
Improvement in Arsenic Adsorption and Calcite Dissolution Kinetics through Size Reduction of a Ferric Hydroxide-Calcite Adsorbent

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Adsorbent Preparation
2.2. Adsorbent Characterization
2.3. Batch Experiment Perfomance
2.4. Aqueous Samples Characterization
2.5. Isotherm Modelling
2.5.1. Freundlich and Langmuir Models
2.5.2. Surface Complexation Model
2.6. Kinetics Modelling
2.6.1. Arsenic Adsorption
2.6.2. Calcite Dissolution
3. Results and Discussion
3.1. Adsorbent Characterization
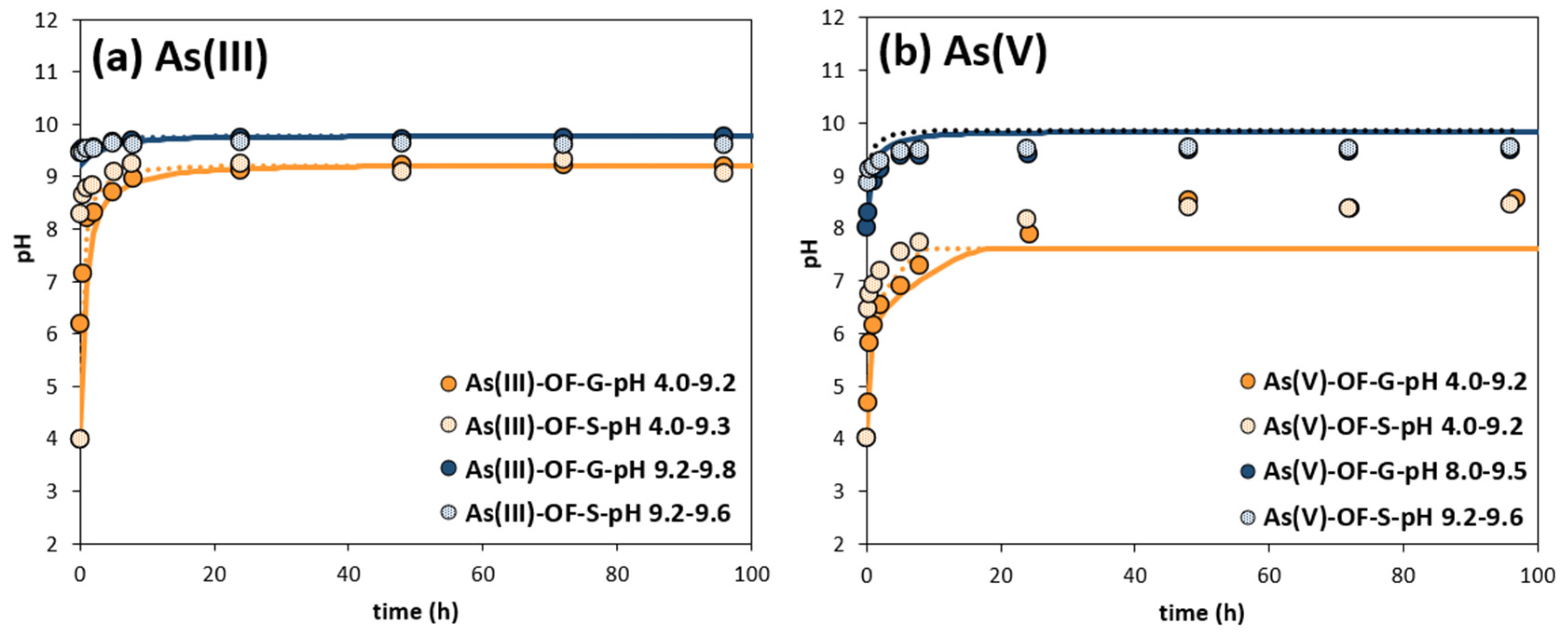
3.2. pH Evolution due to Calcite Dissolution
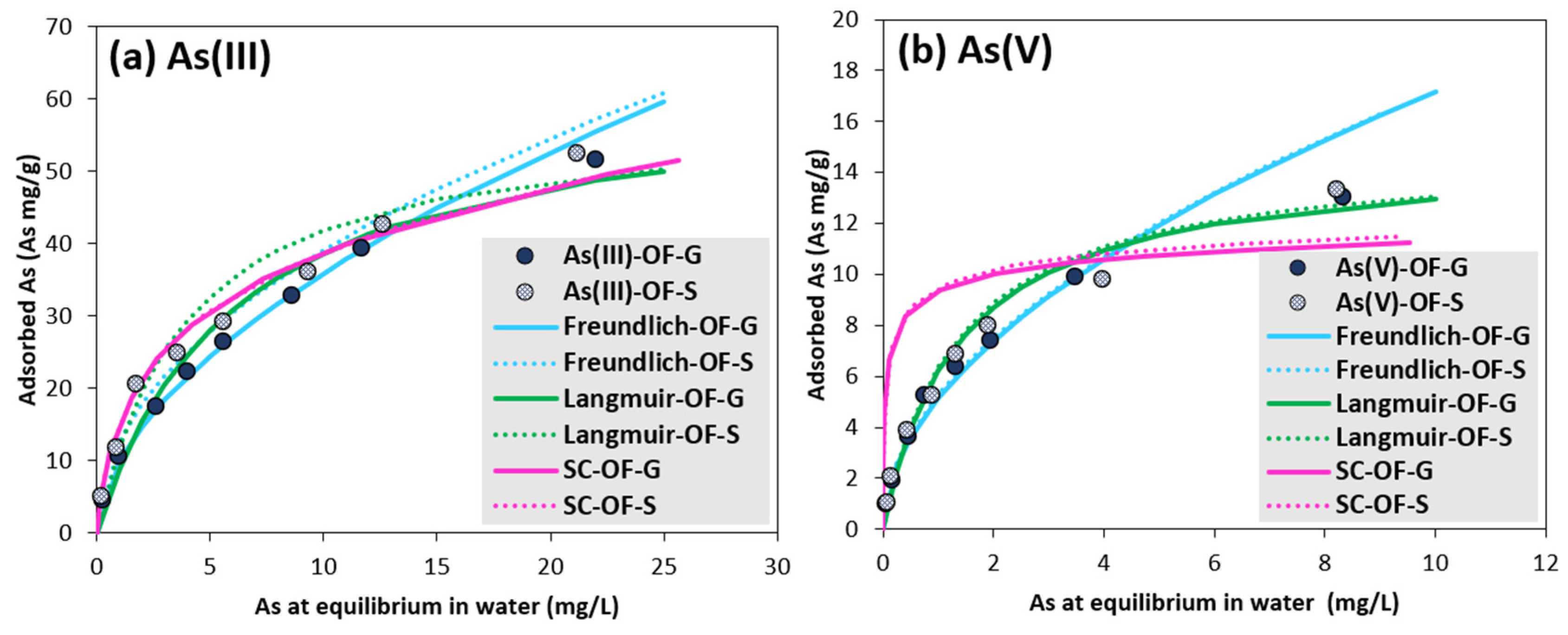
3.3. Arsenic Adsorption Equilibrium
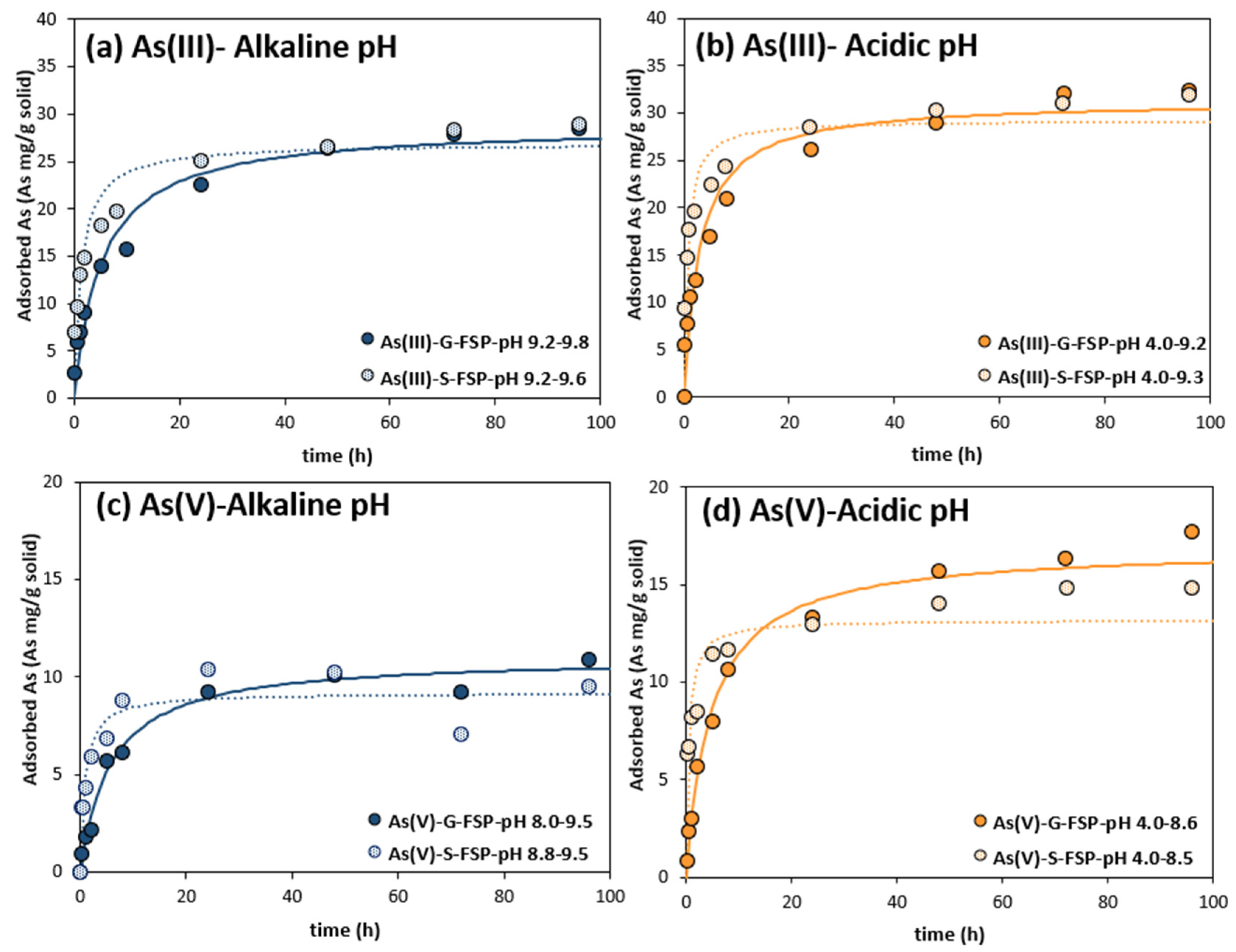
3.4. Arsenic Adsorption Kinetics
3.5. Calcite Dissolution Kinetics
3.6. Environmental Significance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shaji, E.; Santosh, M.; Sarath, K.V.; Prakash, P.; Deepchand, V.; Divya, B.V. Arsenic Contamination of Groundwater: A Global Synopsis with Focus on the Indian Peninsula. Geosci. Front. 2021, 12, 101079. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G. A Review of the Source, Behaviour and Distribution of Arsenic in Natural Waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sengupta, M.K.; Hossain, M.A.; Ahamed, S.; Das, B.; Nayak, B.; Lodh, D.; Rahman, M.M.; Chakraborti, D. Arsenic Contamination in Groundwater: A Global Perspective with Emphasis on the Asian Scenario. J. Health Popul. Nutr. 2006, 24, 142–163. [Google Scholar]
- Baragaño, D.; Boente, C.; Rodríguez-Valdés, E.; Fernández-Braña, A.; Jiménez, A.; Gallego, J.L.R.; González-Fernández, B. Arsenic Release from Pyrite Ash Waste over an Active Hydrogeological System and Its Effects on Water Quality. Environ. Sci. Pollut. Res. 2020, 27, 10672–10684. [Google Scholar] [CrossRef]
- Golab, A.; Peterson, M.; Indraratna, B. Selection of Permeable Reactive Barrier Materials for Treating Acidic Groundwater in Acid Sulphate Soil Terrains Based on Laboratory Column Tests. Environ. Earth Sci. 2009, 59, 241–254. [Google Scholar] [CrossRef]
- Ha, Q.K.; Choi, S.; Phan, N.L.; Kim, K.; Phan, C.N.; Nguyen, V.K.; Ko, K.S. Occurrence of Metal-Rich Acidic Groundwaters around the Mekong Delta (Vietnam): A Phenomenon Linked to Well Installation. Sci. Total Environ. 2019, 654, 1100–1109. [Google Scholar] [CrossRef]
- Angai, J.U.; Ptacek, C.J.; Pakostova, E.; Bain, J.G.; Verbuyst, B.R.; Blowes, D.W. Removal of Arsenic and Metals from Groundwater Impacted by Mine Waste Using Zero-Valent Iron and Organic Carbon: Laboratory Column Experiments. J. Hazard. Mater. 2022, 424, 127295. [Google Scholar] [CrossRef]
- Gibert, O.; Rötting, T.; Cortina, J.L.; de Pablo, J.; Ayora, C.; Carrera, J.; Bolzicco, J. In-Situ Remediation of Acid Mine Drainage Using a Permeable Reactive Barrier in Aznalcóllar (Sw Spain). J. Hazard. Mater. 2011, 191, 287–295. [Google Scholar] [CrossRef]
- Casiot, C.; Leblanc, M.; Bruneel, O.; Personné, J.-C.; Koffi, K.; Elbaz-Poulichet, F. Geochemical Processes Controlling the Formation of As-Rich Waters within a Tailings Impoundment (Carnoulès, France). Aquat. Geochem. 2003, 9, 273–290. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U. Arsenic Removal from Water/Wastewater Using Adsorbents-A Critical Review. J. Hazard. Mater. 2007, 142, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Asere, T.G.; Stevens, C.V.; Du Laing, G. Use of (Modified) Natural Adsorbents for Arsenic Remediation: A Review. Sci. Total Environ. 2019, 676, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, O.S.; Viraraghavan, T.; Subramanian, K.S. Arsenic Removal from Drinking Water Using Granular Ferric Hydroxide. Water SA 2003, 29, 161–170. [Google Scholar] [CrossRef]
- Szlachta, M.; Wójtowicz, P. Treatment of Arsenic-Rich Waters Using Granular Iron Hydroxides. Desalin. Water Treat. 2016, 57, 26376–26381. [Google Scholar] [CrossRef]
- Martí, V.; Jubany, I.; Fernández-Rojo, L.; Ribas, D.; Benito, J.A.; Diéguez, B.; Ginesta, A. Improvement of As(V) Adsorption by Reduction of Granular to Micro-Sized Ferric Hydroxide. Processes 2022, 10, 1029. [Google Scholar] [CrossRef]
- Maity, J.P.; Chen, C.Y.; Bhattacharya, P.; Sharma, R.K.; Ahmad, A.; Patnaik, S.; Bundschuh, J. Advanced Application of Nano-Technological and Biological Processes as Well as Mitigation Options for Arsenic Removal. J. Hazard. Mater. 2021, 405, 123885. [Google Scholar] [CrossRef]
- Usman, M.; Zarebanadkouki, M.; Waseem, M.; Katsoyiannis, I.A.; Ernst, M. Mathematical Modeling of Arsenic(V) Adsorption onto Iron Oxyhydroxides in an Adsorption-Submerged Membrane Hybrid System. J. Hazard. Mater. 2020, 400, 123221. [Google Scholar] [CrossRef]
- Sperlich, A.; Schimmelpfennig, S.; Baumgarten, B.; Genz, A.; Amy, G.; Worch, E.; Jekel, M. Predicting Anion Breakthrough in Granular Ferric Hydroxide (GFH) Adsorption Filters. Water Res. 2008, 42, 2073–2082. [Google Scholar] [CrossRef]
- Badruzzaman, M.; Westerhoff, P.; Knappe, D.R.U. Intraparticle Diffusion and Adsorption of Arsenate onto Granular Ferric Hydroxide (GFH). Water Res. 2004, 38, 4002–4012. [Google Scholar] [CrossRef]
- Mohammadian, S.; Krok, B.; Fritzsche, A.; Bianco, C.; Tosco, T.; Cagigal, E.; Mata, B.; Gonzalez, V.; Diez-Ortiz, M.; Ramos, V.; et al. Field-Scale Demonstration of in Situ Immobilization of Heavy Metals by Injecting Iron Oxide Nanoparticle Adsorption Barriers in Groundwater. J. Contam. Hydrol. 2021, 237, 103741. [Google Scholar] [CrossRef]
- Castaño, A.; Prosenkov, A.; Baragaño, D.; Otaegui, N.; Sastre, H.; Rodríguez-Valdés, E.; Gallego, J.L.R.; Peláez, A.I. Effects of in Situ Remediation with Nanoscale Zero Valence Iron on the Physicochemical Conditions and Bacterial Communities of Groundwater Contaminated with Arsenic. Front. Microbiol. 2021, 12, 643589. [Google Scholar] [CrossRef]
- Montalvo, D.; Vanderschueren, R.; Fritzsche, A.; Meckenstock, R.U.; Smolders, E. Efficient Removal of Arsenate from Oxic Contaminated Water by Colloidal Humic Acid-Coated Goethite: Batch and Column Experiments. J. Clean. Prod. 2018, 189, 510–518. [Google Scholar] [CrossRef]
- Kumar, P.S.; Korving, L.; Keesman, K.J.; van Loosdrecht, M.C.M.; Witkamp, G.J. Effect of Pore Size Distribution and Particle Size of Porous Metal Oxides on Phosphate Adsorption Capacity and Kinetics. Chem. Eng. J. 2019, 358, 160–169. [Google Scholar] [CrossRef]
- Martí, V.; Jubany, I.; Ribas, D.; Benito, J.A.; Ferrer, B. Improvement of Phosphate Adsorption Kinetics onto Ferric Hydroxide by Size Reduction. Water 2021, 13, 1558. [Google Scholar] [CrossRef]
- Kunaschk, M.; Schmalz, V.; Dietrich, N.; Dittmar, T.; Worch, E. Novel Regeneration Method for Phosphate Loaded Granular Ferric (Hydr)Oxide—A Contribution to Phosphorus Recycling. Water Res. 2015, 71, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Amy, G.L.; Prevost, M.; Nour, S.; Jekel, M.; Gallagher, P.M.; Blumenschein, C.D. Kinetic and Thermodynamic Aspects of Adsorption of Arsenic onto Granular Ferric Hydroxide (GFH). Water Res. 2008, 42, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Garau, G.; Silvetti, M.; Castaldi, P.; Mele, E.; Deiana, P.; Deiana, S. Stabilising Metal(Loid)s in Soil with Iron and Aluminium-Based Products: Microbial, Biochemical and Plant Growth Impact. J. Environ. Manage. 2014, 139, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rivero, K.; Bastos-Arrieta, J.; Florido, A.; Martí, V. Potential Use of Precipitates from Acid Mine Drainage (AMD) as Arsenic Adsorbents. Water 2023, 15, 3179. [Google Scholar] [CrossRef]
- Wei, Z.; Kosterman, J.A.; Xiao, R.; Pee, G.Y.; Cai, M.; Weavers, L.K. Designing and Characterizing a Multi-Stepped Ultrasonic Horn for Enhanced Sonochemical Performance. Ultrason. Sonochem. 2015, 27, 325–333. [Google Scholar] [CrossRef]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modeling: Hydrous Ferric Oxide; John Wiley & Sons: Hoboken, NJ, USA, 1990. [Google Scholar]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S. Geological Survey Techniques and Methods, Book 6; U.S. Geological Survey: Reston, VA, USA, 2013; Chapter A43; 497p. [CrossRef]
- Appelo, C.A.J.; Van Der Weiden, M.J.J.; Tournassat, C.; Charlet, L. Surface Complexation of Ferrous Iron and Carbonate on Ferrihydrite and the Mobilization of Arsenic. Environ. Sci. Technol. 2002, 36, 3096–3103. [Google Scholar] [CrossRef]
- Suarez, D.L.; Wood, J.D. Division S-2-Soil Chemistry Simultaneous Determination of Calcite Surface Area and Content in Soils 1. Soil Sci. Soc. Am. J. 1984, 48, 1232–1235. [Google Scholar] [CrossRef]
- Yang, J.H.; Shih, S.M.; Wu, C.I.; Tai, C.Y. Der Preparation of High Surface Area CaCO3 for SO2 Removal by Absorption of CO2 in Aqueous Suspensions of Ca(OH)2. Powder Technol. 2010, 202, 101–110. [Google Scholar] [CrossRef]
- Noiriel, C.; Steefel, C.I.; Yang, L.; Ajo-Franklin, J. Upscaling Calcium Carbonate Precipitation Rates from Pore to Continuum Scale. Chem. Geol. 2012, 318–319, 60–74. [Google Scholar] [CrossRef]
- Ho, Y.S.; McKay, G. Pseudo-Second Order Model for Sorption Processes Y.S. Org. Process Res. Dev. 2017, 21, 866–870. [Google Scholar] [CrossRef]
- Podder, M.S.; Majumder, C.B. Biosorption of As(III) and As(V) on the Surface of TW/MnFe2O4 Composite from Wastewater: Kinetics, Mechanistic and Thermodynamics. Appl. Water Sci. 2017, 7, 2689–2715. [Google Scholar] [CrossRef]
- Plummer, L.N.; Wigley, T.M.L.; Parkhurst, D.L. The kinetics of calcite dissolution in CO2-water systems at 5 °C to 60 °C and 0.0 to 1.0 atm CO2. Am. J. Sci. 1978, 278, 179–216. [Google Scholar] [CrossRef]
- Janneck, E.; Burghardt, D.; Simon, E.; Pfeiffer, S.; Paul, M.; Koch, T. Development of an Adsorbent Comprising Schwertmannite and Its Utilization in Mine Water Treatment. In Proceedings of the International Mine Water Association, Santiago de Chile, Chile, 21–24 April 2015; Paper 215. pp. 1–10. [Google Scholar]
- Usman, M.; Katsoyiannis, I.; Mitrakas, M.; Zouboulis, A.; Ernst, M. Performance Evaluation of Small Sized Powdered Ferric Hydroxide as Arsenic Adsorbent. Water 2018, 10, 957. [Google Scholar] [CrossRef]
- Pierce, M.L.; Moore, C.B. Adsorption of Arsenite and Arsenate on Amorphous Iron Hydroxide. Water Res. 1982, 16, 1247–1253. [Google Scholar] [CrossRef]
- Kim, S.O.; Lee, W.C.; Cho, H.G.; Lee, B.T.; Lee, P.K.; Choi, S.H. Equilibria, Kinetics, and Spectroscopic Analyses on the Uptake of Aqueous Arsenite by Two-Line Ferrihydrite. Environ. Technol. 2014, 35, 251–261. [Google Scholar] [CrossRef]
- Guan, X.H.; Wang, J.; Chusuei, C.C. Removal of Arsenic from Water Using Granular Ferric Hydroxide: Macroscopic and Microscopic Studies. J. Hazard. Mater. 2008, 156, 178–185. [Google Scholar] [CrossRef]
- Karn, S.K.; Pan, X. Simultaneous Application Arsenic Oxidising Bacteria and Biochar for the Reclamation of Arsenic Contaminated Soil. Int. J. Environ. Waste Manag. 2018, 21, 155. [Google Scholar] [CrossRef]
- Hlavay, J.; Polyák, K. Determination of Surface Properties of Iron Hydroxide-Coated Alumina Adsorbent Prepared for Removal of Arsenic from Drinking Water. J. Colloid Interface Sci. 2005, 284, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Sø, H.U.; Postma, D.; Jakobsen, R.; Larsen, F. Sorption and Desorption of Arsenate and Arsenite on Calcite. Geochim. Cosmochim. Acta 2008, 72, 5871–5884. [Google Scholar] [CrossRef]
- Radu, T.; Subacz, J.L.; Phillippi, J.M.; Barnett, M.O. Effects of Dissolved Carbonate on Arsenic Adsorption and Mobility. Environ. Sci. Technol. 2005, 39, 7875–7882. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Yamaguchi, K.; Nishimiya, N. Effect of Amplitude and Frequency of Ultrasonic Irradiation on Morphological Characteristics Control of Calcium Carbonate. Ultrason. Sonochem. 2010, 17, 617–620. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Golubev, S.V.; Schott, J.; Castillo, A. Calcite, Dolomite and Magnesite Dissolution Kinetics in Aqueous Solutions at Acid to Circumneutral PH, 25 to 150 °C and 1 to 55 Atm PCO2: New Constraints on CO2 Sequestration in Sedimentary Basins. Chem. Geol. 2009, 265, 20–32. [Google Scholar] [CrossRef]
- Maree, J.P.; Du Plessis, P. Neutralization of Acid Mine Water with Calcium Carbonate. Water Sci. Technol. 1994, 29, 285–296. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Log K |
|---|---|
| Hfo_sOH + H+ = Hfo_sOH2+ | 7.29 |
| Hfo_wOH + H+ = Hfo_wOH2+ | 7.29 |
| Hfo_sOH = Hfo_sO− + H+ | −8.93 |
| Hfo_wOH = Hfo_wO− + H+ | −8.93 |
| Hfo_sOH + H3AsO3 = Hfo_sH2AsO3 + H2O | 5.41 |
| Hfo_wOH + H3AsO3 = Hfo_wH2AsO3 + H2O | 5.41 |
| Hfo_sOH + H3AsO4 = Hfo_sH2AsO4 + H2O | 8.61 |
| Hfo_wOH + H3AsO4 = Hfo_wH2AsO4 + H2O | 8.61 |
| Hfo_sOH + H3AsO4 = Hfo_sHAsO4− + H2O + H+ | 2.81 |
| Hfo_wOH + H3AsO4 = Hfo_wHAsO4− + H2O + H+ | 2.81 |
| Hfo_sOH + H3AsO4 = Hfo_sOHAsO4−3 + 3H+ | −10.12 |
| Hfo_wOH + H3AsO4 = Hfo_wOHAsO4−3 + 3H+ | −10.12 |
| Hfo_sOH + Ca+2 = Hfo_sOHCa+2 | 4.97 |
| Hfo_wOH + Ca+2 = Hfo_wOCa+ + H+ | −5.85 |
| Hfo_wOH + CO3−2 + H+ = Hfo_wOCO2− + H2O | 12.78 |
| Hfo_wOH + CO3−2 + 2H+ = Hfo_wOCO2H + H2O | 20.37 |
| OF-G | OF-S | |
|---|---|---|
| Size (µm) | 250–1000 | 0.4–50 |
| Median size (µm) | 747 | 7 |
| BET surface area (m2/g) | 218 | 233 |
| Micropore area (m2/g) | 39 | 21 |
| As(III) | As(V) | ||||
|---|---|---|---|---|---|
| OF-G | OF-S | OF-G | OF-S | ||
| Freundlich | KF (As mg1−1/n·L1/n/OF g) | 10.01 | 12.77 | 5.13 | 5.25 |
| n | 1.80 | 2.06 | 1.91 | 1.94 | |
| R2 | 0.995 | 0.984 | 0.982 | 0.977 | |
| SSE (As mg/g)2 | 17 | 34 | 7 | 7 | |
| Langmuir | qmax (As mg/OF g) | 62.11 | 58.14 | 14.75 | 14.77 |
| b (L/As mg) | 0.16 | 0.26 | 0.72 | 0.75 | |
| R2 | 0.954 | 0.962 | 0.981 | 0.977 | |
| SSE (As mg/g)2 | 48 | 190 | 3 | 5 | |
| Surface complexation | Strong sites (sites mol/mol OF) | 0.007 | 0.007 | 0.001 | 0.001 |
| Weak sites (sites mol/mol OF) | 0.120 | 0.120 | 0.060 | 0.060 | |
| Surface area (m2/g) | 211 | 233 | 211 | 233 | |
| SSE (As mg/g)2 | 142 | 101 | 101 | 108 | |
| As(III) | As(V) | ||||
|---|---|---|---|---|---|
| OF-G | OF-S | OF-G | OF-S | ||
| Alkaline pH | qe (As mg/g) | 28.9 | 26.9 | 11.0 | 9.2 |
| K2 (g adsorbent/As mg·h) | 0.007 | 0.028 | 0.016 | 0.123 | |
| Initial rate (As mg/g·h) | 5.5 | 20.6 | 1.9 | 10.4 | |
| SSE | 26.3 | 52.5 | 2.7 | 12.8 | |
| Acidic pH | qe (As mg/g) | 31.4 | 29.3 | 16.9 | 13.2 |
| K2 (g adsorbent/As mg·h) | 0.011 | 0.051 | 0.012 | 0.156 | |
| Initial rate (As mg/g·h) | 10.6 | 45.6 | 3.5 | 27.1 | |
| SSE | 53.2 | 56.4 | 2.5 | 18.8 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Rojo, L.; Martí, V.; Jubany, I.; Bahí, N.; Janer, M.; Martínez-Lladó, X.; Rovira, M. Improvement in Arsenic Adsorption and Calcite Dissolution Kinetics through Size Reduction of a Ferric Hydroxide-Calcite Adsorbent. Water 2024, 16, 30. https://doi.org/10.3390/w16010030
Fernandez-Rojo L, Martí V, Jubany I, Bahí N, Janer M, Martínez-Lladó X, Rovira M. Improvement in Arsenic Adsorption and Calcite Dissolution Kinetics through Size Reduction of a Ferric Hydroxide-Calcite Adsorbent. Water. 2024; 16(1):30. https://doi.org/10.3390/w16010030
Chicago/Turabian StyleFernandez-Rojo, Lidia, Vicenç Martí, Irene Jubany, Neus Bahí, Marcel Janer, Xavier Martínez-Lladó, and Miquel Rovira. 2024. "Improvement in Arsenic Adsorption and Calcite Dissolution Kinetics through Size Reduction of a Ferric Hydroxide-Calcite Adsorbent" Water 16, no. 1: 30. https://doi.org/10.3390/w16010030
APA StyleFernandez-Rojo, L., Martí, V., Jubany, I., Bahí, N., Janer, M., Martínez-Lladó, X., & Rovira, M. (2024). Improvement in Arsenic Adsorption and Calcite Dissolution Kinetics through Size Reduction of a Ferric Hydroxide-Calcite Adsorbent. Water, 16(1), 30. https://doi.org/10.3390/w16010030