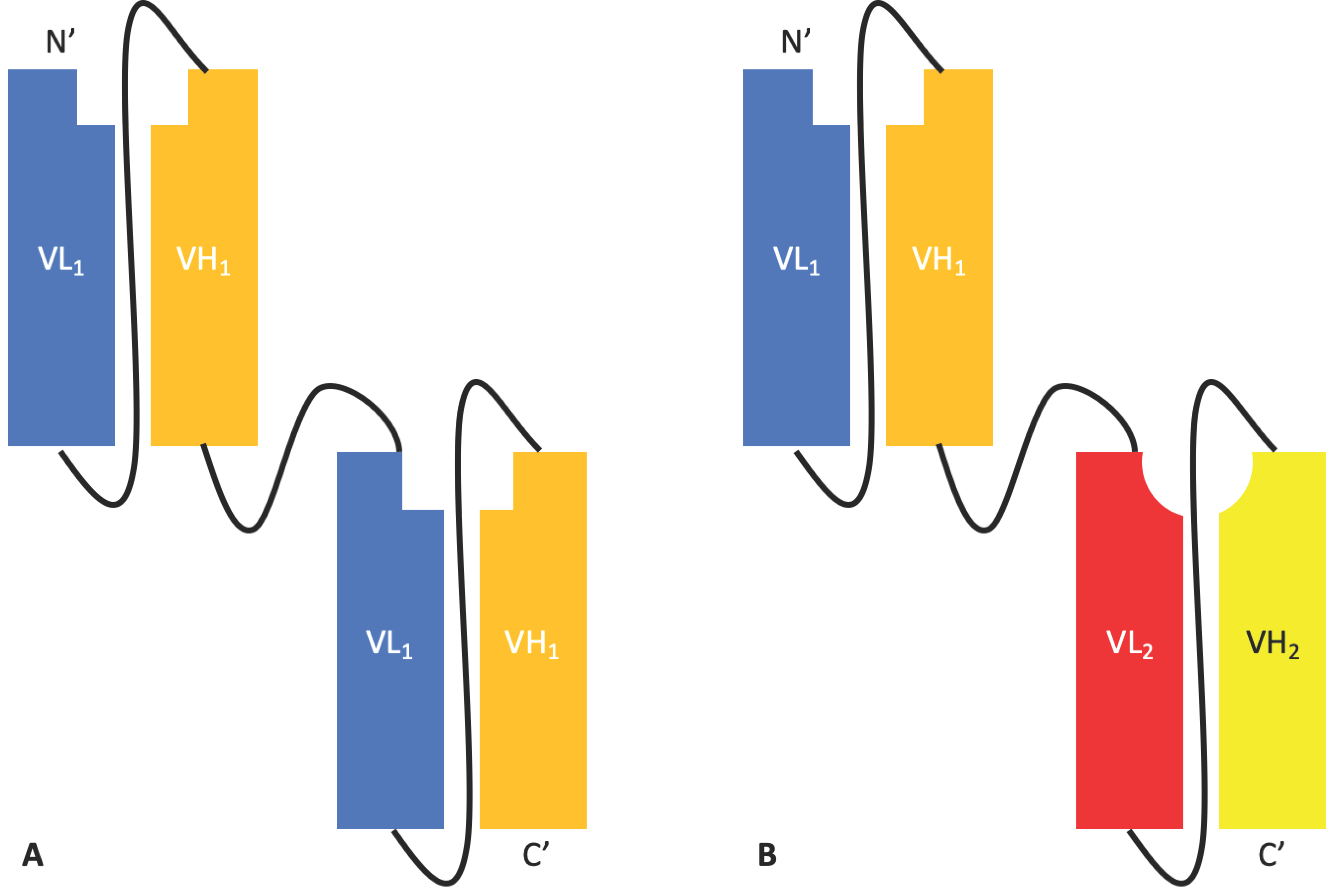
2.1.2. Tandem scFvs
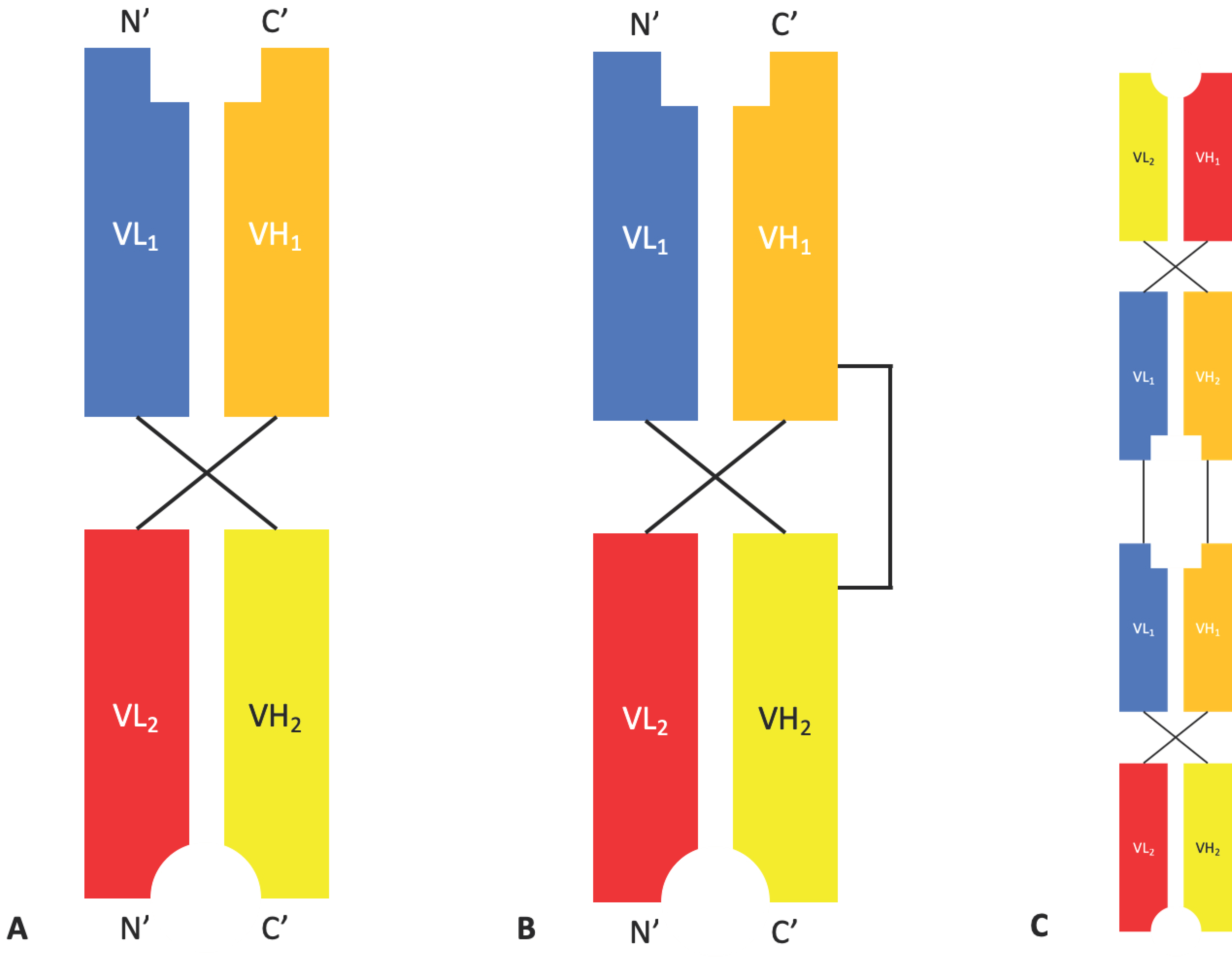
2.1.3. Diabodies, DART®s, and TandAbs
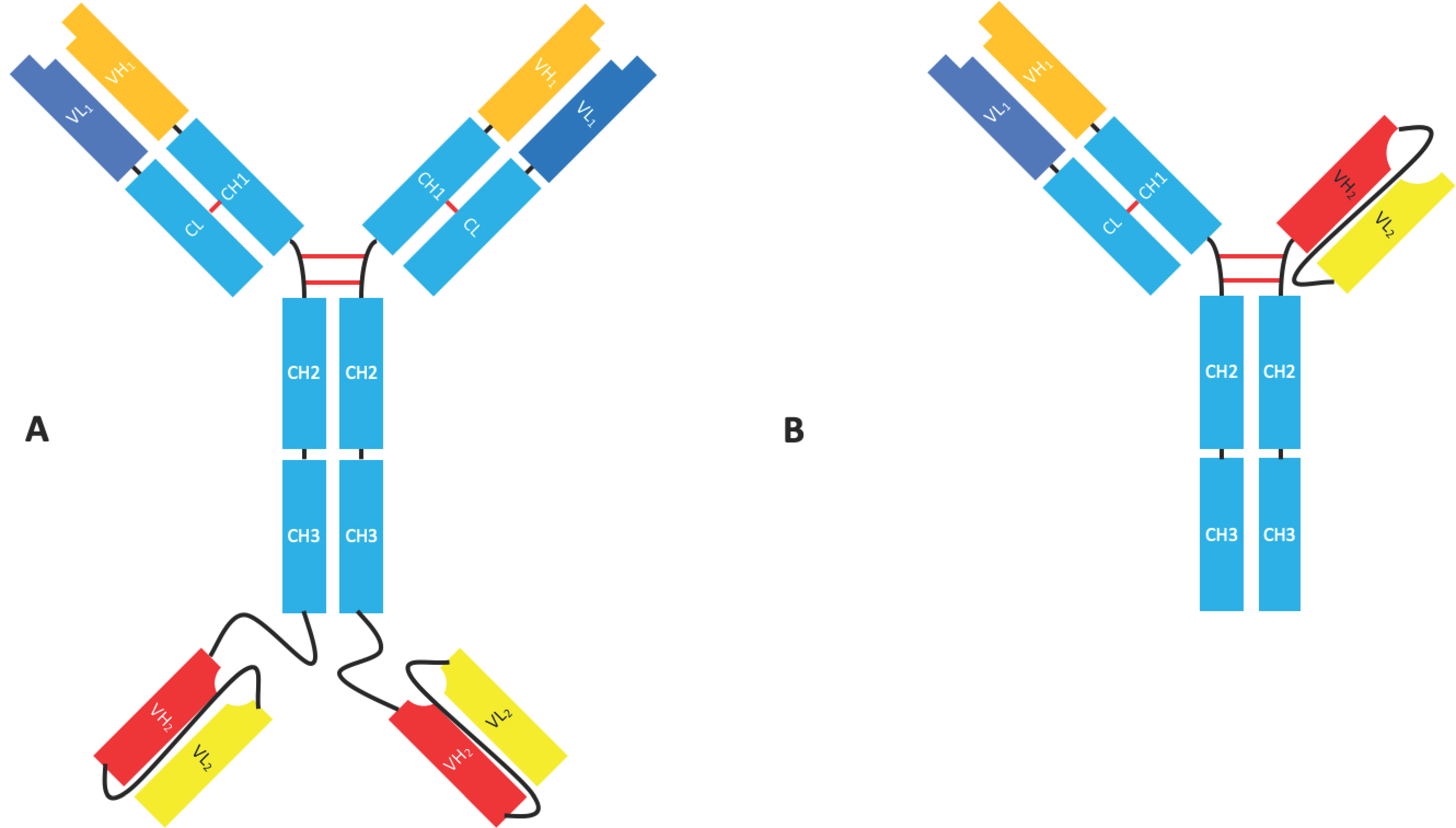
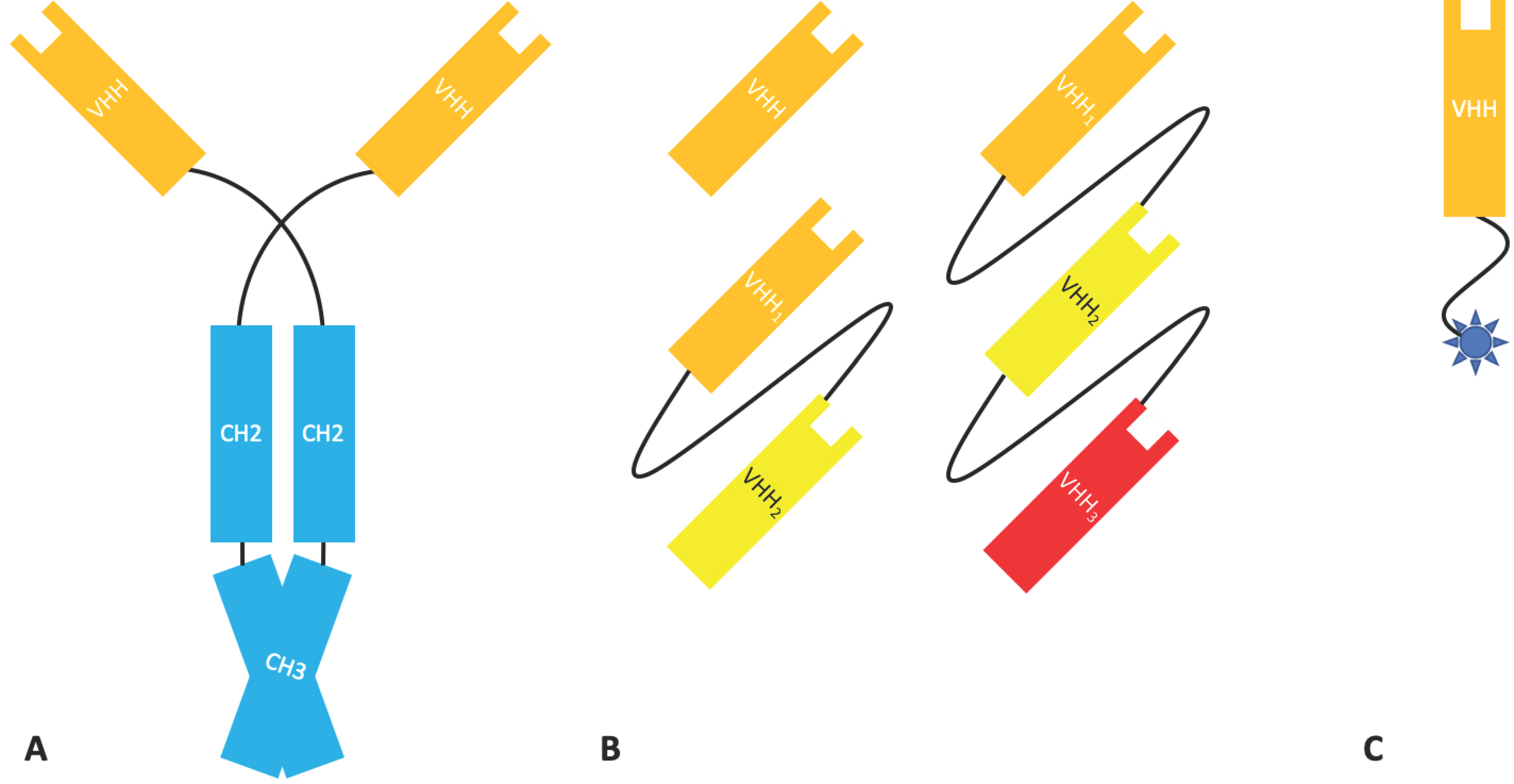
2.1.4. Bispecific Fv Fusion Antibodies with an Fc Domain
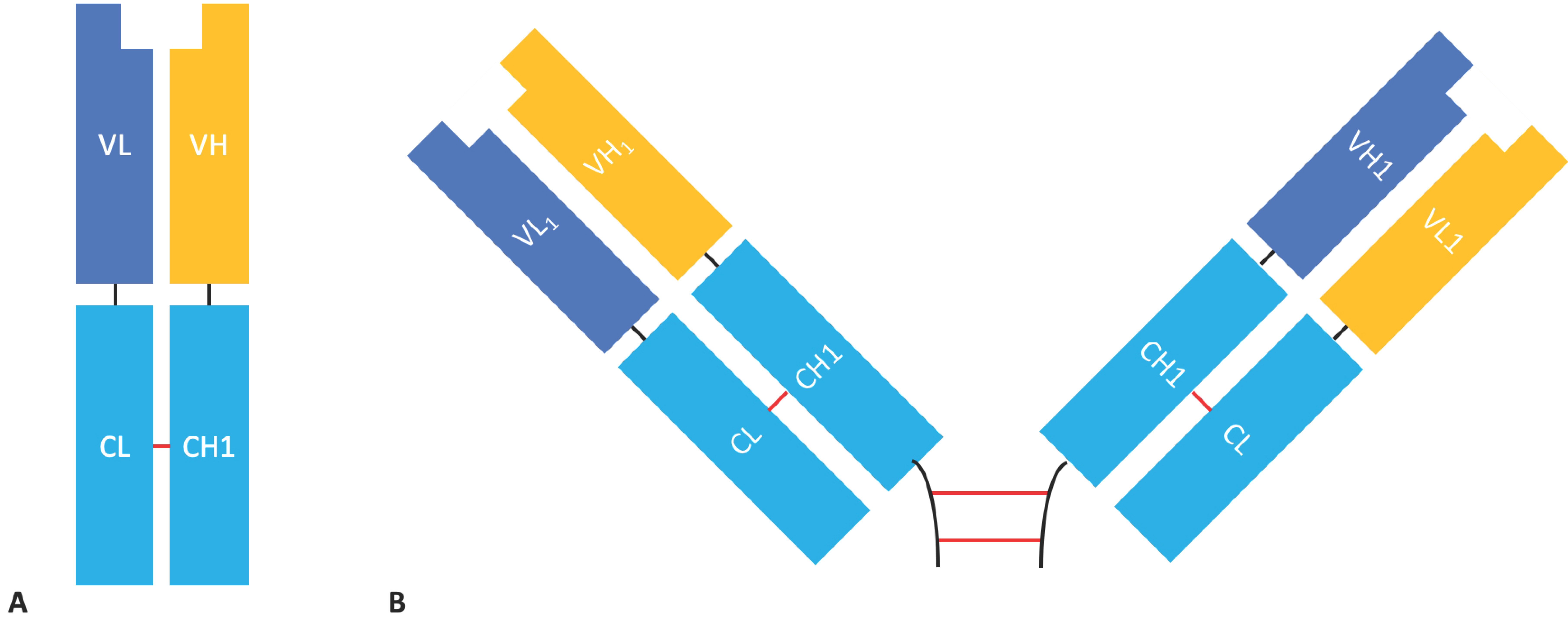
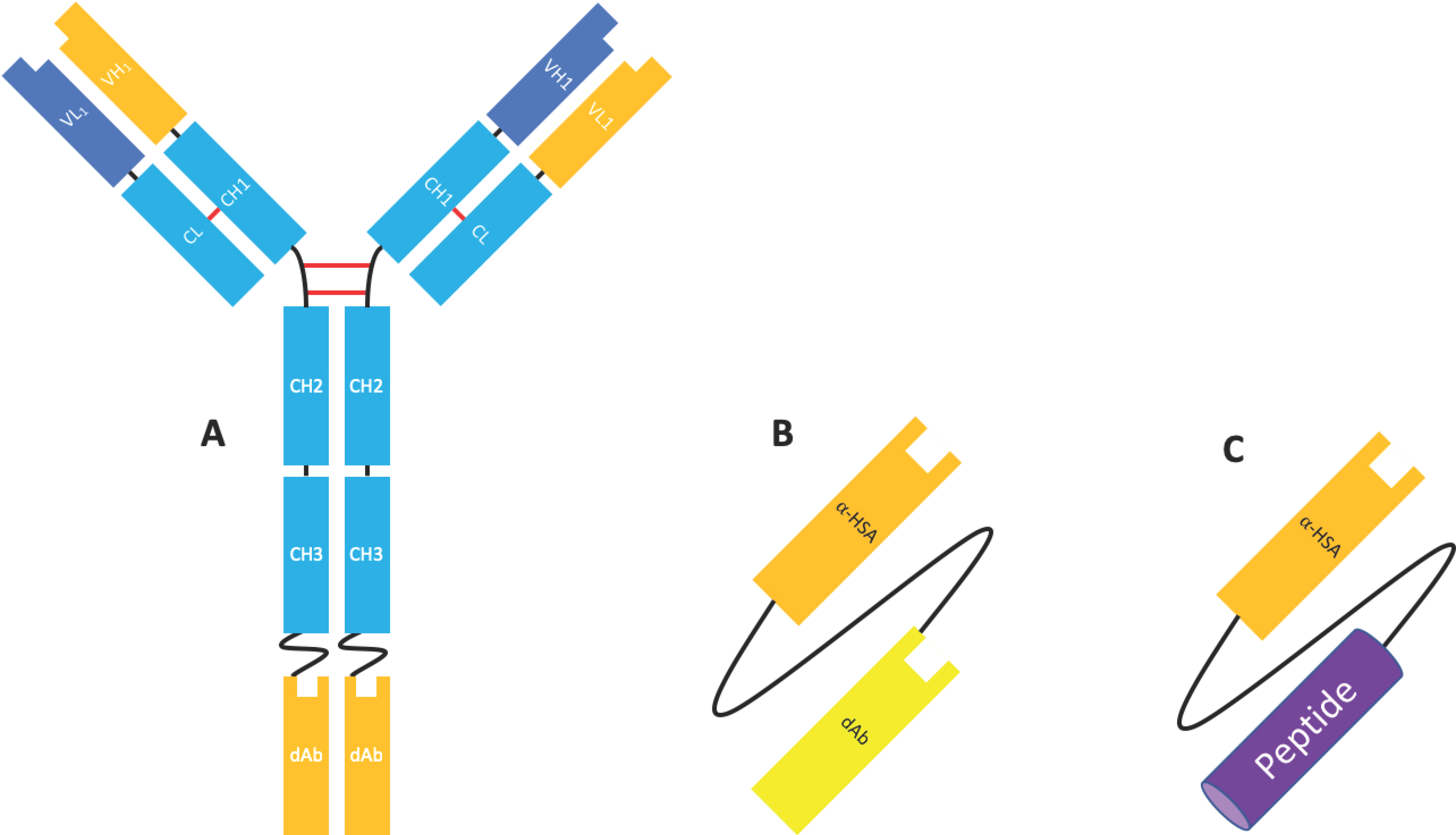
2.2. Fab Based Formats
Fab & F(ab’)2 Formats
2.3. Single-Domain Antibodies
2.3.1. Nanobodies
2.3.2. Domain Antibodies
3. The Production of Antibody Fragments
3.1. Expression
3.1.1. Escherichia coli
3.1.2. Saccharomyces cerevisiae
3.1.3. Pichia pastoris
3.1.4. Cell-Free Expression Systems
3.2. Enzymatic Cleavage
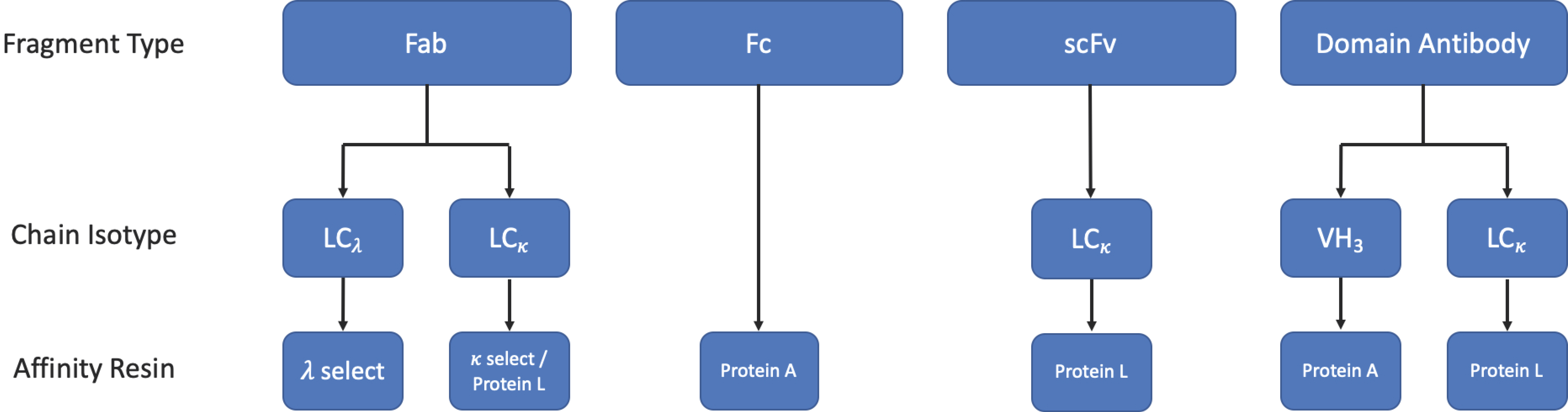
3.3. Purification
3.3.1. Protein L Affinity Chromatography
3.3.2. Affinity Tags
3.3.3. Other Chromatographic Methods
4. Antibody Fragments in the Clinic
4.1. Oncology
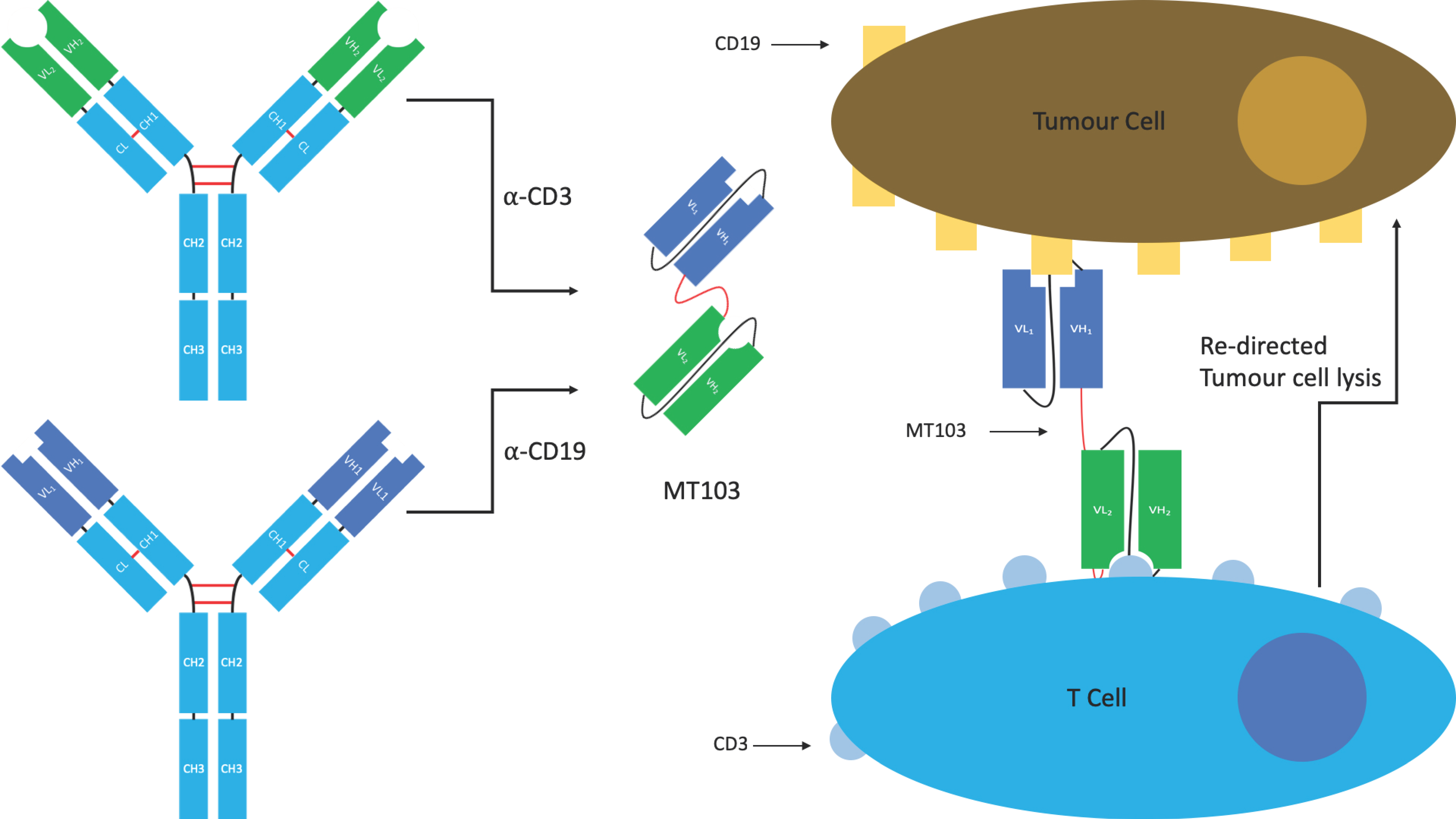
4.1.1. BiTE®s
4.1.2. BiKEs & TriKEs
4.1.3. DART®s
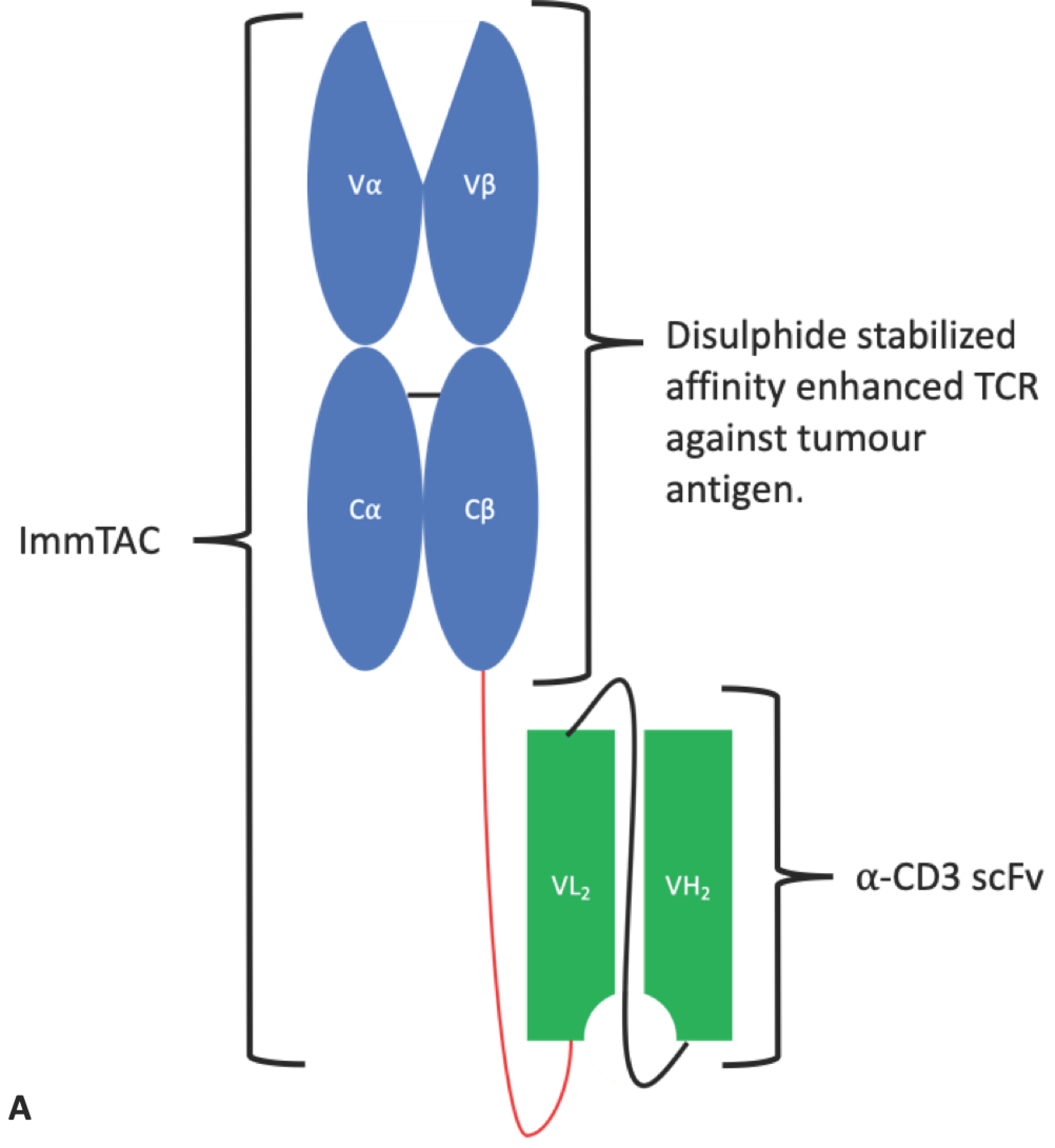
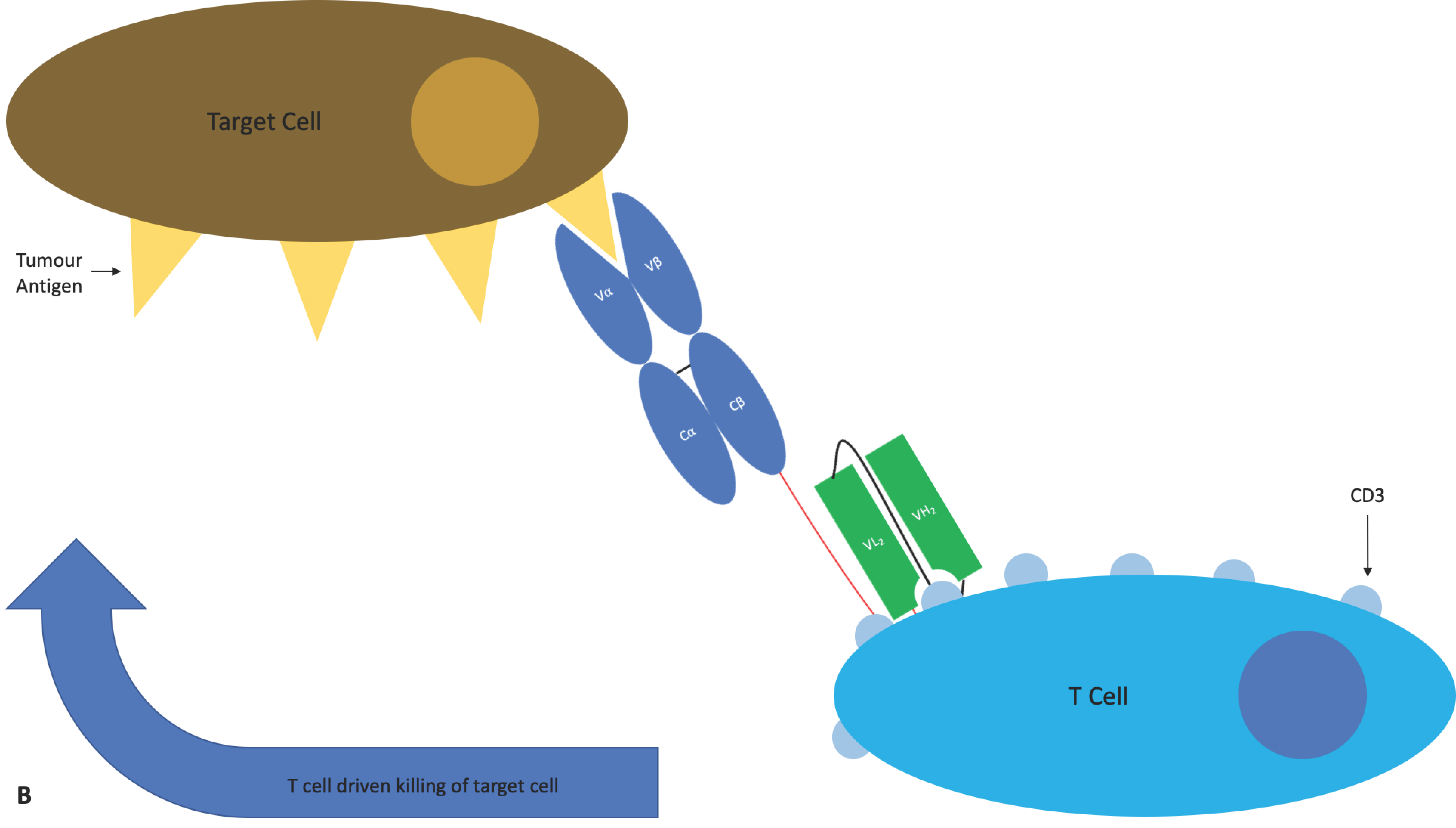
4.1.4. ImmTAC®s
4.1.5. Nanobodies®
4.1.6. Antibody Fragment-Drug Conjugates
4.2. Autoimmune and Inflammatory Diseases
4.3. Other Clinical Applications
4.3.1. Ophthalmic Indications
4.3.2. Infectious Diseases
4.3.3. Anti-Toxins and Anti-Venoms
5. Non-Therapeutic Uses
Imaging & Diagnostics
6. Future Opportunities
6.1. Neurodegenerative Diseases
6.2. Cell and Tissue Specific Antibody Delivery
6.3. Intracellular Targeting
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6HIS | hexa-histidine |
| ADA | anti-drug antibody |
| ADCC | antibody dependant cellular cytotoxicity |
| ADCP | antibody dependant cellular phagocytosis |
| ADH1 | alcohol dehydrogenase 1 |
| AIEX | anion exchange chromatography |
| ALI | acute lung inflammation |
| ARDS | acute respiratory distress syndrome |
| aTTP | acquired thrombotic thrombocytopenic purpura |
| ALL | acute lymphoblastic leukaemia |
| AMD | age-related macular degeneration |
| AML | acute myeloid leukaemia |
| BBB | blood–brain barrier |
| BCMA | B-cell maturation antigen |
| BiKE | bispecific natural killer cell engager |
| BiTE® | bispecific T-cell engager |
| CDC | complement-dependent cytotoxicity |
| CDR | complementarity determining region |
| CEA | carcinoembryonic antigen |
| CHO | Chinese hamster ovary |
| CIEX | cation exchange chromatography |
| CL | constant domain of immunoglobulin light chain |
| CNS | central nervous system |
| CPP | cell penetrating peptide |
| dAb | Domain antibody® |
| DART® | dual affinity re-targeting protein |
| EpCAM | epithelial cell adhesion molecule |
| ER | endoplasmic reticulum |
| Fab | fragment of antigen binding |
| Fc | fragment crystallizable |
| FcRn | neonatal fragment crystallizable receptor |
| Fv | fragment variable |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GFP | green fluorescent protein |
| GM-CSF | granulocyte-monocyte colony stimulating factor |
| GST | glutathione-S transferase |
| HAVH | human anti-VH |
| HEK | human embryonic kidney |
| HER2 | human epidermal growth factor 2 |
| HIC | hydrophobic interaction chromatography |
| HIV | human immunodeficiency virus |
| HLA | human leukocyte antigen |
| IgG | immunoglobulin gamma |
| IMAC | immobilised metal affinity chromatography |
| ImmTAC® | immune mobilising monoclonal t-cell receptors against cancer |
| ImmTAV® | immune mobilising monoclonal t-cell receptors against virus antigens |
| mAb | monoclonal antibody |
| MBP | mannose binding protein |
| MDS | myelodysplastic syndrome |
| MDSC | myeloid derived suppressor cell |
| MG | myasthenia gravis |
| MHC | major histocompatibility complex |
| MS | multiple sclerosis |
| Nb | Nanobody® |
| NK cell | Natural killer cell |
| NY ESO | New York esophageal squamous cell carcinoma |
| PD | pharmacodynamic |
| PEG | polyethylene glycol |
| PGK1 | phosphoglycerate kinase 1 |
| RMT | receptor-mediated endocytosis |
| RSV | respiratory syncytial virus |
| scFv | single chain fragment variable |
| SEC | size exclusion chromatography |
| SLE | systemic lupus erythematosus |
| TBGP | anti-trophoblast glycoprotein 5T4 |
| TCR | T-cell receptor |
| TfR | transferrin receptor |
| TNF | tumour necrosis factor |
| IL | interleukin |
| TNFR1 | tumour necrosis factor receptor 1 |
| TriKE | trispecific natural killer cell engager |
| VEGF | vascular endothelial growth factor |
| VH | variable domain of immunoglobulin heavy chain |
| VL | variable domain of immunoglobulin light chain |
| V-NAR | variable new antigen receptor |
| vWF | von Willebrand factor |
References
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Kontermann, R.E. Bispecific Antibodies. In Handbook of Therapeutic Antibodies, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2014; ISBN 9783527682423. [Google Scholar]
- Drake, P.M.; Rabuka, D. An emerging playbook for antibody-drug conjugates: Lessons from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr. Opin. Chem. Biol. 2015, 28, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L. Antibody fragments: Hope and hype. MAbs 2010, 2, 77–83. [Google Scholar] [CrossRef]
- Fernandes, J.C. Therapeutic application of antibody fragments in autoimmune diseases: Current state and prospects. Drug Discov. Today 2018, 23, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, R.V.; Kalinovsky, D.V.; Doronin, I.I.; Ponomarev, E.D.; Kholodenko, I. V Antibody Fragments as Potential Biopharmaceuticals for Cancer Therapy: Success and Limitations. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988. [Google Scholar] [CrossRef]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotný, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef] [PubMed]
- Montoliu-Gaya, L.; Esquerda-Canals, G.; Bronsoms, S.; Villegas, S. Production of an anti-Aβ antibody fragment in Pichia pastoris and in vitro and in vivo validation of its therapeutic effect. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Spadiut, O.; Capone, S.; Krainer, F.; Glieder, A.; Herwig, C. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol. 2014, 32, 54–60. [Google Scholar] [CrossRef]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid Tumor Penetration of a Single-Chain Fv and Comparison with Other Immunoglobulin Forms. Cancer Res. 1992. [Google Scholar] [CrossRef]
- Li, Z.; Krippendorff, B.F.; Sharma, S.; Walz, A.C.; Lavé, T.; Shah, D.K. Influence of molecular size on tissue distribution of antibody fragments. MAbs 2016. [Google Scholar] [CrossRef]
- Cumber, A.J.; Ward, E.S.; Winter, G.; Parnell, G.D.; Wawrzynczak, E.J. Comparative stabilities in vitro and in vivo of a recombinant mouse antibody FvCys fragment and a bisFvCys conjugate. J. Immunol. 1992, 149, 120–126. [Google Scholar]
- Sanz, L.; Cuesta, Á.M.; Compte, M.; Álvarez-Vallina, L. Antibody engineering: Facing new challenges in cancer therapy. Acta Pharmacol. Sin. 2005, 26, 641–648. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S. PEGylation: An approach for drug delivery. A review. Crit. Rev. Drug Carr. Syst. 2008. [Google Scholar] [CrossRef]
- Müller, D.; Karle, A.; Meißburger, B.; Höfig, I.; Stork, R.; Kontermann, R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007. [Google Scholar] [CrossRef]
- Poiron, C.; Wu, Y.; Ginestoux, C.; Ehrenmann, F.; Duroux, P.; Lefranc, M. IMGT®, the international ImMunoGeneTics information system®. Nucleic Acids Res. 2008, 37 (Suppl. S1), D1006–D1012. [Google Scholar]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Linsley, P.S.; Gayle, M.A.; Bajorath, J.; Brady, W.A.; Norris, N.A.; Fell, H.P.; Ledbetter, J.A.; Gilliland, L.K. Single-chain mono- and bispecific antibody derivatives with novel biological properties and antitumour activity from a COS cell transient expression system. Ther. Immunol. 1994, 1, 3–15. [Google Scholar] [PubMed]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. 2008. [Google Scholar] [CrossRef]
- Holliger, P. “Diabodies”: Small Bivalent and Bispecific Antibody Fragments. Proc. Natl. Acad. Sci. USA 1993. [Google Scholar] [CrossRef]
- Lu, D.; Jimenez, X.; Witte, L.; Zhu, Z. The effect of variable domain orientation and arrangement on the antigen-binding activity of a recombinant human bispecific diabody. Biochem. Biophys. Res. Commun. 2004. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Braunagel, M.; Reusch, U.; Cochlovius, B.; Le Gall, F.; Kouprianova, O.A.; Von Der Lieth, C.W.; Little, M. Effect of domain order on the activity of bacterially produced bispecific single-chain Fv antibodies. J. Mol. Biol. 2003. [Google Scholar] [CrossRef]
- MacroGenics Pipeline. Available online: https://www.macrogenics.com/pipeline/ (accessed on 16 January 2019).
- Affimed Pipeline. Available online: https://www.affimed.com/pipeline/ (accessed on 17 January 2019).
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; Von Der Lieth, C.W.; Matys, E.R.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999. [Google Scholar] [CrossRef]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007. [Google Scholar] [CrossRef]
- Metz, S.; Haas, A.K.; Daub, K.; Croasdale, R.; Stracke, J.; Lau, W.; Georges, G.; Josel, H.-P.; Dziadek, S.; Hopfner, K.-P.; et al. Bispecific digoxigenin-binding antibodies for targeted payload delivery. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef]
- Xencor Pipeline. Available online: https://www.xencor.com/pipeline/ (accessed on 17 January 2019).
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef]
- Bazin-Redureau, M.I.; Renard, C.B.; Scherrmann, J.M.G. Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab’)2and Fab after intravenous administration in the rat. J. Pharm. Pharmacol. 1997. [Google Scholar] [CrossRef]
- Röthlisberger, D.; Honegger, A.; Plückthun, A. Domain interactions in the Fab fragment: A comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J. Mol. Biol. 2005. [Google Scholar] [CrossRef]
- Simister, N.E.; Mostov, K.E. An Fc receptor structurally related to MHC class I antigens. Nature 1989. [Google Scholar] [CrossRef]
- Ober, R.J.; Martinez, C.; Lai, X.; Zhou, J.; Ward, E.S. Exocytosis of IgG as mediated by the receptor, FcRn: An analysis at the single-molecule level. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef]
- Rodewald, R.; Kraehenbuhl, J.P. Receptor-mediated transport of IgG. J. Cell Biol. 1984, 99, 159s–164s. [Google Scholar] [CrossRef]
- Chapman, A.P.; Antoniw, P.; Spitali, M.; West, S.; Stephens, S.; King, D.J. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 1999. [Google Scholar] [CrossRef]
- Schreiber, S. Certolizumab pegol for the treatment of Crohn’s disease. Ther. Adv. Gastroenterol. 2011, 357, 228–238. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient Cancer Therapy with a Nanobody-Based Conjugate. Cancer Res. 2004. [Google Scholar] [CrossRef]
- Van Der Linden, R.H.J.; Frenken, L.G.J.; De Geus, B.; Harmsen, M.M.; Ruuls, R.C.; Stok, W.; De Ron, L.; Wilson, S.; Davis, P.; Verrips, C.T. Comparison of physical chemical properties of llama V(HH) antibody fragments and mouse monoclonal antibodies. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1999. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006. [Google Scholar] [CrossRef]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Lauwereys, M.; Hassanzadeh Gh., G.; Gobert, M.; Conrath, K.; Muyldermans, S.; De Baetselier, P.; Revets, H. Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 2002. [Google Scholar] [CrossRef]
- Shen, J.; Vil, M.D.; Jimenez, X.; Iacolina, M.; Zhang, H.; Zhu, Z. Single variable domain-IgG fusion: A novel recombinant approach to Fc domain-containing bispecific antibodies. J. Biol. Chem. 2006. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Avila, D.; Hughes, M.; Hughes, A.; McKinney, E.C.; Flajnik, M.F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 1995. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Dooley, H.; Verdino, P.; Flajnik, M.F.; Wilson, I.A. Maturation of Shark Single-domain (IgNAR) Antibodies: Evidence for Induced-fit Binding. J. Mol. Biol. 2007. [Google Scholar] [CrossRef]
- Zielonka, S.; Empting, M.; Grzeschik, J.; Könning, D.; Barelle, C.J.; Kolmar, H. Structural insights and biomedical potential of IgNAR scaffolds from sharks. MAbs 2015, 7, 15–25. [Google Scholar] [CrossRef]
- Davies, J.; Riechmann, L. “Camelising” human antibody fragments: NMR studies on VH domains. FEBS Lett. 1994. [Google Scholar] [CrossRef]
- Holt, L.J.; Herring, C.; Jespers, L.S.; Woolven, B.P.; Tomlinson, I.M. Domain antibodies: Proteins for therapy. Trends Biotechnol. 2003, 21, 484–490. [Google Scholar] [CrossRef]
- Scott, M.J.; Lee, J.A.; Wake, M.S.; Batt, K.V.; Wattam, T.A.; Hiles, I.D.; Batuwangala, T.D.; Ashman, C.I.; Steward, M. ‘In-Format’ screening of a novel bispecific antibody format reveals significant potency improvements relative to unformatted molecules. MAbs 2017. [Google Scholar] [CrossRef]
- O’Connor-Semmes, R.L.; Lin, J.; Hodge, R.J.; Andrews, S.; Chism, J.; Choudhury, A.; Nunez, D.J. GSK2374697, a Novel Albumin-Binding Domain Antibody (AlbudAb), Extends Systemic Exposure of Exendin-4: First Study in Humans—PK/PD and Safety. Clin. Pharmacol. Ther. 2014. [Google Scholar] [CrossRef]
- Jefferis, R. Recombinant antibody therapeutics: The impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 2009, 30, 356–362. [Google Scholar] [CrossRef]
- Li, F.; Vijayasankaran, N.; Shen, A.; Kiss, R.; Amanullah, A. Cell culture processes for monoclonal antibody production. MAbs 2010, 2, 466–479. [Google Scholar] [CrossRef]
- Striedner, G.; Pfaffenzeller, I.; Markus, L.; Nemecek, S.; Grabherr, R.; Bayer, K. Plasmid-free T7-based Escherichia coli expression systems. Biotechnol. Bioeng. 2010. [Google Scholar] [CrossRef]
- Mairhofer, J.; Cserjan-Puschmann, M.; Striedner, G.; Nöbauer, K.; Razzazi-Fazeli, E.; Grabherr, R. Marker-free plasmids for gene therapeutic applications—Lack of antibiotic resistance gene substantially improves the manufacturing process. J. Biotechnol. 2010. [Google Scholar] [CrossRef]
- Sonoda, H.; Kumada, Y.; Katsuda, T.; Yamaji, H. Effects of cytoplasmic and periplasmic chaperones on secretory production of single-chain Fv antibody in Escherichia coli. J. Biosci. Bioeng. 2011. [Google Scholar] [CrossRef]
- Yuan, J.; Zweers, J.C.; Van Dijl, J.M.; Dalbey, R.E. Protein transport across and into cell membranes in bacteria and archaea. Cell. Mol. Life Sci. 2010, 67, 179–199. [Google Scholar] [CrossRef]
- Levy, R.; Ahluwalia, K.; Bohmann, D.J.; Giang, H.M.; Schwimmer, L.J.; Issafras, H.; Reddy, N.B.; Chan, C.; Horwitz, A.H.; Takeuchi, T. Enhancement of antibody fragment secretion into the Escherichia coli periplasm by co-expression with the peptidyl prolyl isomerase, FkpA, in the cytoplasm. J. Immunol. Methods 2013. [Google Scholar] [CrossRef]
- Jalalirad, R. Production of antibody fragment (Fab) throughout Escherichia coli fed-batch fermentation process: Changes in titre, location and form of product. Electron. J. Biotechnol. 2013. [Google Scholar] [CrossRef]
- Gorlani, A.; De Haard, H.; Verrips, T. Expression of VHHs in saccharomyces cerevisiae. Methods Mol. Biol. 2012. [Google Scholar] [CrossRef]
- Chee, M.K.; Haase, S.B. New and Redesigned pRS Plasmid Shuttle Vectors for Genetic Manipulation of Saccharomyces cerevisiae. G3 Genes Genomes Genet. 2012. [Google Scholar] [CrossRef]
- Leite, F.C.B.; dos Anjos, R.S.G.; Basilio, A.C.M.; Leal, G.F.C.; Simões, D.A.; de Morais, M.A. Construction of integrative plasmids suitable for genetic modification of industrial strains of Saccharomyces cerevisiae. Plasmid 2013. [Google Scholar] [CrossRef]
- Partow, S.; Siewers, V.; Bjørn, S.; Nielsen, J.; Maury, J. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast 2010. [Google Scholar] [CrossRef]
- Maury, J.; Asadollahi, M.A.; Møller, K.; Schalk, M.; Clark, A.; Formenti, L.R.; Nielsen, J. Reconstruction of a bacterial isoprenoid biosynthetic pathway in Saccharomyces cerevisiae. FEBS Lett. 2008. [Google Scholar] [CrossRef]
- Joosten, V.; Lokman, C.; van den Hondel, C.A.M.J.J.; Punt, P.J. The production of antibody fragments and antibody fusion proteins by yeasts and filamentous fungi. Microb. Cell Fact. 2003, 2. [Google Scholar] [CrossRef]
- Xu, P.; Raden, D.; Doyle, F.J.; Robinson, A.S. Analysis of unfolded protein response during single-chain antibody expression in Saccaromyces cerevisiae reveals different roles for BiP and PDI in folding. Metab. Eng. 2005. [Google Scholar] [CrossRef]
- Ferndahl, C.; Bonander, N.; Logez, C.; Wagner, R.; Gustafsson, L.; Larsson, C.; Hedfalk, K.; Darby, R.A.J.; Bill, R.M. Increasing cell biomass in Saccharomyces cerevisiae increases recombinant protein yield: The use of a respiratory strain as a microbial cell factory. Microb. Cell Fact. 2010. [Google Scholar] [CrossRef]
- Delic, M.; Mattanovich, D.; Gasser, B. Repressible promoters—A novel tool to generate conditional mutants in Pichia pastoris. Microb. Cell Fact. 2013. [Google Scholar] [CrossRef]
- Sohn, S.B.; Graf, A.B.; Kim, T.Y.; Gasser, B.; Maurer, M.; Ferrer, P.; Mattanovich, D.; Lee, S.Y. Genome-scale metabolic model of methylotrophic yeast Pichia pastoris and its use for in silico analysis of heterologous protein production. Biotechnol. J. 2010. [Google Scholar] [CrossRef]
- Jahic, M.; Rotticci-Mulder, J.; Martinelle, M.; Hult, K.; Enfors, S.O. Modeling of growth and energy metabolism of Pichia pastoris producing a fusion protein. Bioprocess Biosyst. Eng. 2001. [Google Scholar] [CrossRef]
- Detalle, L.; Stohr, T.; Palomo, C.; Piedra, P.A.; Gilbert, B.E.; Mas, V.; Millar, A.; Power, U.F.; Stortelers, C.; Allosery, K.; et al. Generation and characterization of ALX-0171, a potent novel therapeutic nanobody for the treatment of respiratory syncytial virus infection. Antimicrob. Agents Chemother. 2016. [Google Scholar] [CrossRef]
- Vallet-Courbin, A.; Larivière, M.; Hocquellet, A.; Hemadou, A.; Parimala, S.N.; Laroche-Traineau, J.; Santarelli, X.; Clofent-Sanchez, G.; Jacobin-Valat, M.J.; Noubhani, A. A recombinant human anti-platelet SCFV antibody produced in pichia pastoris for atheroma targeting. PLoS ONE 2017. [Google Scholar] [CrossRef]
- Oh, I.S.; Lee, J.C.; Lee, M.S.; Chung, J.H.; Kim, D.M. Cell-free production of functional antibody fragments. Bioprocess Biosyst. Eng. 2010. [Google Scholar] [CrossRef]
- Stech, M.; Nikolaeva, O.; Thoring, L.; Stöcklein, W.F.M.; Wüstenhagen, D.A.; Hust, M.; Dübel, S.; Kubick, S. Cell-free synthesis of functional antibodies using a coupled in vitro transcription-Translation system based on CHO cell lysates. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Stech, M.; Hust, M.; Schulze, C.; Dübel, S.; Kubick, S. Cell-free eukaryotic systems for the production, engineering, and modification of scFv antibody fragments. Eng. Life Sci. 2014. [Google Scholar] [CrossRef]
- Stech, M.; Kubick, S. Cell-Free Synthesis Meets Antibody Production: A Review. Antibodies 2015. [Google Scholar] [CrossRef]
- Ryabova, L.A.; Desplancq, D.; Spirin, A.S.; Plückthun, A. Functional antibody production using cell-free translation: Effects of protein disulfide isomerase and chaperones. Nat. Biotechnol. 1997. [Google Scholar] [CrossRef]
- Jiang, X.; Ookubo, Y.; Fujii, I.; Nakano, H.; Yamane, T. Expression of Fab fragment of catalytic antibody 6D9 in an Escherichia coli in vitro coupled transcription/translation system. FEBS Lett. 2002. [Google Scholar] [CrossRef]
- Kanter, G.; Yang, J.; Voloshin, A.; Levy, S.; Swartz, J.R.; Levy, R. Cell-free production of scFv fusion proteins: An efficient approach for personalized lymphoma vaccines. Blood 2007. [Google Scholar] [CrossRef]
- Shimizu, Y.; Kuruma, Y.; Ying, B.W.; Umekage, S.; Ueda, T. Cell-free translation systems for protein engineering. FEBS J. 2006, 273, 4133–4140. [Google Scholar] [CrossRef]
- STR001—Clinical Trial: NCT03424603. Available online: https://clinicaltrials.gov/ct2/show/NCT03424603 (accessed on 15 January 2019).
- Wang, A.C.; Wang, I.Y. Cleavage sites of human IgGl immunoglobulin by papain. Immunochemistry 1977. [Google Scholar] [CrossRef]
- Jones, R.G.A.; Landon, J. Enhanced pepsin digestion: A novel process for purifying antibody F(ab′)2 fragments in high yield from serum. J. Immunol. Methods 2002. [Google Scholar] [CrossRef]
- Genovis Website. Available online: https://www.genovis.com (accessed on 17 January 2019).
- Hober, S.; Nord, K.; Linhult, M. Protein A chromatography for antibody purification. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 848, 40–47. [Google Scholar] [CrossRef]
- Grodzki, A.C.; Berenstein, E. Antibody Purification: Affinity Chromatography—Protein A and Protein G Sepharose. In Immunocytochemical Methods and Protocols; Humana Press: New York, NY, USA, 2009. [Google Scholar]
- Roben, P.W.; Salem, A.N.; Silverman, G.J. VH3 family antibodies bind domain D of staphylococcal protein A. J. Immunol. 1995, 154, 6437–6445. [Google Scholar]
- Rodrigo, G.; Gruvegård, M.; Van Alstine, J. Antibody Fragments and Their Purification by Protein L Affinity Chromatography. Antibodies 2015. [Google Scholar] [CrossRef]
- Nascimento, A.; Pinto, I.F.; Chu, V.; Aires-Barros, M.R.; Conde, J.P.; Azevedo, A.M. Studies on the purification of antibody fragments. Sep. Purif. Technol. 2018. [Google Scholar] [CrossRef]
- Björck, L. A Novel Bacterial Cell Wall Protein with Affinity for Ig L Chains. J. Immunol. 1988, 140, 1194–1197. [Google Scholar]
- De Château, M.; Nilson, B.H.K.; Erntell, M.; Myhre, E.; Magnusson, C.G.M.; Åkerström, B.; Björck, L. On the Interaction between Protein L and Immunoglobulins of Various Mammalian Species. Scand. J. Immunol. 1993. [Google Scholar] [CrossRef]
- Lichty, J.J.; Malecki, J.L.; Agnew, H.D.; Michelson-Horowitz, D.J.; Tan, S. Comparison of affinity tags for protein purification. Protein Expr. Purif. 2005. [Google Scholar] [CrossRef]
- Goel, A.; Colcher, D.; Koo, J.S.; Booth, B.J.M.; Pavlinkova, G.; Batra, S.K. Relative position of the hexahistidine tag effects binding properties of a tumor-associated single-chain Fv construct. Biochim. Biophys. Acta Gen. Subj. 2000. [Google Scholar] [CrossRef]
- Schmeisser, H.; Kontsek, P.; Esposito, D.; Gillette, W.; Schreiber, G.; Zoon, K.C. Binding Characteristics of IFN-alpha Subvariants to IFNAR2-EC and Influence of the 6-Histidine Tag. J. Interferon Cytokine Res. 2006. [Google Scholar] [CrossRef]
- Das, D.; Allen, T.M.; Suresh, M.R. Comparative evaluation of two purification methods of anti-CD19-c-myc- His6-Cys scFv. Protein Expr. Purif. 2005. [Google Scholar] [CrossRef]
- Liu, H.; Gaza-Bulseco, G.; Chumsae, C. Analysis of Reduced Monoclonal Antibodies Using Size Exclusion Chromatography Coupled with Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2009. [Google Scholar] [CrossRef]
- Ljunglöf, A.; Lacki, K.M.; Mueller, J.; Harinarayan, C.; van Reis, R.; Fahrner, R.; Van Alstine, J.M. Ion exchange chromatography of antibody fragments. Biotechnol. Bioeng. 2007. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, C.M.; Kim, K.; Yoo, J.M.; Kang, S.M.; Ha, G.S.; Park, M.K.; Choi, M.A.; Lee, D.E.; Seong, B.L. Purification of antibody fragments for the reduction of charge variants using cation exchange chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018. [Google Scholar] [CrossRef]
- Karkov, H.S.; Krogh, B.O.; Woo, J.; Parimal, S.; Ahmadian, H.; Cramer, S.M. Investigation of protein selectivity in multimodal chromatography using in silico designed Fab fragment variants. Biotechnol. Bioeng. 2015. [Google Scholar] [CrossRef]
- Wu, Z.; Cheung, N.V. T-cell engaging bispecific antibody (T-BsAb): From technology to therapeutics. Pharmacol. Ther. 2018, 182, 161–175. [Google Scholar] [CrossRef]
- Velasquez, M.P.; Bonifant, C.L.; Gottschalk, S. Redirecting T-cells to hematological malignancies with bispecific antibodies. Blood 2018, 131, 30–38. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T-cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006. [Google Scholar] [CrossRef]
- Bispecific Antibodies Technology: NK Cells Engagers—Innate Pharma. Available online: https://www.innate-pharma.com/en/pipeline/bispecific-antibodies-technology-nk-cells-engagers (accessed on 18 January 2019).
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014. [Google Scholar] [CrossRef]
- Felices, M.; Sarhan, D.; Brandt, L.; Guldevall, K.; McElmurry, R.; Lenvik, A.; Chu, S.; Tolar, J.; Taras, E.; Spellman, S.R.; et al. CD16-IL15-CD33 Trispecific Killer Engager (TriKE) Overcomes Cancer-Induced Immune Suppression and Induces Natural Killer Cell-Mediated Control of MDS and AML Via Enhanced Killing Kinetics. Blood 2016, 128, 4291. [Google Scholar]
- Lichtenegger, F.S.; Rothe, M.; Schnorfeil, F.M.; Deiser, K.; Krupka, C.; Augsberger, C.; Schlüter, M.; Neitz, J.; Subklewe, M. Targeting LAG-3 and PD-1 to enhance T-cell activation by antigen-presenting cells. Front. Immunol. 2018. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011. [Google Scholar] [CrossRef]
- Oates, J.; Jakobsen, B.K. ImmTACs: Novel bi-specific agents for targeted cancer therapy. Oncoimmunology 2013, 2, e22891. [Google Scholar] [CrossRef]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol. Immunol. 2015, 67, 67–74. [Google Scholar] [CrossRef]
- Immunocore Pipeline. Available online: https://www.immunocore.com/pipeline (accessed on 15 January 2019).
- Zaia, J.A. A new agent in the strategy to cure AIDS. Mol. Ther. 2016, 24, 1894–1896. [Google Scholar] [CrossRef]
- Safdari, Y.; Ahmadzadeh, V. Use of Single-Chain Antibody Derivatives for Targeted Drug Delivery. Mol. Med. 2016. [Google Scholar] [CrossRef]
- Lu, Z.R.; Kopekov, P.; Kopeek, J. Polymerizable Fab’ antibody fragments for targeting of anticancer drugs. Nat. Biotechnol. 1999. [Google Scholar] [CrossRef]
- Philogen Pipeline. Available online: http://www.philogen.com/en/products/pipeline/pipeline_16.html (accessed on 17 January 2019).
- Lemaire, M.; D’Huyvetter, M.; Lahoutte, T.; Van Valckenborgh, E.; Menu, E.; De Bruyne, E.; Kronenberger, P.; Wernery, U.; Muyldermans, S.; Devoogdt, N.; et al. Imaging and radioimmunotherapy of multiple myeloma with anti-idiotypic Nanobodies. Leukemia 2014, 28, 444–447. [Google Scholar] [CrossRef]
- Pruszynski, M.; Koumarianou, E.; Vaidyanathan, G.; Revets, H.; Devoogdt, N.; Lahoutte, T.; Zalutsky, M.R. Targeting breast carcinoma with radioiodinated anti-HER2 Nanobody. Nucl. Med. Biol. 2013. [Google Scholar] [CrossRef]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- ALX-0061—Clinical Trial: NCT02287922. Available online: https://clinicaltrials.gov/ct2/show/NCT02287922 (accessed on 21 January 2019).
- ALX-0061—Clinical Trial: NCT02437890. Available online: https://clinicaltrials.gov/ct2/show/NCT02437890 (accessed on 21 January 2019).
- ALX-0761—Clinical Trial: NCT02156466. Available online: https://clinicaltrials.gov/ct2/show/NCT02156466 (accessed on 21 January 2019).
- ATN-103—Clinical trial: NCT01063803. Available online: https://clinicaltrials.gov/ct2/show/NCT01063803 (accessed on 21 January 2019).
- ATN-192—Clinical Trial: NCT01284036. Available online: https://clinicaltrials.gov/ct2/show/NCT01284036 (accessed on 21 January 2019).
- Philogen Website. Available online: http://www.philogen.com/en/ (accessed on 21 January 2019).
- Ulrichts, P.; Cousin, T.; Dreier, T.; de Haard, H.; Leupin, N. Argx-113, a novel Fc-based approach for antibody-induced pathologies such as primary immune thrombocytopenia. Blood 2016, 128, 4919. [Google Scholar]
- ARGX-113—Clinical Trial NCT03669588. Available online: https://clinicaltrials.gov/ct2/show/NCT03669588 (accessed on 29 January 2019).
- Merrill, J.T.; Shevell, D.E.; Duchesne, D.; Nowak, M.; Kundu, S.; Girgis, I.G.; Hu, Y.S.; Nadler, S.G.; Banerjee, S.; Throup, J. An Anti-CD28 Domain Antibody, Lulizumab, in Systemic Lupus Erythematosus: Results of a Phase II Study. In Arthritis & Rheumatology; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Cordy, J.C.; Morley, P.J.; Wright, T.J.; Birchler, M.A.; Lewis, A.P.; Emmins, R.; Chen, Y.Z.; Powley, W.M.; Bareille, P.J.; Wilson, R.; et al. Specificity of human anti-variable heavy (VH) chain autoantibodies and impact on the design and clinical testing of a VH domain antibody antagonist of tumour necrosis factor-α receptor 1. Clin. Exp. Immunol. 2015. [Google Scholar] [CrossRef]
- GSK286227—Clinical Trial: NCT02221037. Available online: https://clinicaltrials.gov/ct2/show/NCT02221037 (accessed on 29 January 2019).
- Thiel, M.A.; Coster, D.J.; Standfield, S.D.; Brereton, H.M.; Mavrangelos, C.; Zola, H.; Taylor, S.; Yusim, A.; Williams, K.A. Penetration of engineered antibody fragments into the eye. Clin. Exp. Immunol. 2002. [Google Scholar] [CrossRef]
- Lampalizumab—Clinical Trial: NCT02247531. Available online: https://clinicaltrials.gov/ct2/show/NCT02247531 (accessed on 24 January 2019).
- Lampalizumab—Clinical Trial: NCT02247479. Available online: https://clinicaltrials.gov/ct2/show/NCT02247479 (accessed on 24 January 2019).
- Holz, F.G.; Sadda, S.R.; Busbee, B.; Chew, E.Y.; Mitchell, P.; Tufail, A.; Brittain, C.; Ferrara, D.; Gray, S.; Honigberg, L.; et al. Efficacy and safety of lampalizumab for geographic atrophy due to age-related macular degeneration: Chroma and spectri phase 3 randomized clinical trials. JAMA Ophthalmol. 2018. [Google Scholar] [CrossRef]
- RTH 258—Clinical Trial: NCT02307682. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=2ahUKEwiNsqmMi5PgAhUS2qQKHbi1BZoQFjAAegQICRAB&url=https%3A%2F%2Fclinicaltrials.gov%2Fct2%2Fshow%2FNCT02307682&usg=AOvVaw2oDAJKgexjOb5qZHcOVqJy (accessed on 24 January 2018).
- RTH 258—Clinical Trial: NCT02434328. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=2ahUKEwiMjoKbi5PgAhVO3KQKHa2cDKoQFjAAegQIChAB&url=https%3A%2F%2Fclinicaltrials.gov%2Fct2%2Fshow%2FNCT02434328&usg=AOvVaw1Uobk9ivR8iGx_t6gl-Lec (accessed on 24 January 2019).
- Camacho-Villegas, T.A.; Mata-González, M.T.; García-Ubbelohd, W.; Núñez-García, L.; Elosua, C.; Paniagua-Solis, J.F.; Licea-Navarro, A.F. Intraocular penetration of a vNAR: In vivo and in vitro VEGF165 neutralization. Mar. Drugs 2018. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, M.; Johnson, K.; Steven, J.; Barelle, C.J.; Porter, A. Therapeutic potential of shark anti-ICOSL VNAR domains is exemplified in a murine model of autoimmune non-infectious uveitis. Front. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Elasmogen Website. Available online: www.elasmogen.com (accessed on 24 January 2019).
- Wilken, L.; McPherson, A. Application of camelid heavy-chain variable domains (VHHs) in prevention and treatment of bacterial and viral infections. Int. Rev. Immunol. 2018, 37, 69–76. [Google Scholar] [CrossRef] [PubMed]
- AdisInsight ALX-0171. Available online: https://adisinsight.springer.com/drugs/800035341 (accessed on 24 January 2019).
- Laustsen, A.H.; María Gutiérrez, J.; Knudsen, C.; Johansen, K.H.; Bermúdez-Méndez, E.; Cerni, F.A.; Jürgensen, J.A.; Ledsgaard, L.; Martos-Esteban, A.; Øhlenschlæger, M.; et al. Pros and cons of different therapeutic antibody formats for recombinant antivenom development. Toxicon 2018. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.M.; Mukherjee, J.; Leysath, C.E.; Debatis, M.; Ofori, K.; Baldwin, K.; Boucher, C.; Peters, R.; Beamer, G.; Sheoran, A.; et al. A single VHH-based toxin-neutralizing agent and an effector antibody protect mice against challenge with Shiga toxins 1 and 2. Infect. Immun. 2013. [Google Scholar] [CrossRef]
- Freise, A.C.; Wu, A.M. In vivo imaging with antibodies and engineered fragments. Mol. Immunol. 2015, 67, 142–152. [Google Scholar] [CrossRef] [PubMed]
- van Brussel, A.S.A.; Adams, A.; Oliveira, S.; Dorresteijn, B.; El Khattabi, M.; Vermeulen, J.F.; van der Wall, E.; Mali, W.P.T.M.; Derksen, P.W.B.; van Diest, P.J.; et al. Hypoxia-Targeting Fluorescent Nanobodies for Optical Molecular Imaging of Pre-Invasive Breast Cancer. Mol. Imaging Biol. 2016. [Google Scholar] [CrossRef]
- Kijanka, M.M.; van Brussel, A.S.A.; van der Wall, E.; Mali, W.P.T.M.; van Diest, P.J.; van Bergen en Henegouwen, P.M.P.; Oliveira, S. Optical imaging of pre-invasive breast cancer with a combination of VHHs targeting CAIX and HER2 increases contrast and facilitates tumour characterization. EJNMMI Res. 2016. [Google Scholar] [CrossRef]
- Viola-Villegas, N.T.; Sevak, K.K.; Carlin, S.D.; Doran, M.G.; Evans, H.W.; Bartlett, D.W.; Wu, A.M.; Lewis, J.S. Noninvasive imaging of PSMA in prostate tumors with89Zr-Labeled huJ591 engineered antibody fragments: The faster alternatives. Mol. Pharm. 2014. [Google Scholar] [CrossRef]
- Raubitschek, A.A.; Tsai, S.-W.; Shively, J.E.; Yazaki, P.J.; Williams, L.E.; Ikle’, D.N.; Wu, A.M.; Wong, J.Y.C. Tumor Targeting of Radiometal Labeled Anti-CEA Recombinant T84.66 Diabody and T84.66 Minibody: Comparison to Radioiodinated Fragments. Bioconjug. Chem. 2002. [Google Scholar] [CrossRef]
- Maier, J.; Traenkle, B.; Rothbauer, U. Real-time analysis of epithelial-mesenchymal transition using fluorescent single-domain antibodies. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Broisat, A.; Hernot, S.; Toczek, J.; De Vos, J.; Riou, L.M.; Martin, S.; Ahmadi, M.; Thielens, N.; Wernery, U.; Caveliers, V.; et al. Nanobodies targeting mouse/human VCAM1 for the nuclear imaging of atherosclerotic lesions. Circ. Res. 2012. [Google Scholar] [CrossRef]
- Broisat, A.; Toczek, J.; Dumas, L.S.; Ahmadi, M.; Bacot, S.; Perret, P.; Slimani, L.; Barone-Rochette, G.; Soubies, A.; Devoogdt, N.; et al. 99mTc-cAbVCAM1-5 Imaging Is a Sensitive and Reproducible Tool for the Detection of Inflamed Atherosclerotic Lesions in Mice. J. Nucl. Med. 2014. [Google Scholar] [CrossRef]
- Yu, Y.J.; Watts, R.J. Developing Therapeutic Antibodies for Neurodegenerative Disease. Neurotherapeutics 2013, 10, 459–472. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the “high-hanging fruit”. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004. [Google Scholar] [CrossRef]
- Messer, A.; Lynch, S.M.; Butler, D.C. Developing intrabodies for the therapeutic suppression of neurodegenerative pathology. Expert Opin. Biol. Ther. 2009. [Google Scholar] [CrossRef]
- Pardridge, W.M. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 347–361. [Google Scholar] [CrossRef]
- Abzyme Website. Available online: www. abzymetx.com (accessed on 21 January 2019).
- Li, T.; Bourgeois, J.P.; Celli, S.; Glacial, F.; Le Sourd, A.M.; Mecheri, S.; Weksler, B.; Romero, I.; Couraud, P.O.; Rougeon, F.; et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: Application to brain imaging. FASEB J. 2012. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Ramakrishnan, M.; Holasek, S.S.; Ramirez-Alvarado, M.; Kandimalla, K.K.; Gilles, E.J.; Curran, G.L.; Wengenack, T.M. In vivo targeting of antibody fragments to the nervous system for Alzheimer’s disease immunotherapy and molecular imaging of amyloid plaques. J. Neurochem. 2007. [Google Scholar] [CrossRef]
- Caljon, G.; Caveliers, V.; Lahoutte, T.; Stijlemans, B.; Ghassabeh, G.H.; Van Den Abbeele, J.; Smolders, I.; De Baetselier, P.; Michotte, Y.; Muyldermans, S.; et al. Using microdialysis to analyse the passage of monovalent nanobodies through the blood-brain barrier. Br. J. Pharmacol. 2012. [Google Scholar] [CrossRef]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002. [Google Scholar] [CrossRef]
- Farrington, G.K.; Caram-Salas, N.; Haqqani, A.S.; Brunette, E.; Eldredge, J.; Pepinsky, B.; Antognetti, G.; Baumann, E.; Ding, W.; Garber, E.; et al. A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 2014. [Google Scholar] [CrossRef]
- Slastnikova, T.A.; Ulasov, A.V.; Rosenkranz, A.A.; Sobolev, A.S. Targeted intracellular delivery of antibodies: The state of the art. Front. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Lim, K.J.; Sung, B.H.; Shin, J.R.; Lee, Y.W.; Kim, D.J.; Yang, K.S.; Kim, S.C. A Cancer Specific Cell-Penetrating Peptide, BR2, for the Efficient Delivery of an scFv into Cancer Cells. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Bruce, V.J.; Lopez-Islas, M.; McNaughton, B.R. Resurfaced cell-penetrating nanobodies: A potentially general scaffold for intracellularly targeted protein discovery. Protein Sci. 2016. [Google Scholar] [CrossRef]
- Shin, S.M.; Choi, D.K.; Jung, K.; Bae, J.; Kim, J.S.; Park, S.W.; Song, K.H.; Kim, Y.S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bates, A.; Power, C.A. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies 2019, 8, 28. https://doi.org/10.3390/antib8020028
Bates A, Power CA. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies. 2019; 8(2):28. https://doi.org/10.3390/antib8020028
Chicago/Turabian StyleBates, Adam, and Christine A. Power. 2019. "David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments" Antibodies 8, no. 2: 28. https://doi.org/10.3390/antib8020028
APA StyleBates, A., & Power, C. A. (2019). David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies, 8(2), 28. https://doi.org/10.3390/antib8020028