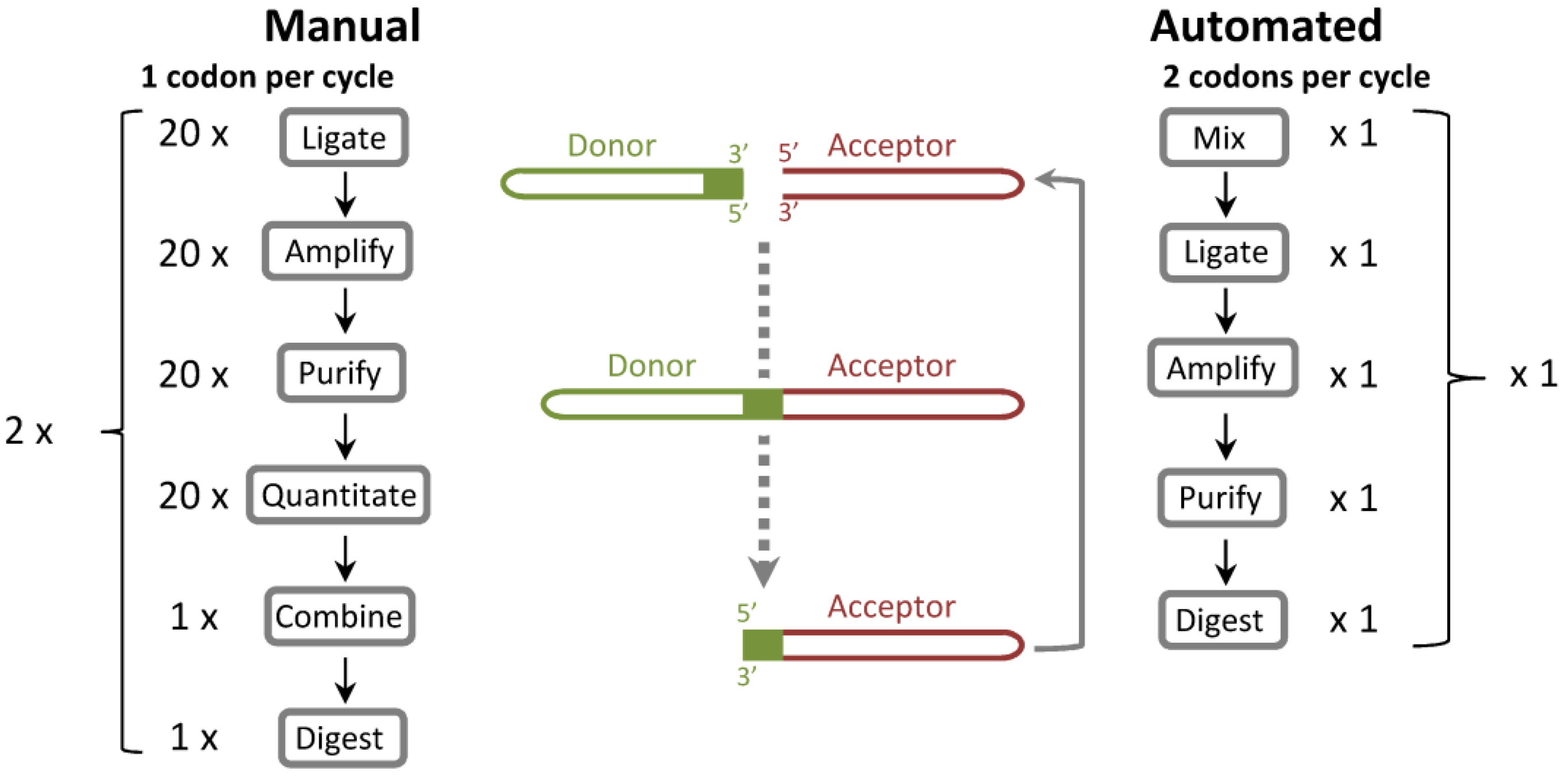
ProxiMAX randomization is a mutagenesis technique that can saturate at defined positions within a protein with any specified mixture of codons in any specified ratio. Gene fragments are built one codon at a time, via a process of enzymatic saturation cycling [
7] (
Figure 1). In our original publication of the methodology, we described the manual construction of a model CDR-H3 library, in which we were able to exclusively encode mixtures of selected amino acids, in user-defined, 5% incremental ratios (for example, CDR-H3 loop Kabat position 100B: 20% Asp; 20% Ser; 20% Tyr; 10% Gly; 10% Thr; 10% Val; 5% Ala; 5% Arg). In every saturation cycle of this manual process, up to 20 donor sequences were individually ligated, amplified, purified and quantified, then combined and digested. The methodology was ideal for constructing the highest quality libraries as exceptional agreement between design specifications and observed library composition was previously demonstrated, but it was relatively expensive and labor intensive as a commercial process.
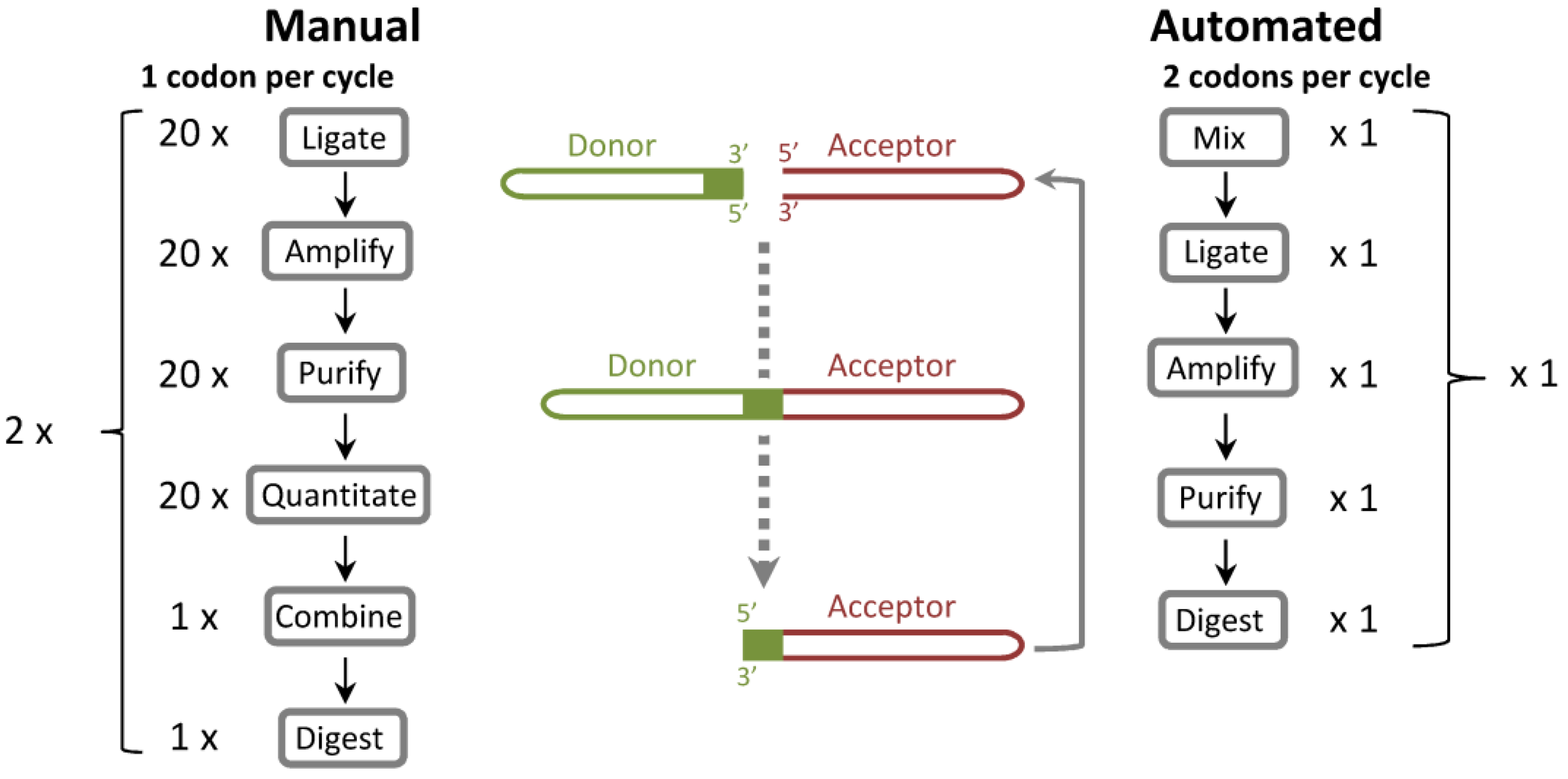
Figure 1.
Comparison of ProxiMAX and automated, hexamer-based ProxiMAX (Colibra™) processes. In the manual process, codons are ligated separately, amplified, purified, quantified, mixed into a pool of ligated products and then digested with MlyI. This procedure transfers the codon from a donor molecule to an acceptor library element. In an automated hexamer assembly, all hexamer components are premixed, ligated as a pool, PCR amplified, purified and then cleaved with MlyI. One hexamer cycle is equivalent to two manual trimer cycles.
2.1. Colibra Development: Manual Hexamer Method Validation
To determine process feasibility, scFv light chain CDR regions were constructed using manual hexamer addition. Initially, as in the published ProxiMAX protocol [
7], the hexamers were ligated separately and individual components were purified and then mixed at the intended ratios to generate a library that reflected the design of the CDRs. Individual ligations were undertaken owing to previous observed biases caused by sequence preferences of T4 ligase during trimer addition, such as for the His codon, CAT [
7]. We assumed that similar biases might be present at the ligation junctions of the hexamers which could skew the library synthesis towards sequences for which T4 ligase had a greater preference. A semi-minimalist design was chosen that would be amenable to manual pipetting, with codon frequencies rounded to the nearest 5%.
The longer light chain CDRs were fabricated using the bi-directional multi-cassette approach previously described [
7], whereby a randomized region can be constructed as multiple cassettes that are then joined together to obtain the desired length. A major advantage of the hexameric approach, when compared to the single codon/trimeric methodology, is the capacity to achieve greater lengths whilst decreasing the number of saturation cycling steps required. Moreover, since the quality of the final product is related to the repeating cycles of additions, decreasing the number of steps by the use of hexamers has the potential of improving the quality of the library by reducing the accumulation of spurious ligation products and codon deletions or base deletions resulting from oligonucleotide impurities. Base deletions in the trimer stocks were measured to be 1.30% ± 0.42% whereas those for hexamers were 1.16% ± 0.13%. For CDRs of shorter length, there is an advantage in using hexamers since the number of cassettes can be minimized, reducing synthesis time. Furthermore, when hexamers cassettes are used, much longer CDR loops can be attained.
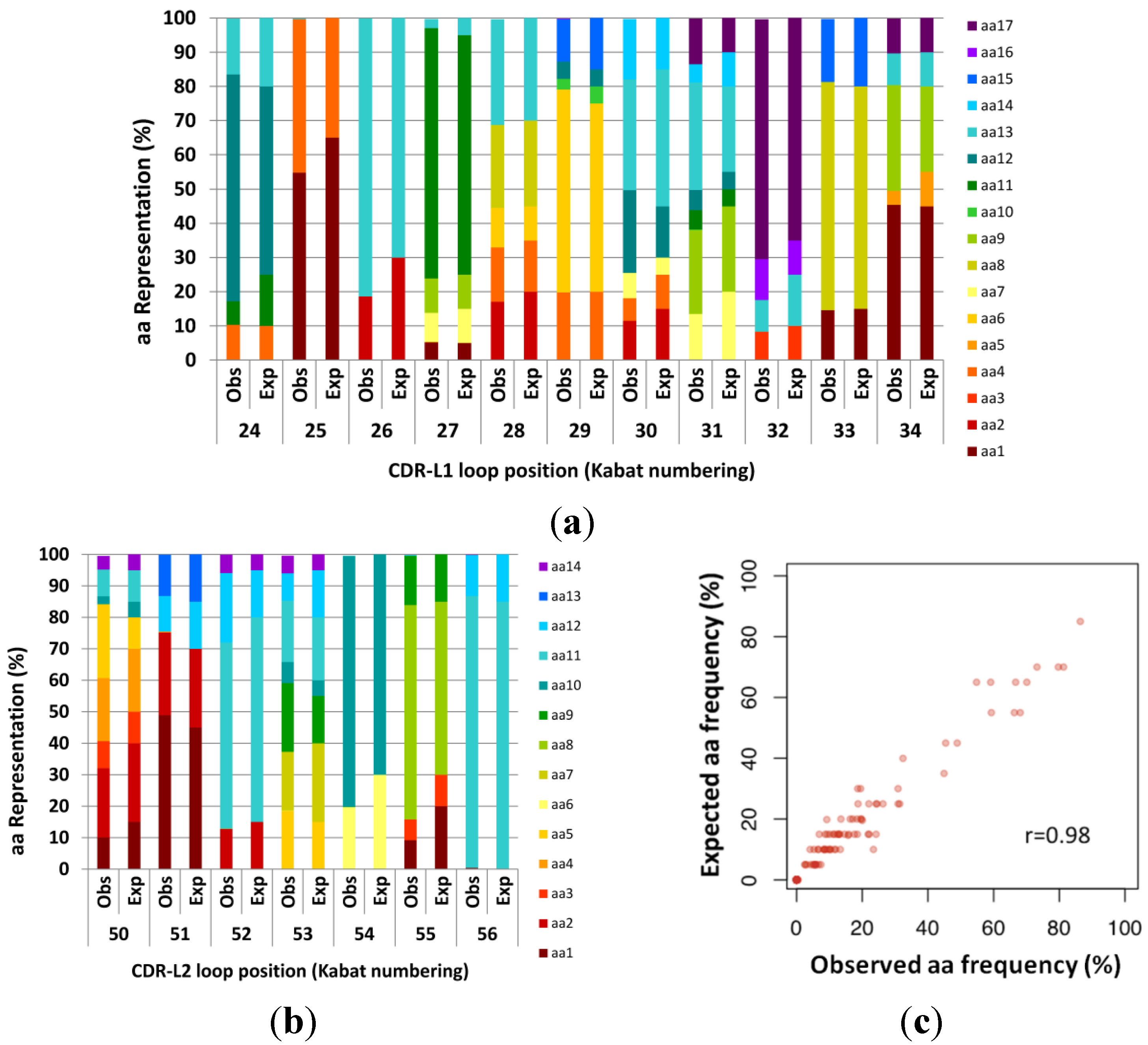
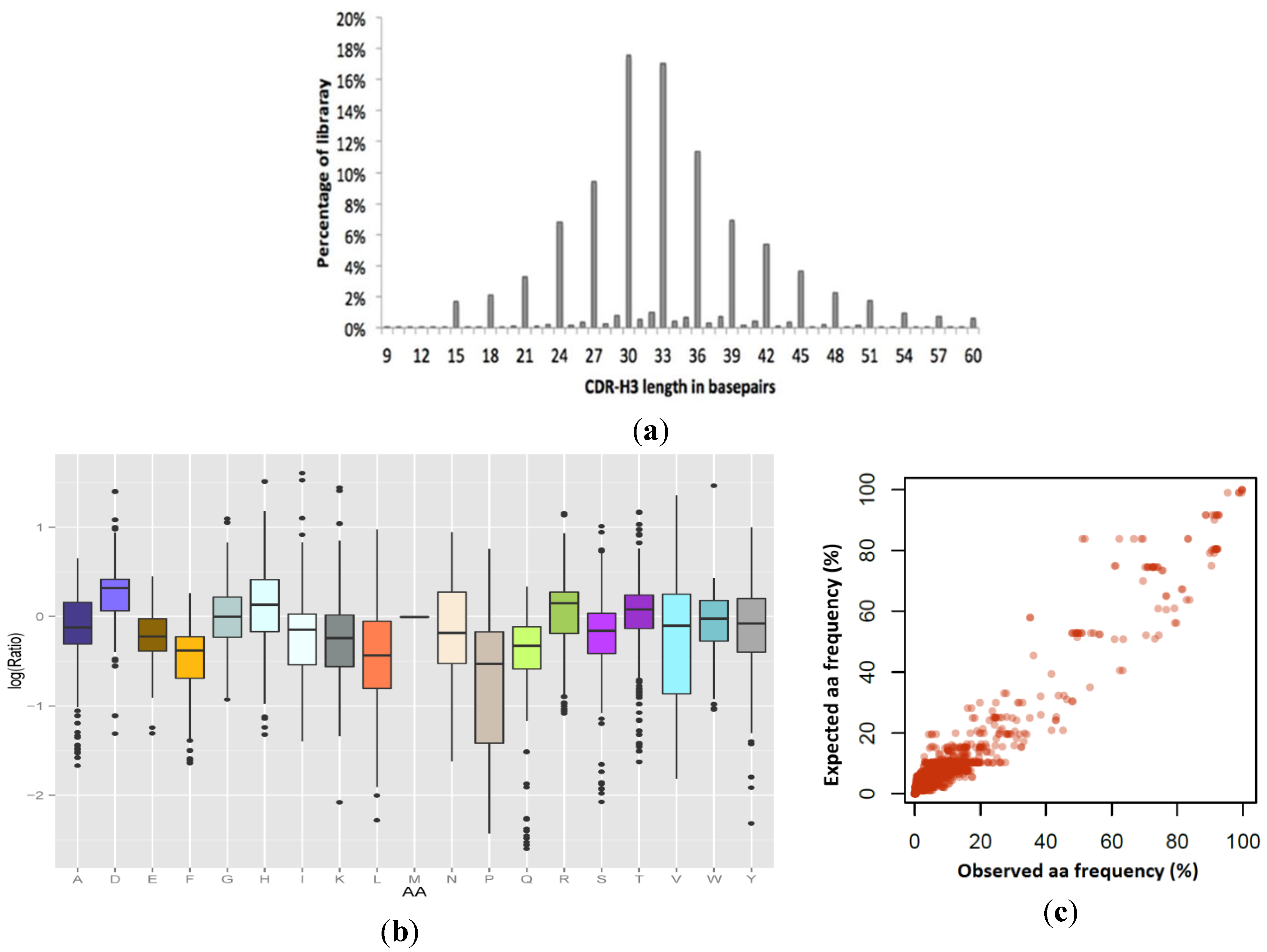
Figure 2.
Expected (Exp) versus observed (Obs) amino acid frequencies in complementarity determining regions (CDR) fragment libraries created using manual hexamer additions. Codon frequencies were determined by Illumina Miseq analysis of amplicons of expected length. Designed (expected) codons ranged from 5% to 85% per position. (a) CDR-L1. (b) CDR-L2. (c) Expected design frequencies plotted versus Observed frequencies calculated from next generation sequencing (NGS) (n = 130399). The resulting library was highly correlated to the design (Pearson’s r = 0.984).
Figure 2.
Expected (Exp) versus observed (Obs) amino acid frequencies in complementarity determining regions (CDR) fragment libraries created using manual hexamer additions. Codon frequencies were determined by Illumina Miseq analysis of amplicons of expected length. Designed (expected) codons ranged from 5% to 85% per position. (a) CDR-L1. (b) CDR-L2. (c) Expected design frequencies plotted versus Observed frequencies calculated from next generation sequencing (NGS) (n = 130399). The resulting library was highly correlated to the design (Pearson’s r = 0.984).
Whilst the synthesis of our designed CDR-L2 did not require multiple cassettes, we fabricated CDR-L1 by adding 3 hexamers on one cassette (6 codons), and 2 hexamers and a trimer (5 codons) on a second cassette, in order to obtain the desired 11 codon loop. The two cassettes were then ligated together using the same approach as during synthesis, as described in the
Experimental Section. The fabrication of scFv light chain CDR-L1 and CDR-L2 demonstrate the fine control of hexamer codon additions across the CDR length with all additions being maintained close to the ratios that were designed (
Figure 2).
In theory, hexamers should result in greater library purity when compared with multiple trimer additions, since impurities accumulate per addition. Comparison with a scFv CDR-H2 loop built by trimer addition (unpublished data) indeed suggests some improved product purity (cf. 88.3% correct length from trimer addition with 92.1% by using hexamers). To generate a scFv library, CDR-L1 and CDR-L2 fragments described in
Figure 2 were combined with exemplar CDR-L3 loops that had been previously fabricated. The resulting light chain CDR segments were then ligated to framework regions to generate V
L domains and the percentage of products with the expected length was analyzed (
Table 1). Finally, the V
L domains were ligated to a V
H library previously constructed using ProxiMAX trimer methodology, to generate complete scFv libraries.
Table 1.
Length accuracy of synthetic scFv loops and domains. Light chain CDR loops were analysed by Illumina Miseq sequencing (n = 351021 for CDR-L1 and 345614 for CDR-L2) and the results verified by Sanger sequencing (n = 102). CDR-L1 and CDR-L2 were then combined with exemplar CDR-L3 domains encoding loop lengths of both 9 and 10 aa’s, to generate light chain libraries VL 3-9 and VL 3-10 respectively. The percentage of functional (in frame) fully-assembled light-chain VL sequences was examined by Sanger sequencing.
Table 1.
Length accuracy of synthetic scFv loops and domains. Light chain CDR loops were analysed by Illumina Miseq sequencing (n = 351021 for CDR-L1 and 345614 for CDR-L2) and the results verified by Sanger sequencing (n = 102). CDR-L1 and CDR-L2 were then combined with exemplar CDR-L3 domains encoding loop lengths of both 9 and 10 aa’s, to generate light chain libraries VL 3-9 and VL 3-10 respectively. The percentage of functional (in frame) fully-assembled light-chain VL sequences was examined by Sanger sequencing.
| Domain | Correct length | n-1 | n-3 | Sequencing methodology |
|---|
| CDR-L1 | 92% | 7% | 0.5% | Ilumina NGS |
| 92% | 8% | 0.0% | Sanger |
| CDR-L2 | 96% | 3% | 0.5% | Ilumina NGS |
| 98% | 2% | 0.0% | Sanger |
| VL 3-9 | 77% | n/a | n/a | Sanger |
| VL 3-10 | 77% | n/a | n/a | Sanger |
2.2. Automated Hexamer Method Validation
Having determined that hexamer addition was a viable process, we next turned attention to automation. In order to make the process more economical, manageable and more flexible in terms of codon use and frequency, we developed a mixed pot procedure to reduce both the number of process steps and the requirement for manual manipulation of the products within each cycle. Specifically, liquid-handling robots were utilized to enable a pool of 400 hexameric donor sequences to be employed (as compared with 20 trimeric donors) at each cycle and 3 sets of such oligonucleotides (total 1200 oligonucleotides) were used over three sequential ligations, which also required three sets of post-ligation PCR recovery oligonucleotides. Whilst we used 20 standard codons, one coding for each amino acid, others have been tested and this provides an additional advantage over chemical synthesis methods where the complete codon set has not been manufactured. The expense in reagents was compensated by improved throughput in the process: for the randomization of two consecutive codons, this constituted a reduction from 164 to just 5 steps (albeit with up to 400 automated pipetting actions for each randomized hexamer position,
Figure 1).
To ‘pressure test’ the relative quality of this automated hexamer process, we sought to challenge the technology by synthesising multiple, long V
HH CDR loops at user-defined mixtures of codons in sometimes less than 1% ratios. The design was based on camel, llama and alpaca repertoires and from bioinformatical analyses of next generation sequencing data sets. Camelid CDR3s are generally longer than those of other characterized vertebrate repertoires (with the exception of bovine CDR3 domains derived from a specialized DH2 region) and potentially have different target preferences from shorter CDR3s [
16]. Therefore a range of V
HH CDR3 domains, with loop lengths of between 7 and 24 saturated residues in our design (5–22 amino acids within CDR3 region 95–102 as defined by Kabat [
17]) were created (
Figure 3).
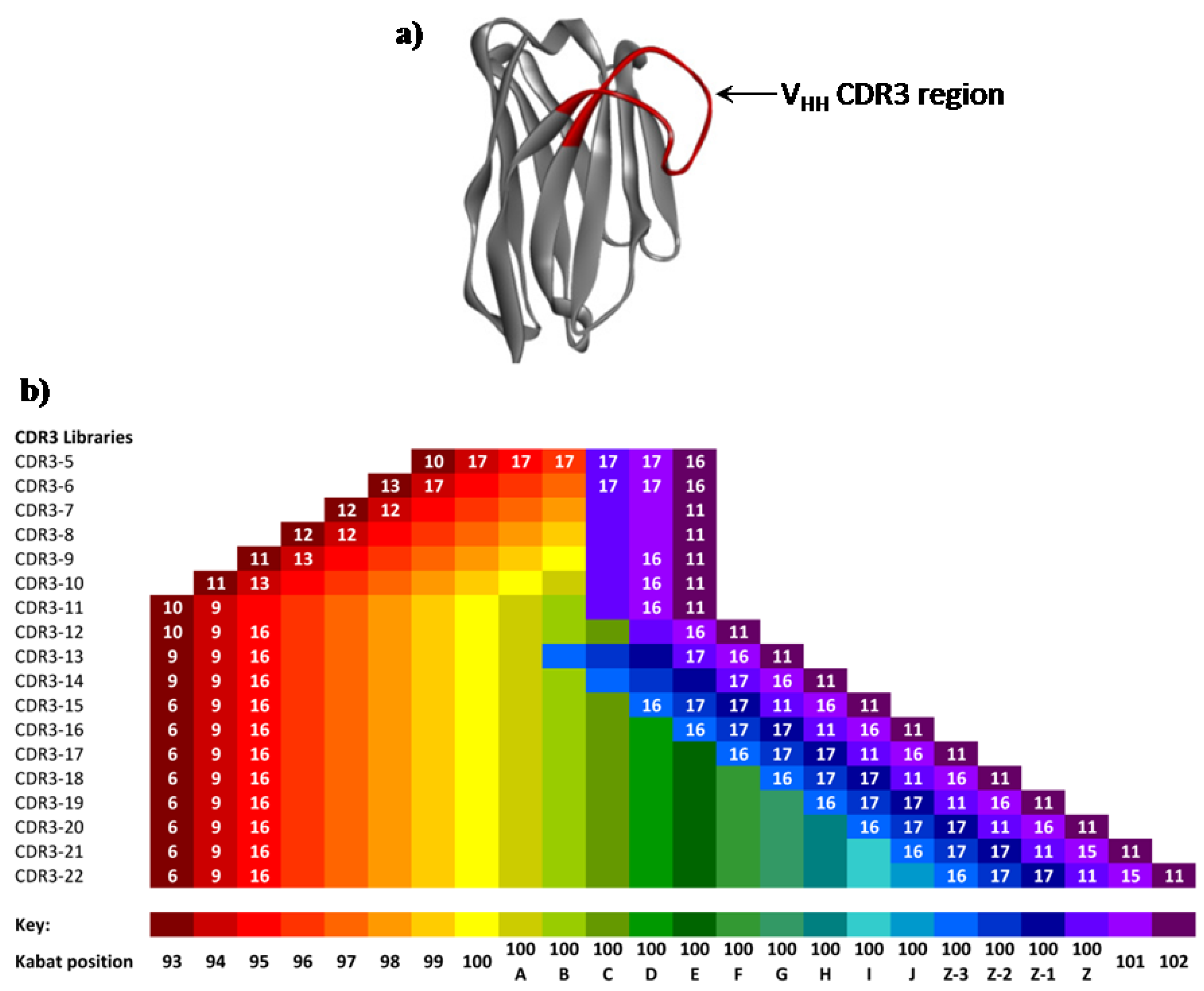
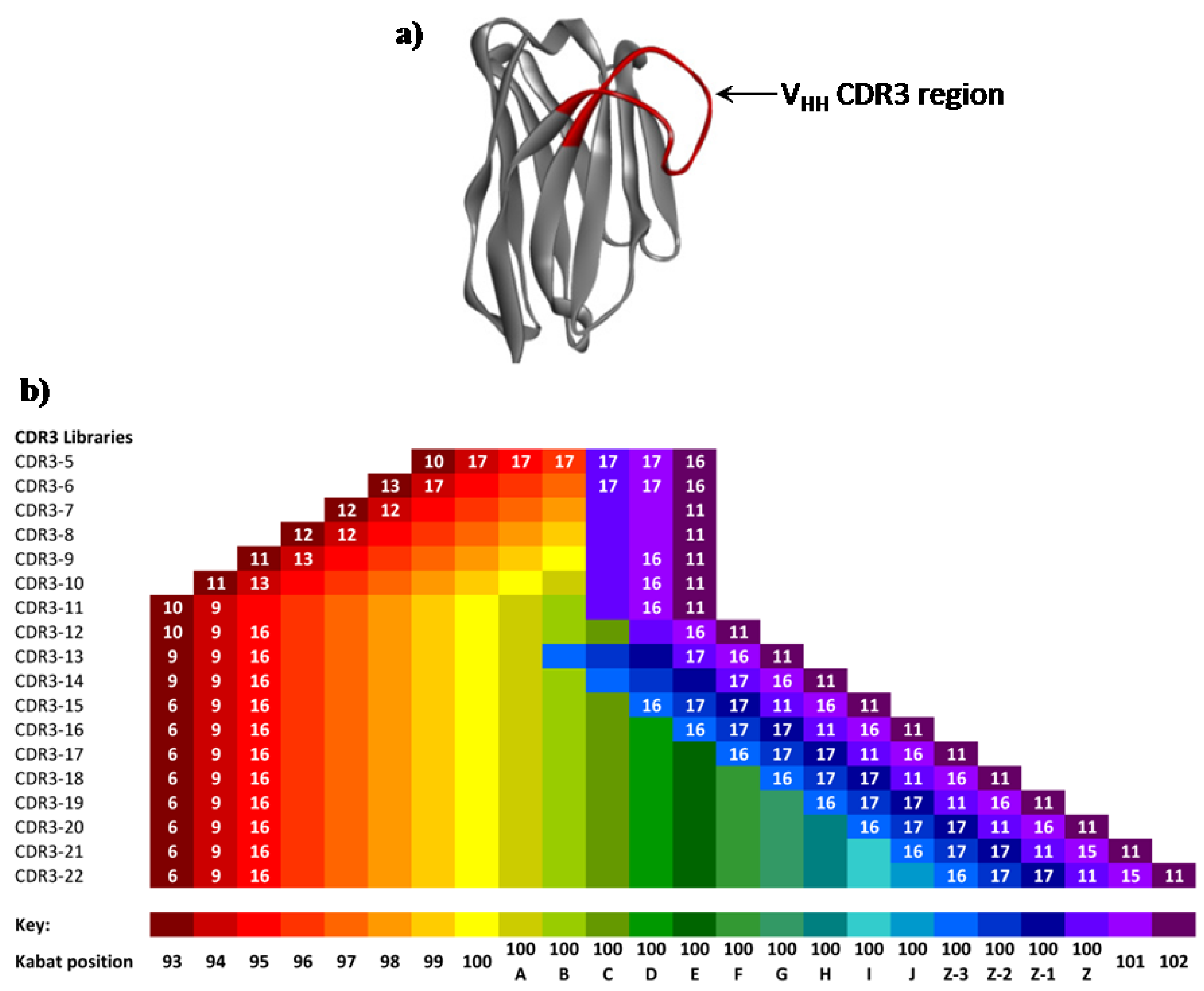
Figure 3.
(a) Ribbon representation of an exemplar llama VHH domain (pdb1I3V16), showing the extended CDR3 domain targeted for saturation mutagenesis. (b) Illustration of the Kabat positions (black font) and numbers of encoded amino acids at each Kabat position (white font, <18 encoded amino acids) within each library segment. Absence of a white number indicates that all 18 amino acids (no Cys or Met) were encoded.
Figure 3.
(a) Ribbon representation of an exemplar llama VHH domain (pdb1I3V16), showing the extended CDR3 domain targeted for saturation mutagenesis. (b) Illustration of the Kabat positions (black font) and numbers of encoded amino acids at each Kabat position (white font, <18 encoded amino acids) within each library segment. Absence of a white number indicates that all 18 amino acids (no Cys or Met) were encoded.
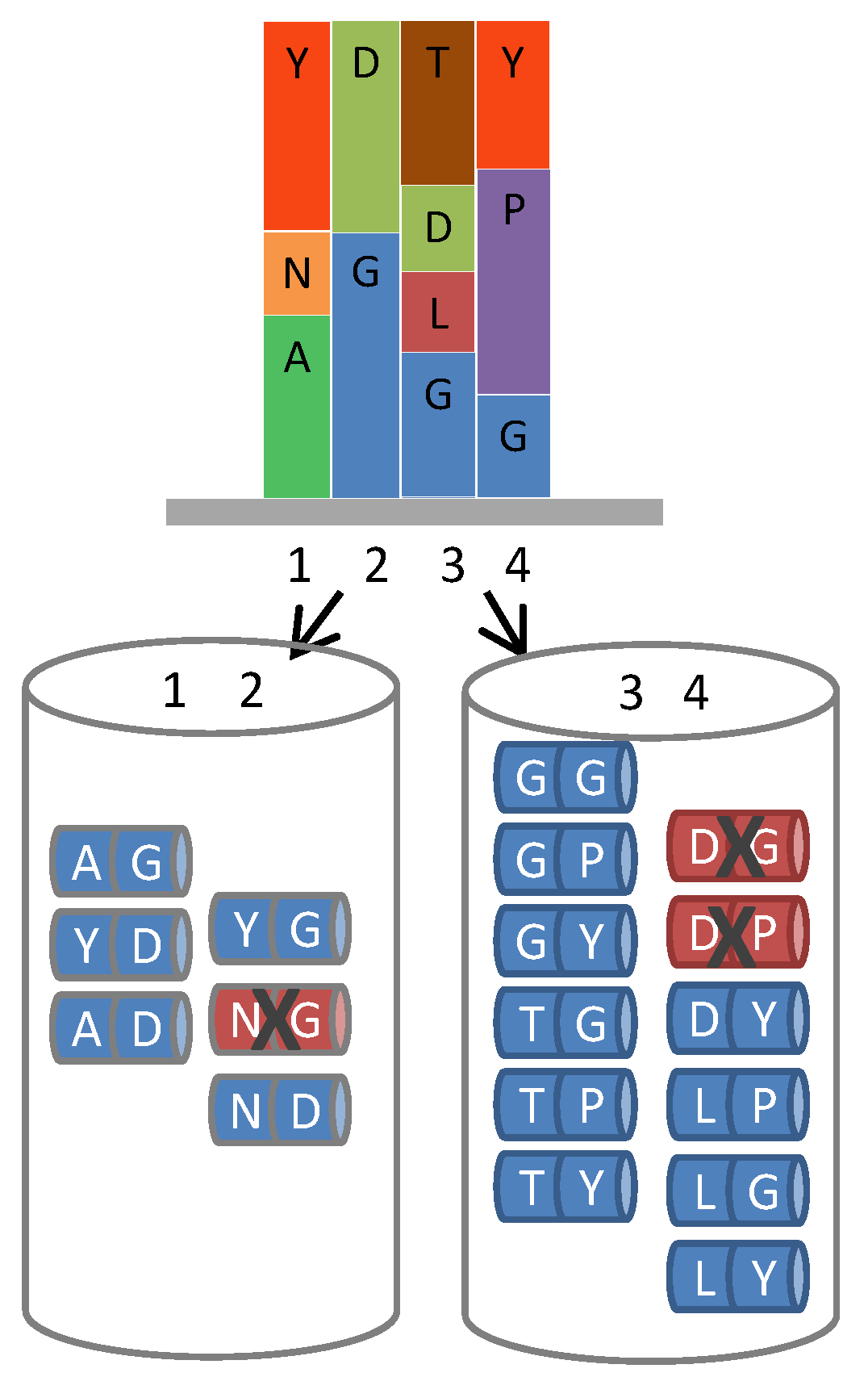
In the synthesis of these loops, it was possible to remove codons for methionine and cysteine from all saturated positions. It was also possible to eliminate the encoding of the undesirable amino acid combinations NG, NS, NA, DG, DP, DS (which render the protein product susceptible to hydrolysis, deamidation and isomerisation [
4]) within hexanucleotide additions (
Figure 4), by simple omission of relevant hexanucleotide donors (Note that such elimination
between hexanucleotide additions was not attempted in the present study but is achieveable, if required, via a split-pot synthetic approach).
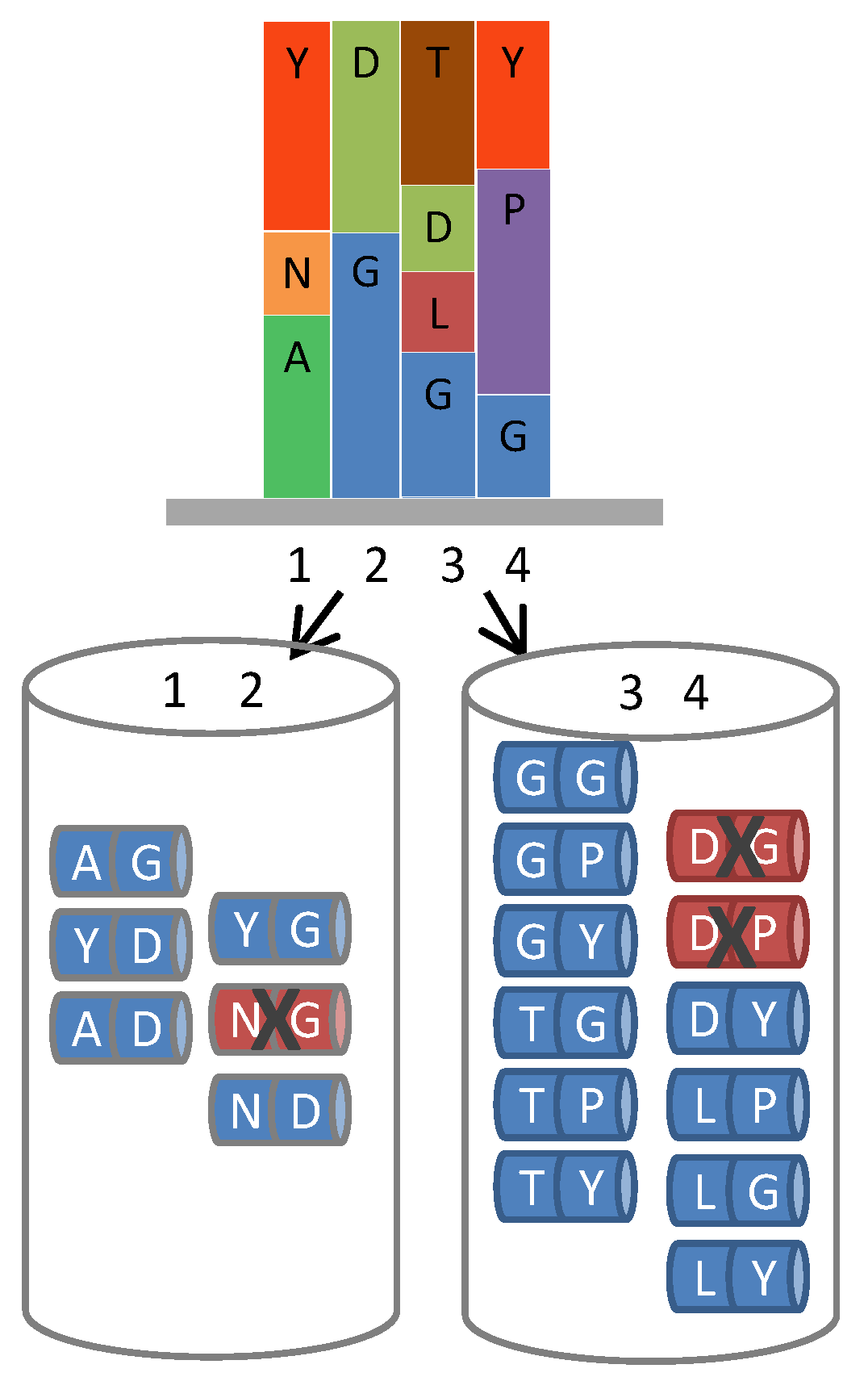
Figure 4.
Removal of liabilities from hexameric ProxiMAX. A hypothetical gene region requiring 4 positions of saturation mutagenesis (1 to 4, top diagram), each position having varying codon number, identity and frequency, can be fabricated by automated hexameric ProxiMAX by sequential addition of two pools of MAXMAX codons (1–2 and 3–4, bottom diagram) containing the required mixture of double-codons at a defined percentage, dictated by the specific library design. Any unfavorable codon pair can be selectively removed from the mixes, increasing the functionality of the library.
Figure 4.
Removal of liabilities from hexameric ProxiMAX. A hypothetical gene region requiring 4 positions of saturation mutagenesis (1 to 4, top diagram), each position having varying codon number, identity and frequency, can be fabricated by automated hexameric ProxiMAX by sequential addition of two pools of MAXMAX codons (1–2 and 3–4, bottom diagram) containing the required mixture of double-codons at a defined percentage, dictated by the specific library design. Any unfavorable codon pair can be selectively removed from the mixes, increasing the functionality of the library.
In order to achieve precision in the mixing and addition of the hexamers, liquid handling robots were employed to mix dilutions of annealed oligonucleotide stocks with individual volumes ranging from 1 to 96 μL. Automation equipment was calibrated for each liquid class and where a concentrated oligonucleotide stock was required, the mix was performed in duplicate and combined into a stock solution of hexamers to reduce pipetting inaccuracies and mistakes in the aspiration or dispense steps.
Prior to their use in the automated ProxiMAX, the hexameric mixes were analysed by NGS to verify both the presence and the proportion of all intended donors (up to 400 per mix). Acceptance criteria dictated that all desired hexameric oligos were present, but excluding those that were not designed. However, adjustments for instrumentation error rates, batch-to-batch and sampling variations, as well as allowances for some deviation of the observed frequencies from the expected levels, similar to those described in
Table 1 determined a ‘QC pass’. On average, greater than 94% of hexameric components in each mix complied with such criteria, and those mixes that fell below this level were repeated.
Variable regions CDR3-5 to CDR3-22 (
Figure 3b) were synthesized individually. Where longer CDR3 regions were required, these were generated in two cassettes and subsequently ligated together (CDR3-13 to CDR3-22). The 18 individual libraries were then combined in a Gaussian distribution of length and ligated to CDR1 and CDR2 regions built similarly, to generate a library of V
HH domains. The distribution of the combined pool, the encoded amino acid identity and the fidelity of designed ratios in the saturated positions were examined by Illumina NGS and subsequent analyses (
Figure 5). The CDR3 loop length in this library was skewed towards the distribution of mouse and human CDR3s for a comparative study of this library against equivalent targets from other synthetic libraries, (although alternative distributions could be prepared). Furthermore, a combination analysis of the amino acid incorporation efficiencies at 255 independent positions in the V
HH library was performed. The expected incorporation of each amino acid in the resulting library design was compared to the observed incorporation and the fidelity per each amino acid found to be high (Pearson’s r = 0.963).
Overall, the synthesis yielded CDR3 regions that were 92% pure (by size) and 99.7% of all amino acid additions passed the QC criteria (
Table 2). In virtually all positions for all amino acids in the design, the designed frequency was achieved within our QC boundaries and each loop was synthesized to greater than 85% purity.
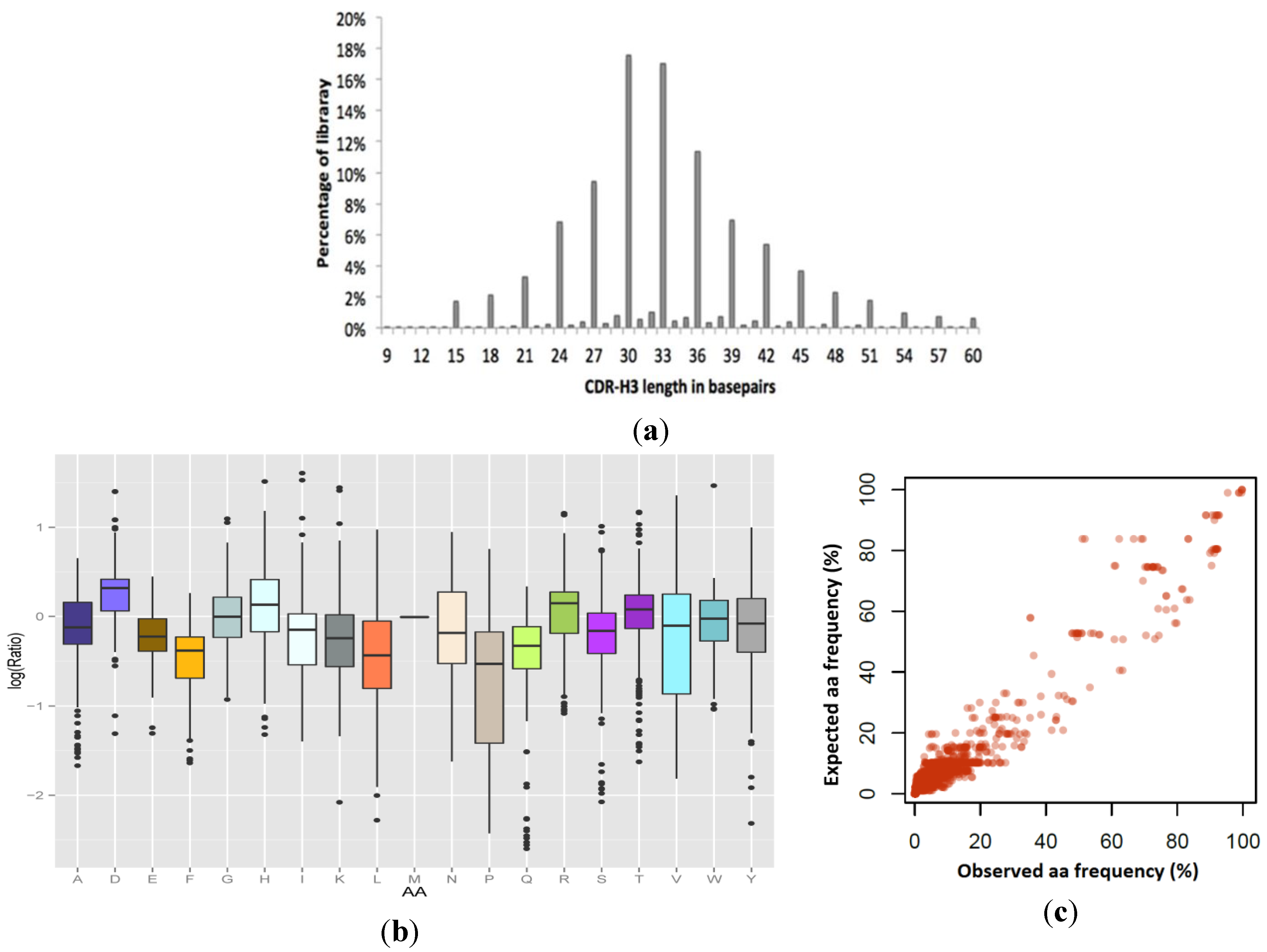
Figure 5.
Colibra design fidelity in the final synthesized VHH library. (a) Observed CDR3 length distribution: 92% of the resulting library was observed in-frame. (b) and (c) Combination analysis of the amino acid incorporation efficiencies at 255 independent positions in the VHH library, as determined by high throughput sequencing, compared to expected values by design. (b) Per-amino acid log (observed/expected) incorporation fidelity, showing the median, 25th percentile, 75th percentile, and outliers. (c) Data for all amino acid incorporation frequencies compared with design displayed in linear-scale (Pearson’s r = 0.963).
Figure 5.
Colibra design fidelity in the final synthesized VHH library. (a) Observed CDR3 length distribution: 92% of the resulting library was observed in-frame. (b) and (c) Combination analysis of the amino acid incorporation efficiencies at 255 independent positions in the VHH library, as determined by high throughput sequencing, compared to expected values by design. (b) Per-amino acid log (observed/expected) incorporation fidelity, showing the median, 25th percentile, 75th percentile, and outliers. (c) Data for all amino acid incorporation frequencies compared with design displayed in linear-scale (Pearson’s r = 0.963).
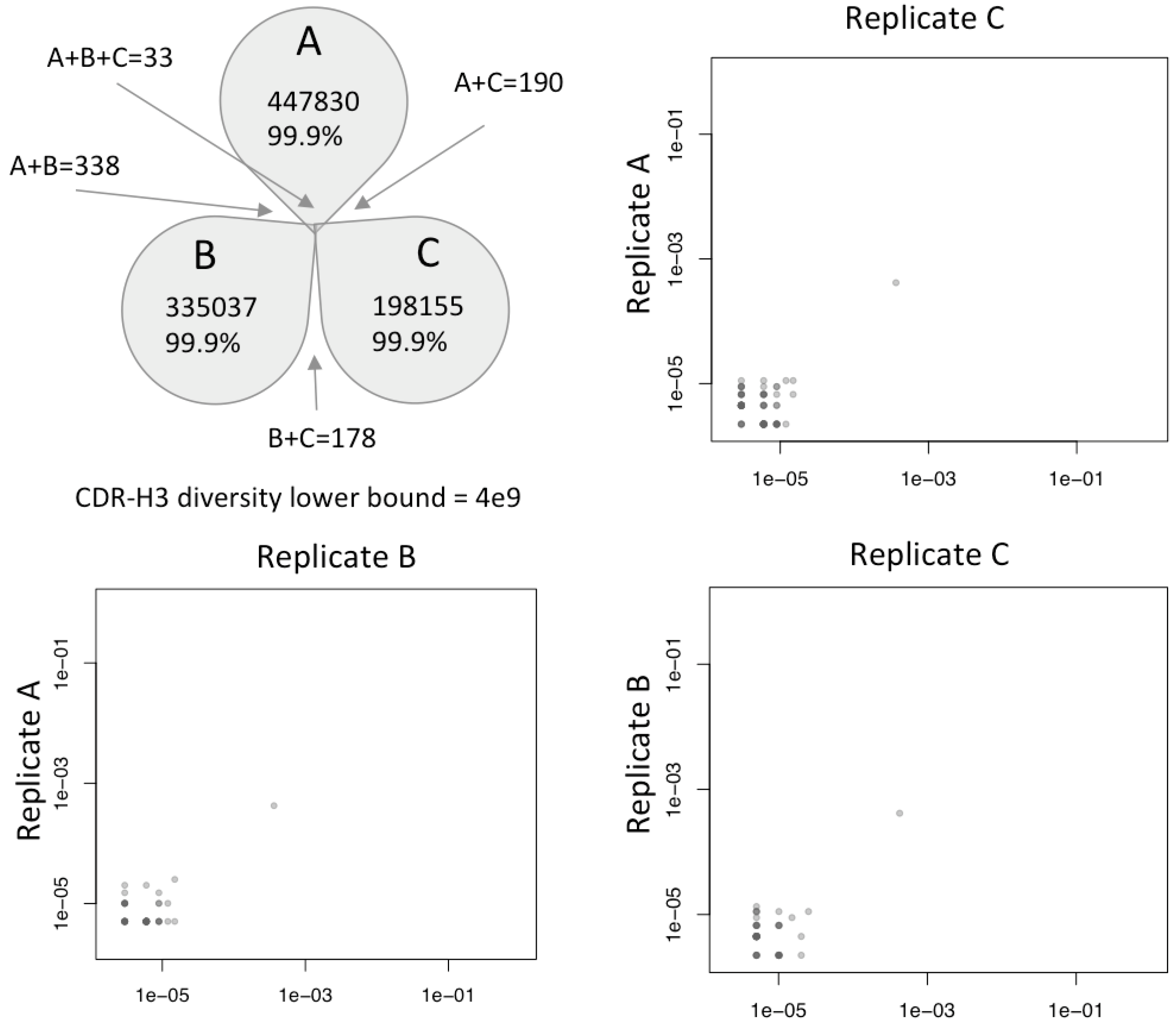
NGS data analysis of three sample replicates of the V
HH CDR3s showed that the synthesis produced a very diverse library such that 99.9% of all sequences observed, even at this great depth, were unique (
Figure 6). A control 2e-2 clone existed to calibrate enrichment during selection. A rare population of shared clones began to emerge in the shorter CDRs at 1e-5 (one in one hundred thousand sequences), although only 33 of such clones were reliably recovered in all three replicates. Using Fischer’s capture recapture, the diversity of the resulting synthesized material was calculated to be minimally 4 billion molecules, and potentially much higher. The results validate our prediction that no physical synthesis steps were restricting the library through any diversity bottlenecks that would adversely affect the resulting library quality.
Table 2.
Quality assessment of VHH libraries. NGS sequence data was assessed to determine the percentage of CDR regions of the designed length.
Table 2.
Quality assessment of VHH libraries. NGS sequence data was assessed to determine the percentage of CDR regions of the designed length.
| Domain | Designed length % | Correct aa identity % | QC Passa % |
|---|
| CDR1 | 93.92 | 99.7 | 100 |
| CDR2 | 87.87 | 99.7 | 100 |
| CDR3-Total | 92.34 | 99.7 | 99.7 |
Figure 6.
Synthesized molecular diversity of VHH CDR-H3. Three replicates were sequenced to depths of 447830, 335037 and 198155 reads, respectively. Over 99.9% of all reads in each replicate were unique and specific to the replicate in which they were generated, with only 33 clones observed across all three replicates at this depth of sequencing. A minimum CDR3 diversity of 4 billion unique CDR3s is calculated by capture-recapture. Plots show clone frequency overlap between replicates. A single 5e-3 enrichment control clone is observed in all libraries, with shared clones becoming evident with frequencies 1e-5 or below.
Figure 6.
Synthesized molecular diversity of VHH CDR-H3. Three replicates were sequenced to depths of 447830, 335037 and 198155 reads, respectively. Over 99.9% of all reads in each replicate were unique and specific to the replicate in which they were generated, with only 33 clones observed across all three replicates at this depth of sequencing. A minimum CDR3 diversity of 4 billion unique CDR3s is calculated by capture-recapture. Plots show clone frequency overlap between replicates. A single 5e-3 enrichment control clone is observed in all libraries, with shared clones becoming evident with frequencies 1e-5 or below.
2.3. Discussion
In this study, we have extended the use of ProxiMAX beyond trimer addition to accommodate hexamer additions both for manual and automated mixing. We initially used the hexamers for manual addition in a semi-minimalist design for VL CDRs in which the most frequent amino acids in our database of VL sequences were simplified to the nearest 5%, with a minimal threshold of 5%. We observed a very close correlation of the observed to the expected (or designed) frequency for every position across the CDRs in this library, demonstrating the utility of this method.
We subsequently refined the technique for an automated platform which was used to construct the CDR3 loops of a camelid V
HH library. These loops differ from the canonical structures typical of antibodies with paired V
H and V
L domains [
15]—an adaptation that probably evolved to compensate for the lack of diversity which can result from pairing V
H and V
L domains. In general the CDR3 loops from V
HH domains are longer than those of HCDR3s from human and mouse repertoires [
3,
4,
15,
18]. Fabrication of the V
HH CDR3 loop lengths demonstrated a further enhancement over the trimer method in that loop lengths greater than 15 amino acids, which present difficulties in the assemblies with single trimer additions, were comfortably tackled with the hexamer components by building the CDR3 in two halves of up to 12 amino acids each and then assembling the completed loop. Hence, CDR3 regions up to 24 randomized amino acids were created. At this length, it is even more critical to be able to dictate functional amino acids as the theoretical diversity extends to 20
24 (1.7 × 10
31) combinations, but through judicious choice of amino acids and their frequencies, this can be distilled to achieve greater sampling of the functional space. This concept has been previously exemplified, where aggregate frequencies of V
H and V
κ CDR3 regions were incorporated into a Fab library design that resulted in 93% of clones displaying correctly folded heavy and light chains [
6]. However, despite extreme examples of minimalist designs, incorporating amino acids which are over-represented in CDRs (primarily Tyr and Ser), being able to generate high affinity hits [
19], severely restricted designs (Y/S) have been shown to lead to lower affinity binders than a fuller repertoire of amino acids [
20]. Nevertheless, synthesis methods that allow programmable design of the CDR composition will permit better interrogation of the capacity of this site for antigen binding [
5].
Whilst MAX codon libraries have been effectively utilized in our antibody engineering to produce libraries that are highly functional, to our knowledge the process described herein is the first report of a saturation mutagenesis method that accommodates a full set double codon hexamer blocks (20 × 20 codons) that can precisely randomize two neighboring positions in parallel in a single step. Clearly the work described herein is not for use at the standard laboratory bench, where the purchase of so many oligonucleotides (and the necessary automation to handle them) would be a prohibitive expense. Rather, we present the achievements of Colibra™ in order to allow prospective users to compare the results achieved using Colibra™ with previously-published achievements of SlonoMAX and TRIM technologies. It is for the accomplished antibody engineers to decide whether Colibra™ offers distinct advantages. However, we also believe that besides the performance enhancement in terms of speed and final purity, there are additional advantages that can be obtained through enzymatic ligation methods such as reducing problematic motifs in the randomized region. Specifically, there are problematic paired residue motifs that have a propensity to cause issues during manufacturing or long-term storage of antibodies which include NG, NS, NA (deamidation) and DG (isomerization) [
4]. These biochemical liabilities can be reduced within the initial library by excluding the corresponding hexamers; however certain pairs may exist at the ligation junctions. To eliminate these completely, depending upon design, a split-pot synthesis method could be employed. It is also conceivable that specific glycosylation signals can be removed through a similar approach so that asparagine can be safely encoded within the CDR.
Therefore, we have demonstrated that the ProxiMAX technique can be adapted to accommodate additions of paired randomized codons. The ProxiMAX method is particularly suited to this purpose as utilizes blunt-ended ligation of codon blocks so that complementary sticky-ends do not have to be created which then require precise pairing. Advantageously, using enzymatic ligations in preference to solid-phase chemical synthesis method, such as TRIM, provides the possibility of including most codons, which can be readily synthesized and used immediately within library fabrication. Some codons that encode a MlyI recognition site (GAGTC) are excluded from our process, therefore precluding the use of certain codon pairs such as GAG-TCN, although other synonymous codons can be used to encode the amino acids. Having a wide choice of available codons is relevant when specifically designing codon optimized libraries for production strains used in the manufacture of biologics. Consequently, the modified ProxiMAX method is particularly applicable to the discovery and development of biologics from hit identification to potentially optimizing the expression of sequences for manufacture.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}