
Depth–Sequential Investigation of Major Ions, δ18O, δ2H and δ13C in Fractured Aquifers of the St. Lawrence Lowlands (Quebec, Canada) Using Passive Samplers

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Geological and Hydrogeochemical Settings
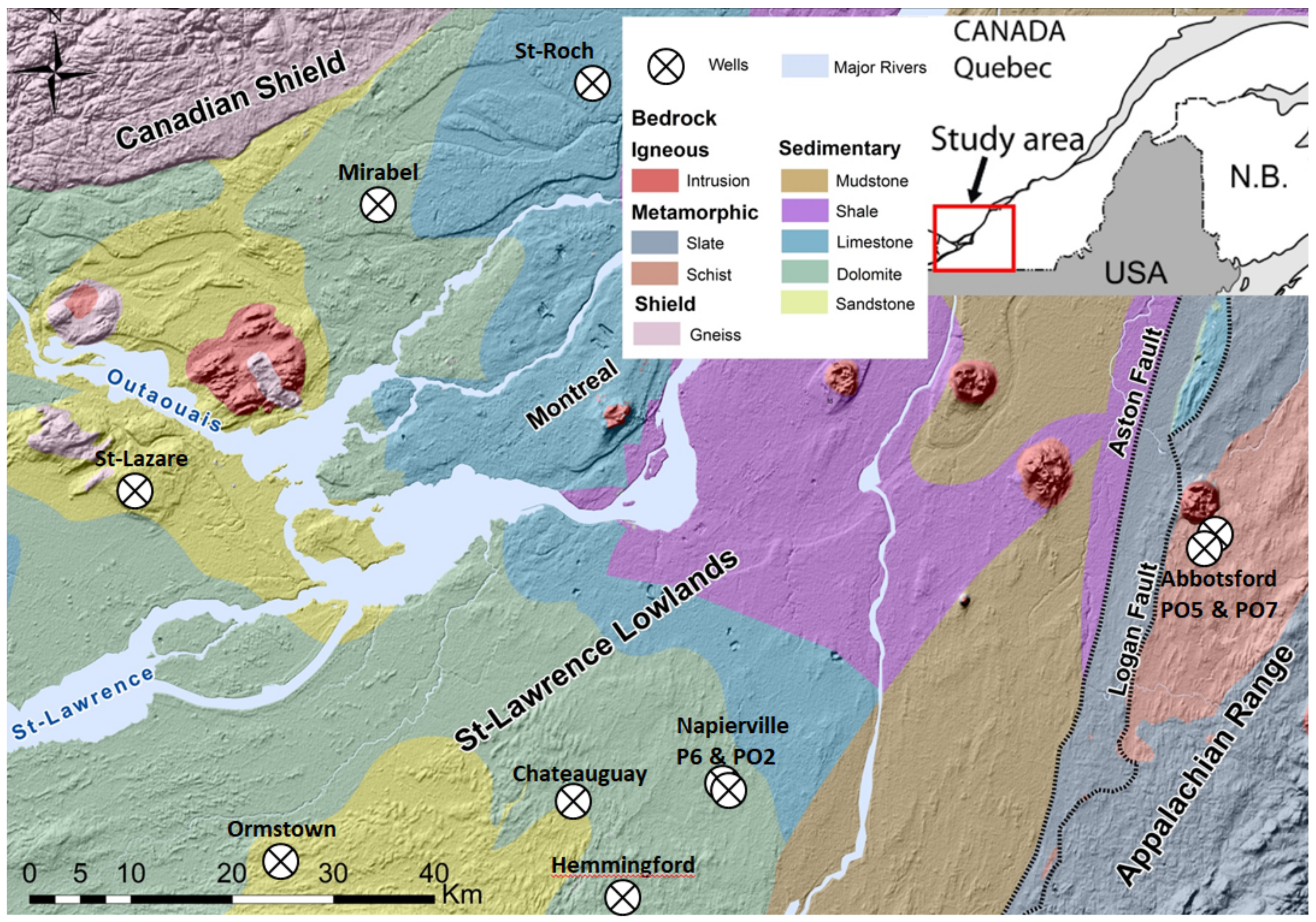
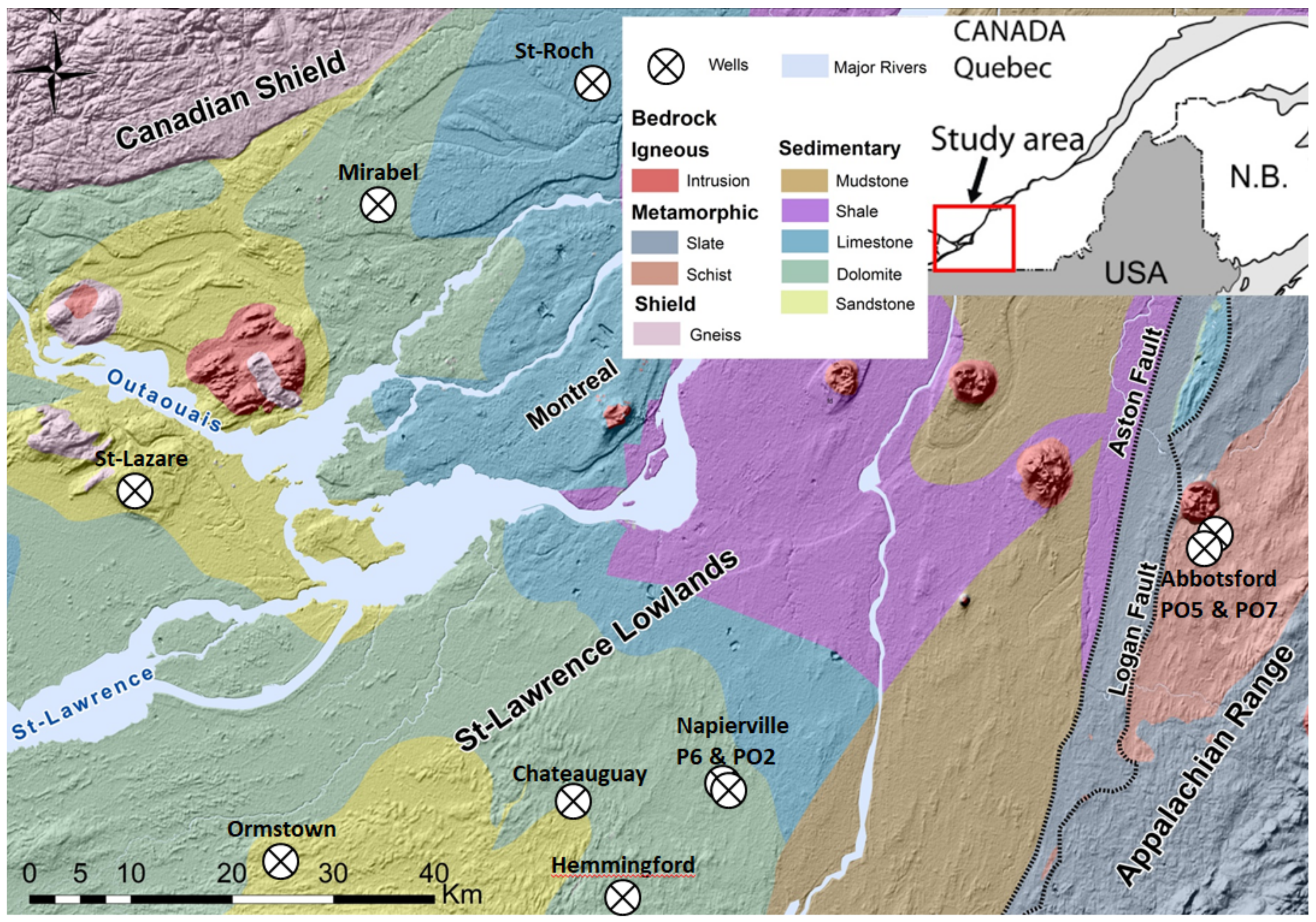
2.1. Geology of the Studied Area and Lithology of the Sampled Wellbores
2.2. Hydrogeochemical Processes of Interest within the Studied Area
3. Methods
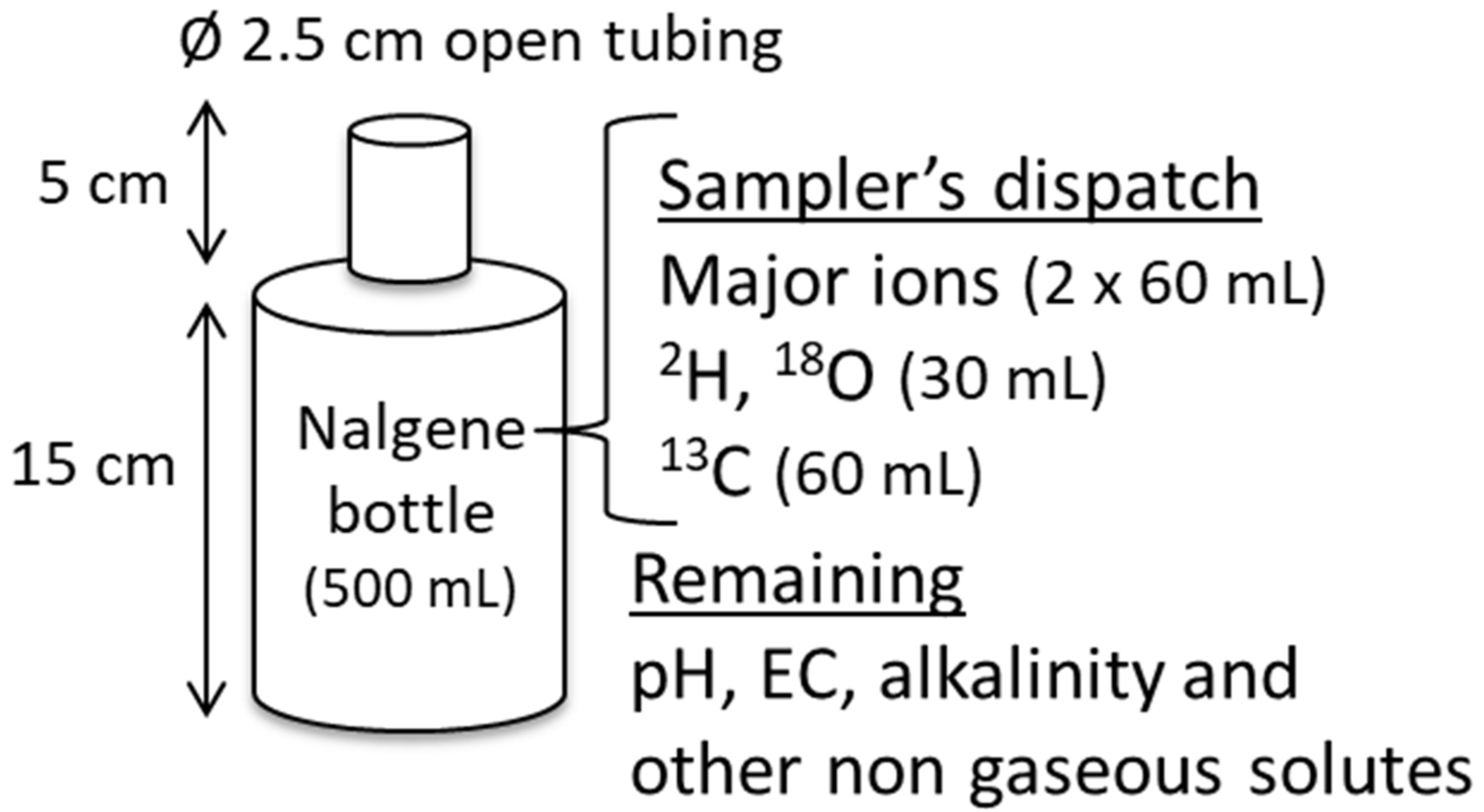
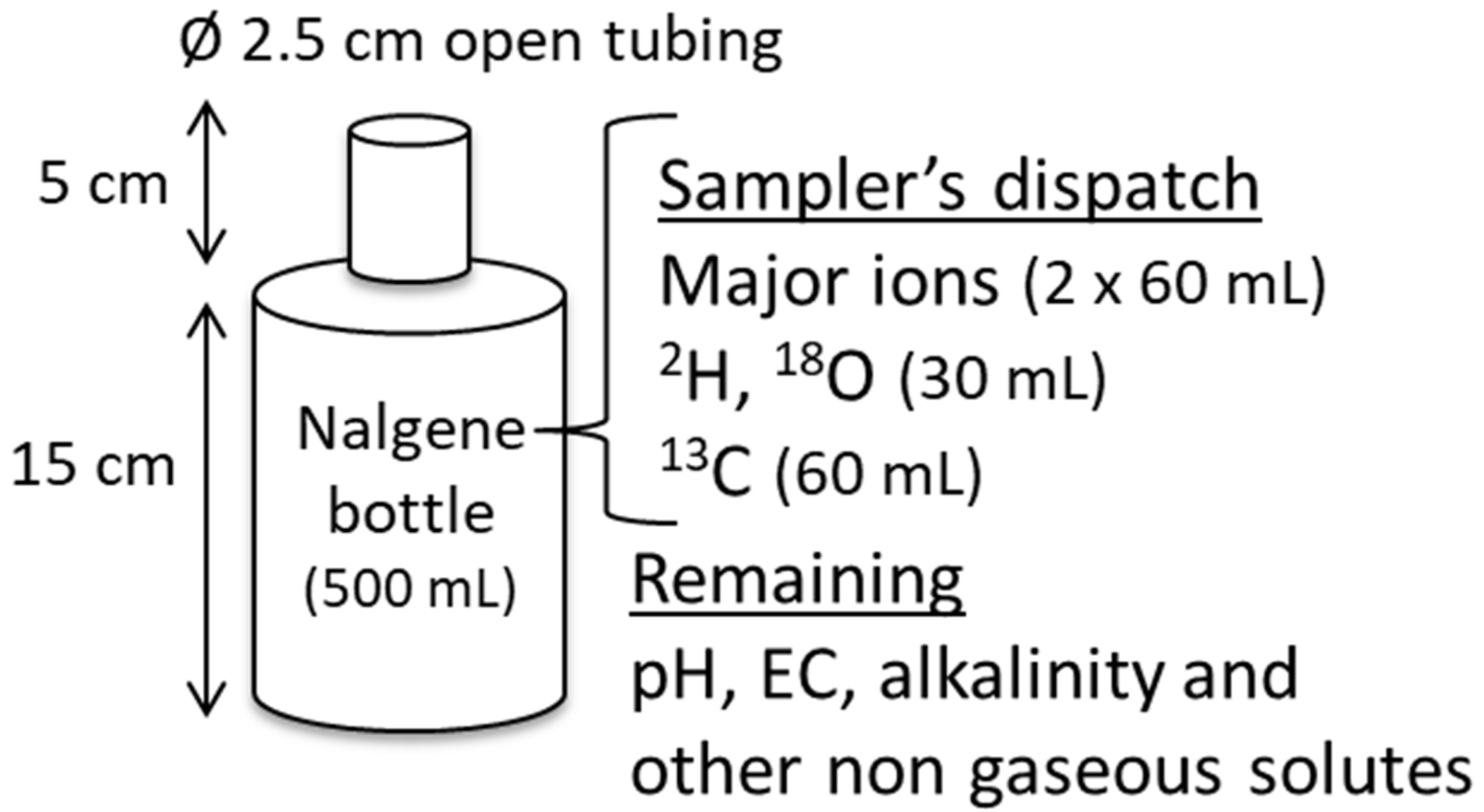
3.1. Passive Sampler Description and Design
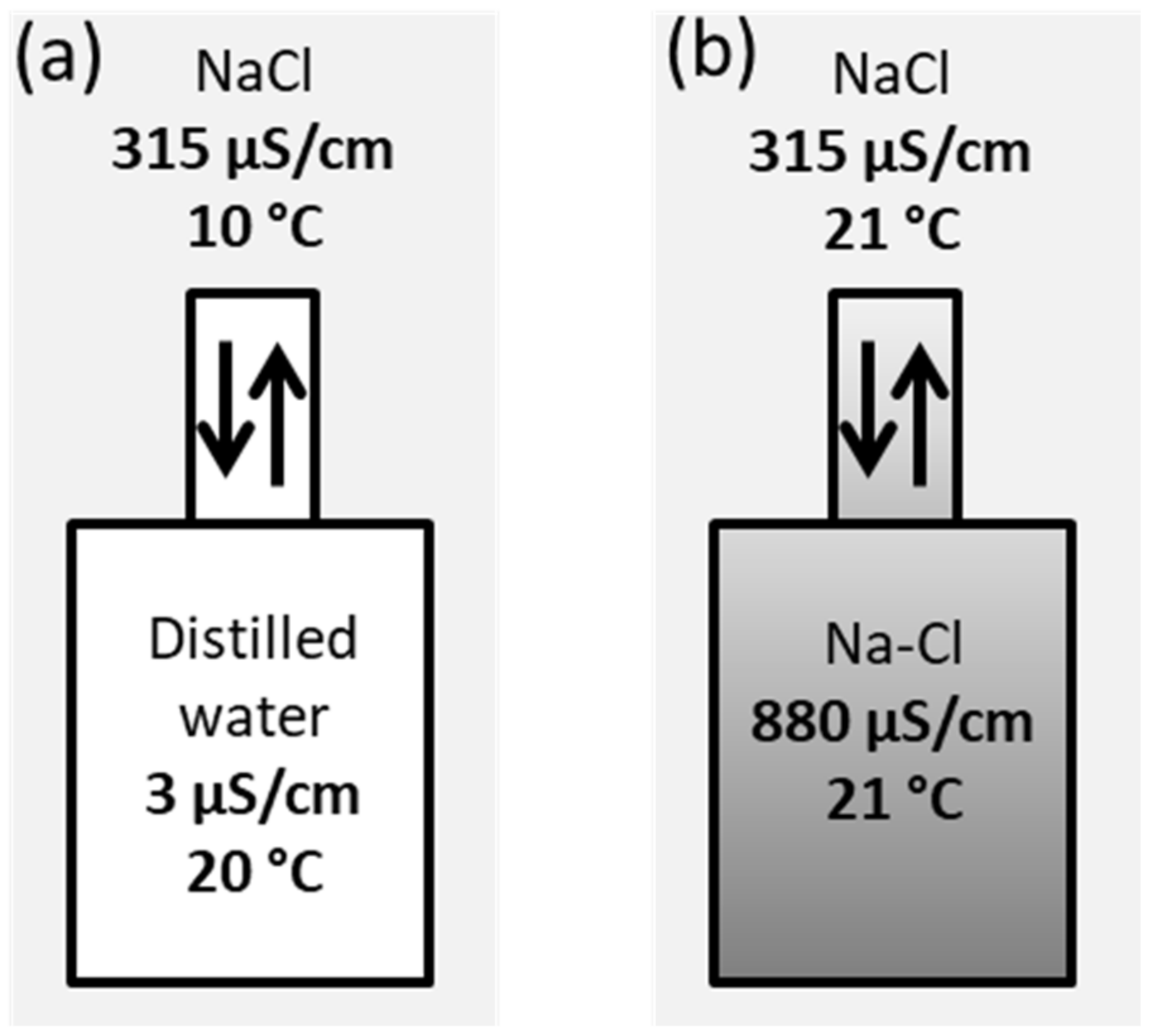
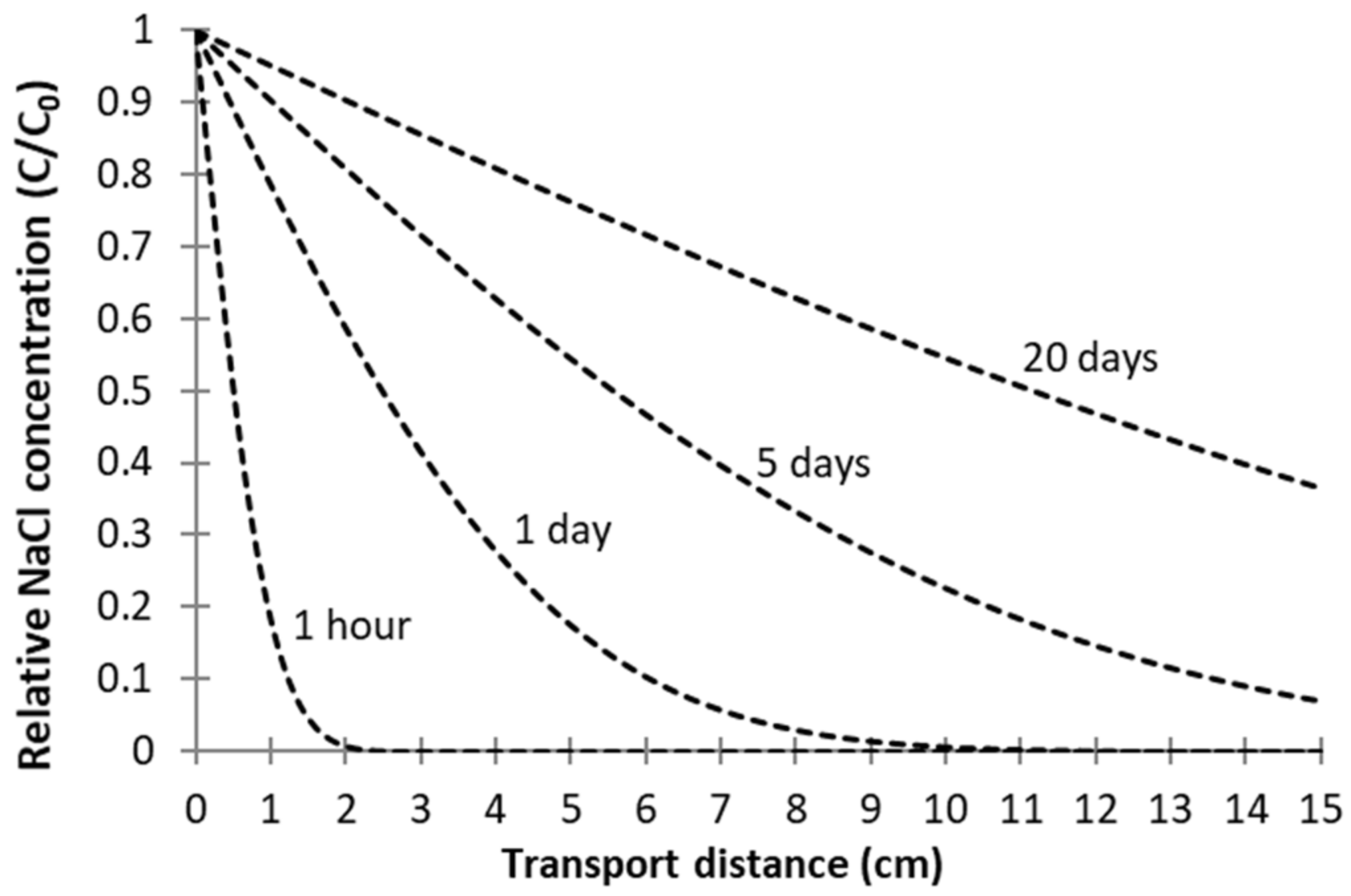
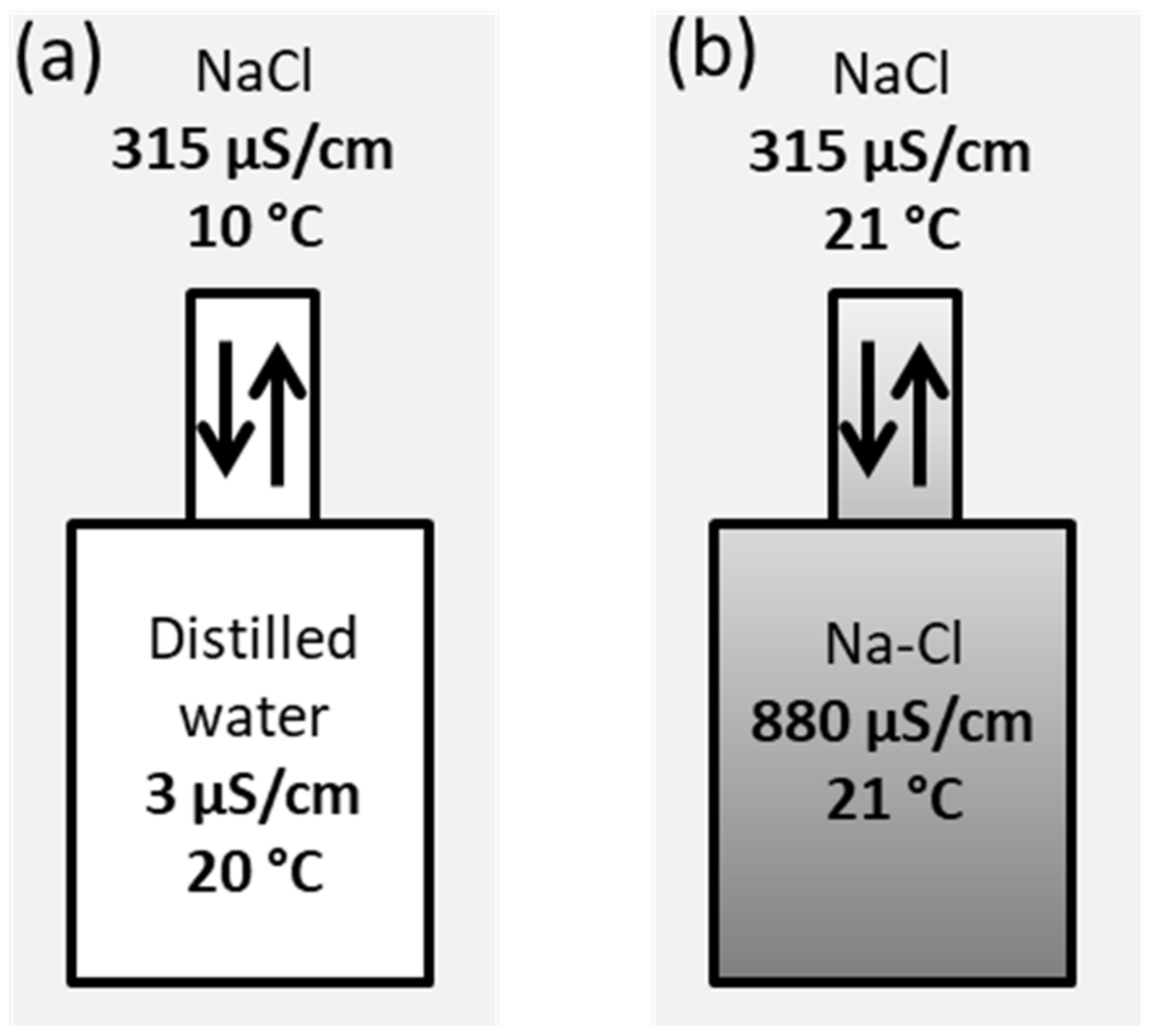
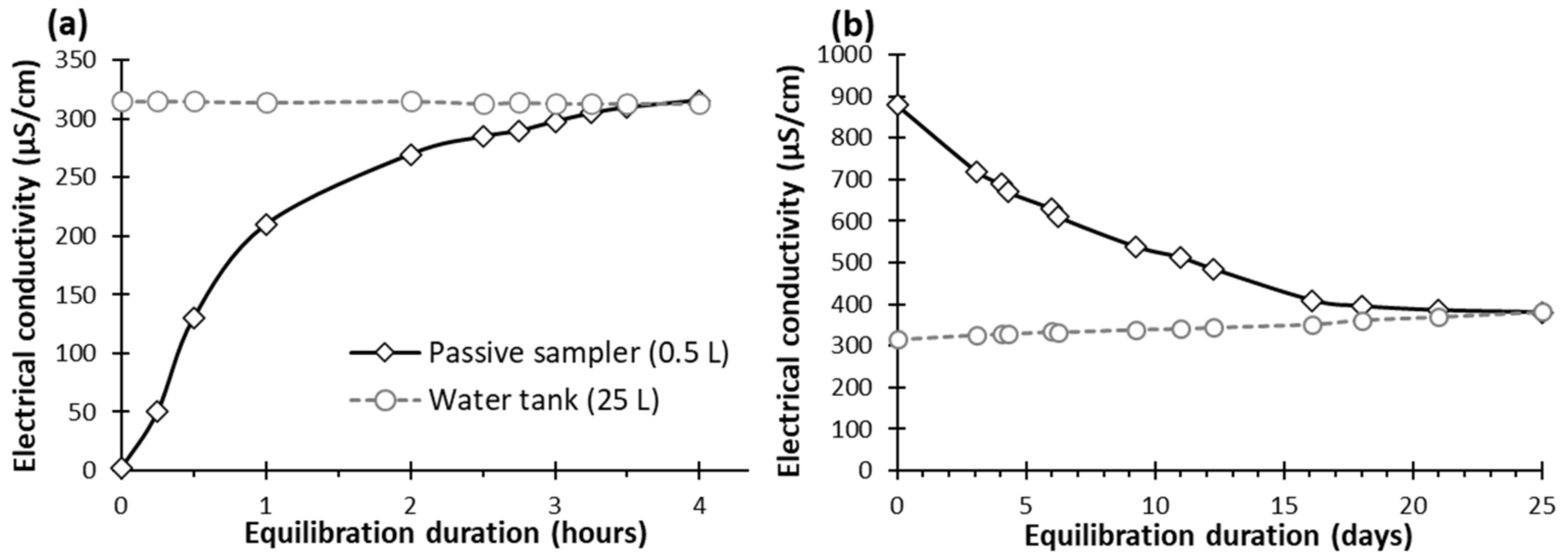
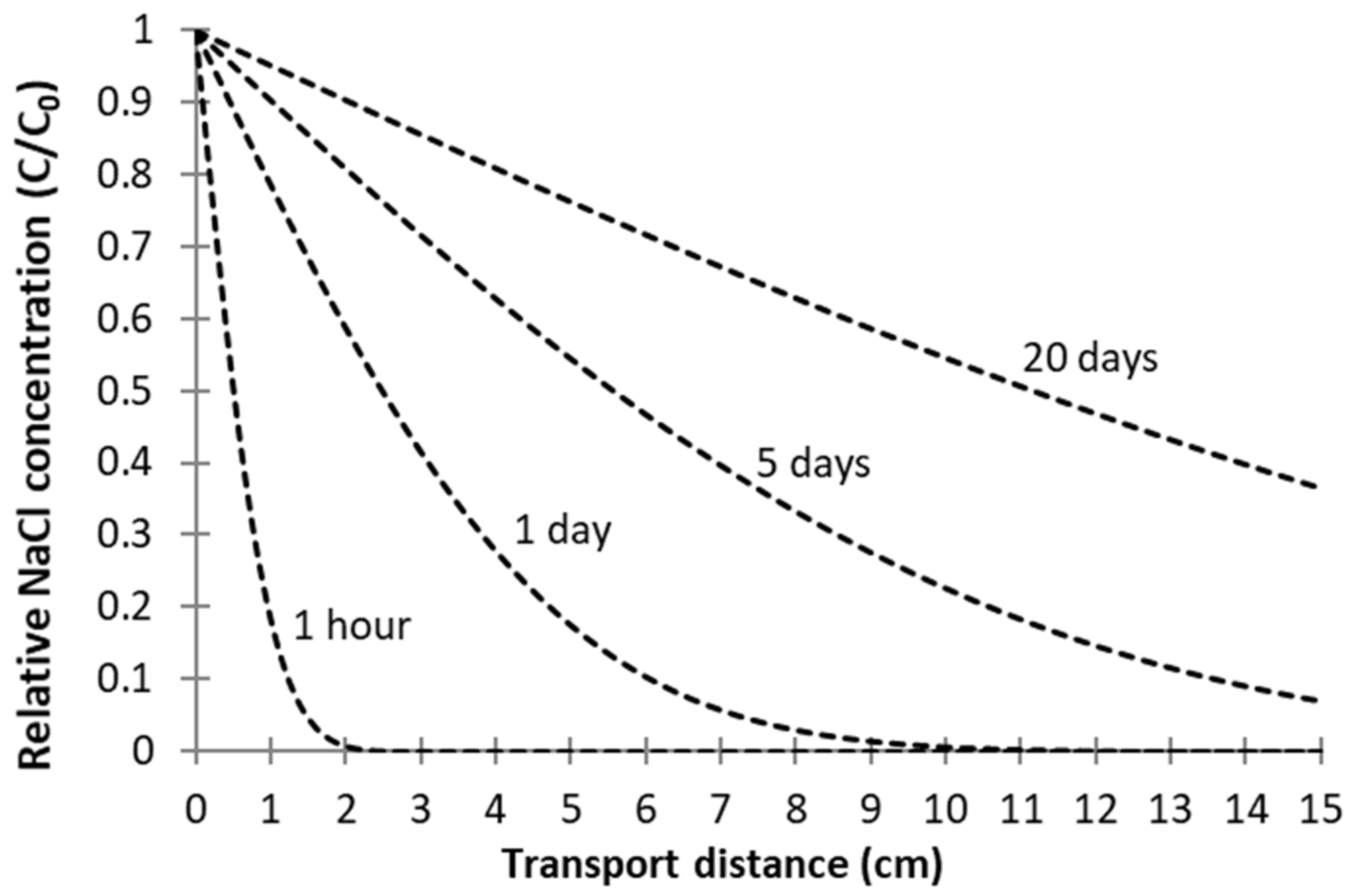
3.2. Passive Sampler Laboratory Testing
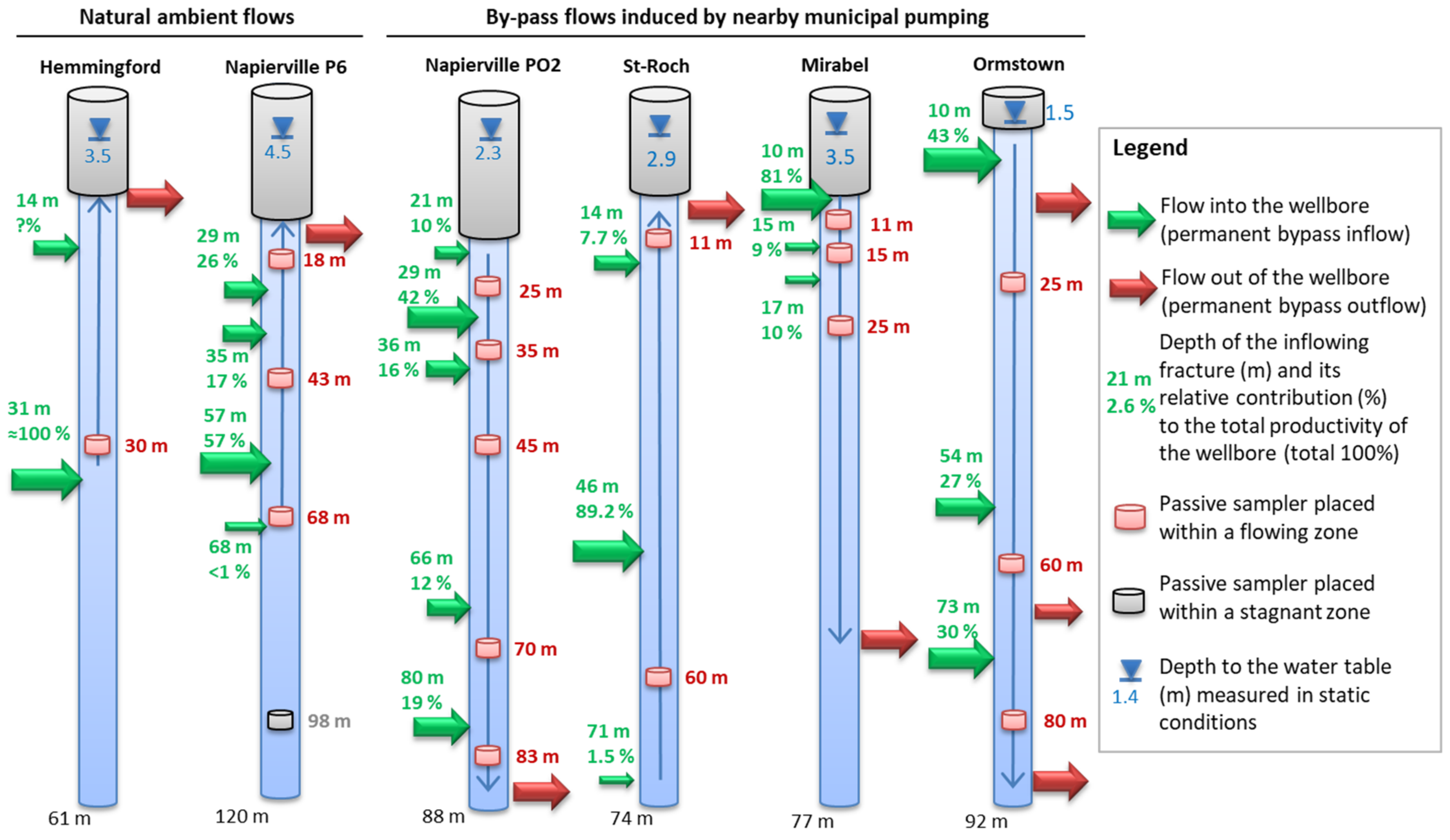
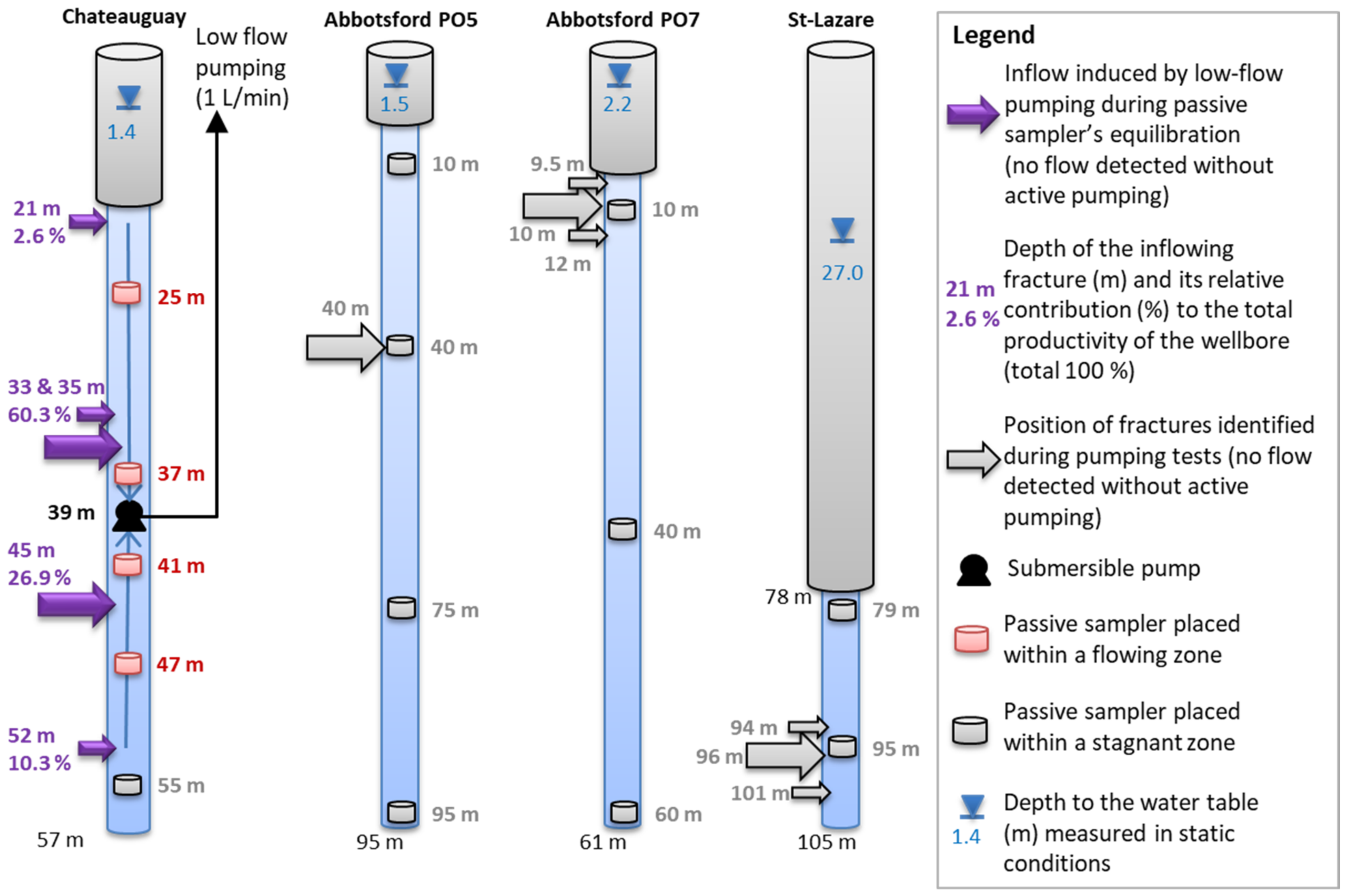
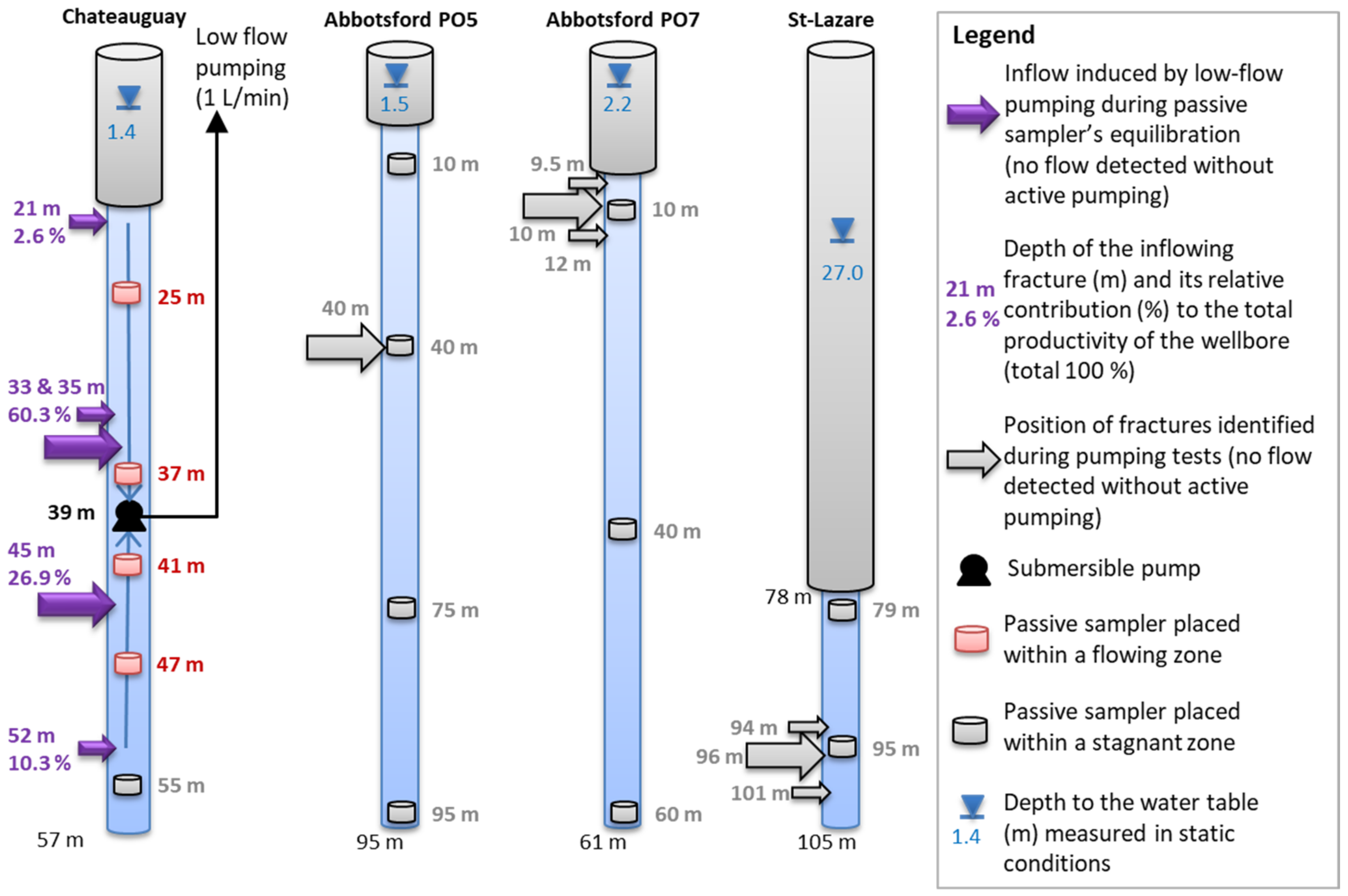
3.3. Passive Samplers’ Placement Determined from Previous Physical Borehole Surveys
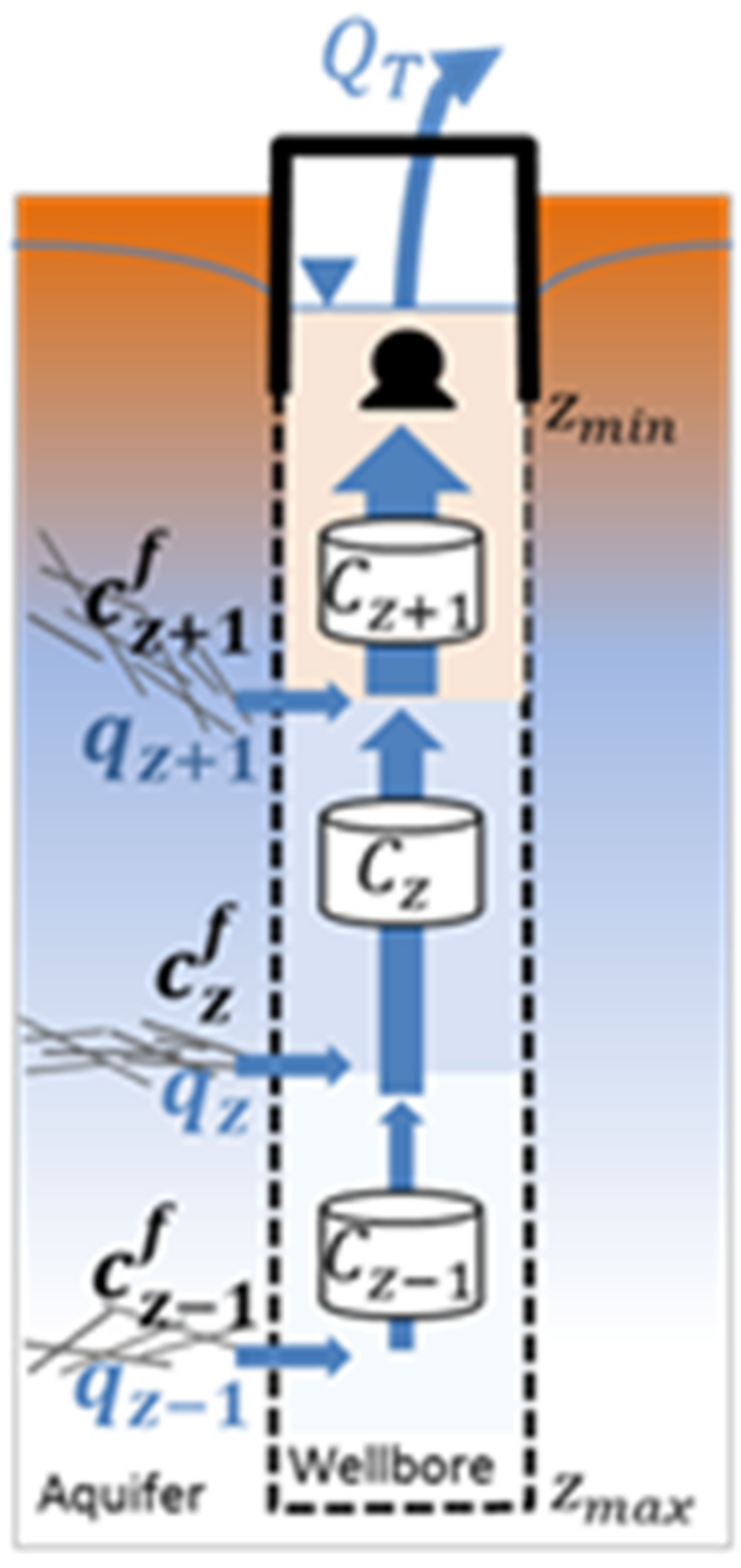
3.4. Low Flow Sampling at Depth
3.5. General Sampling and Analytical Procedures
4. Results
4.1. Passive Sampler Testing in Laboratory
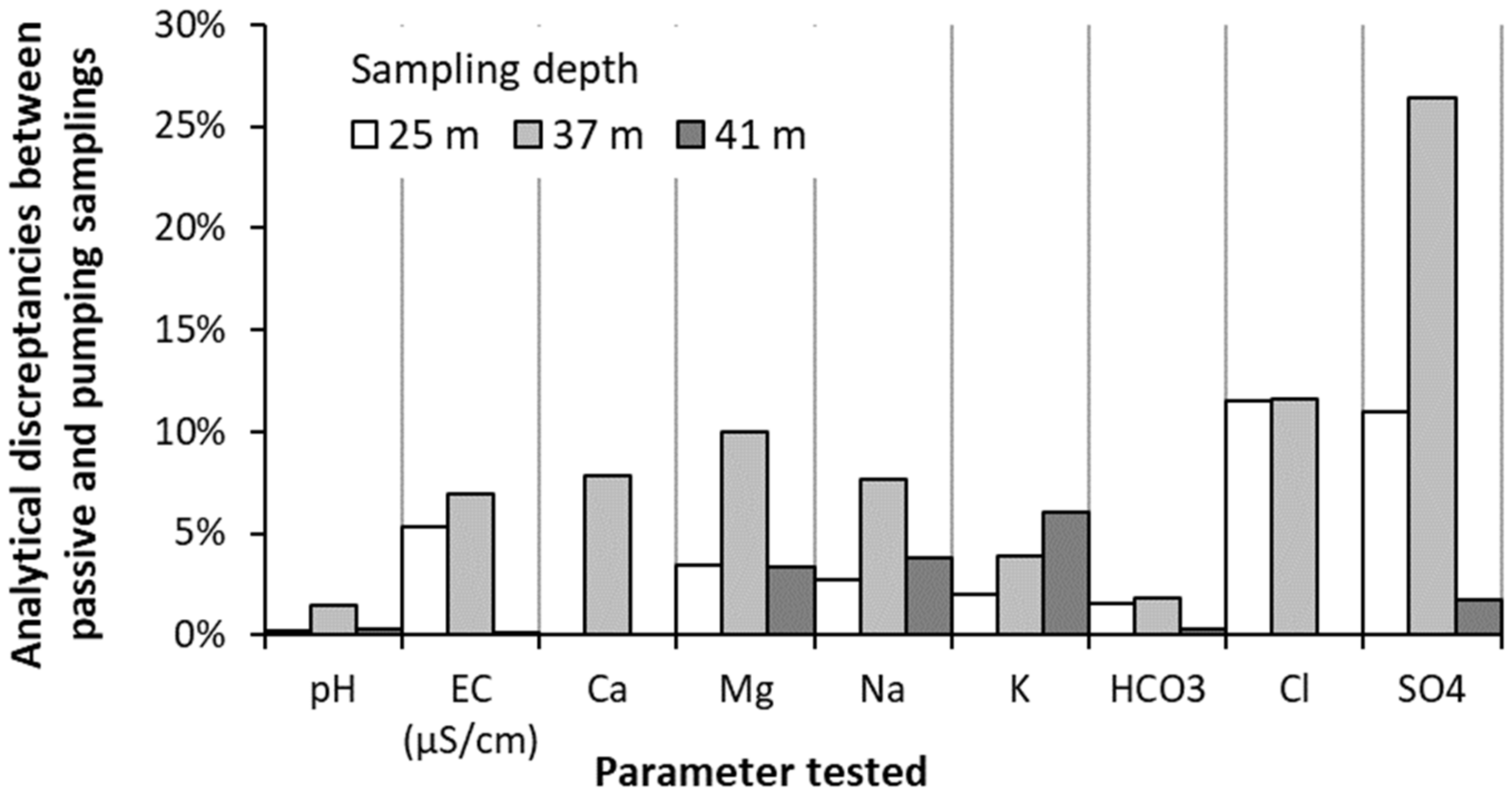
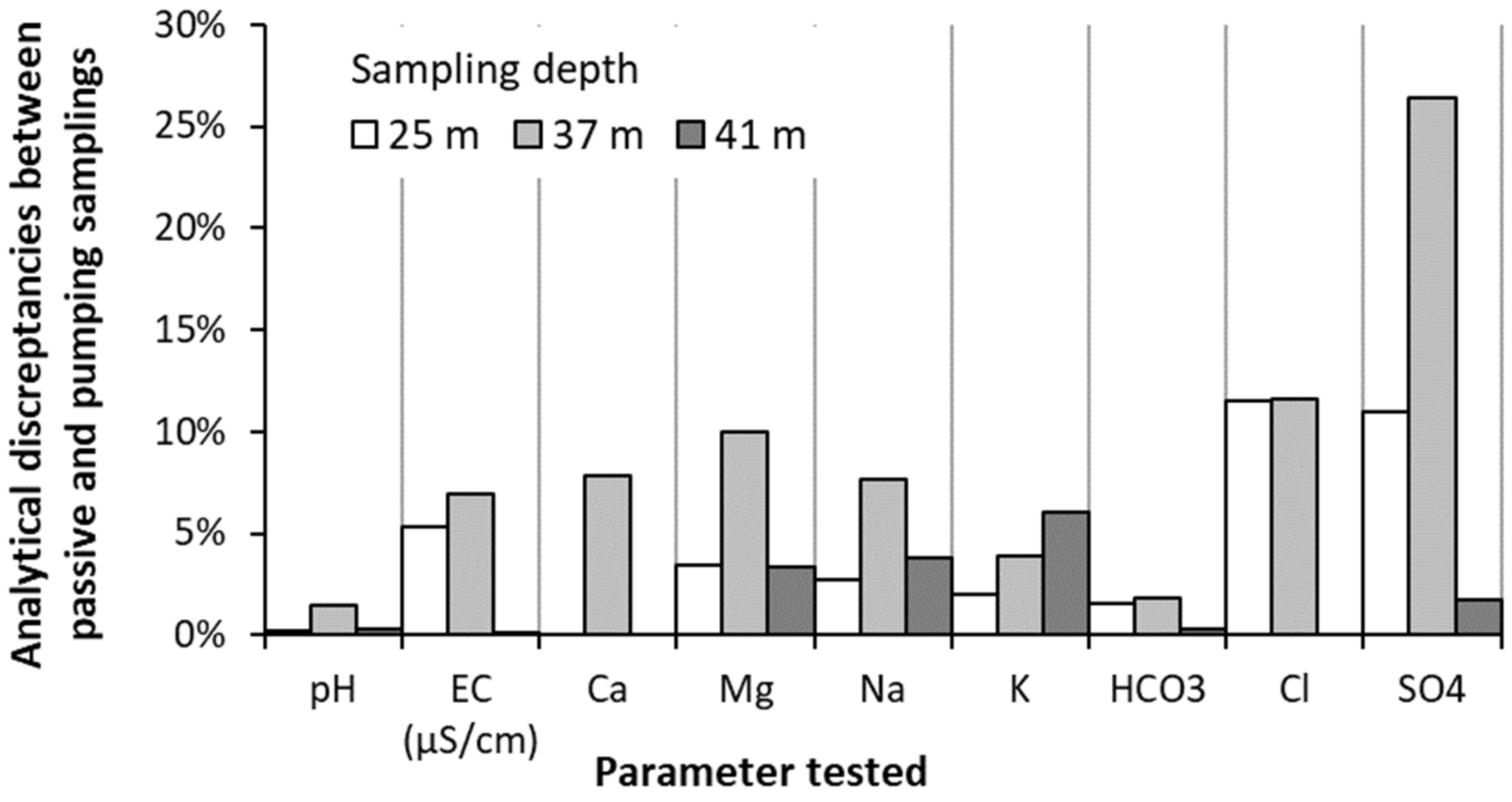
4.2. Validation of Passive Sampling at Depth and Deconvolution at Fractures
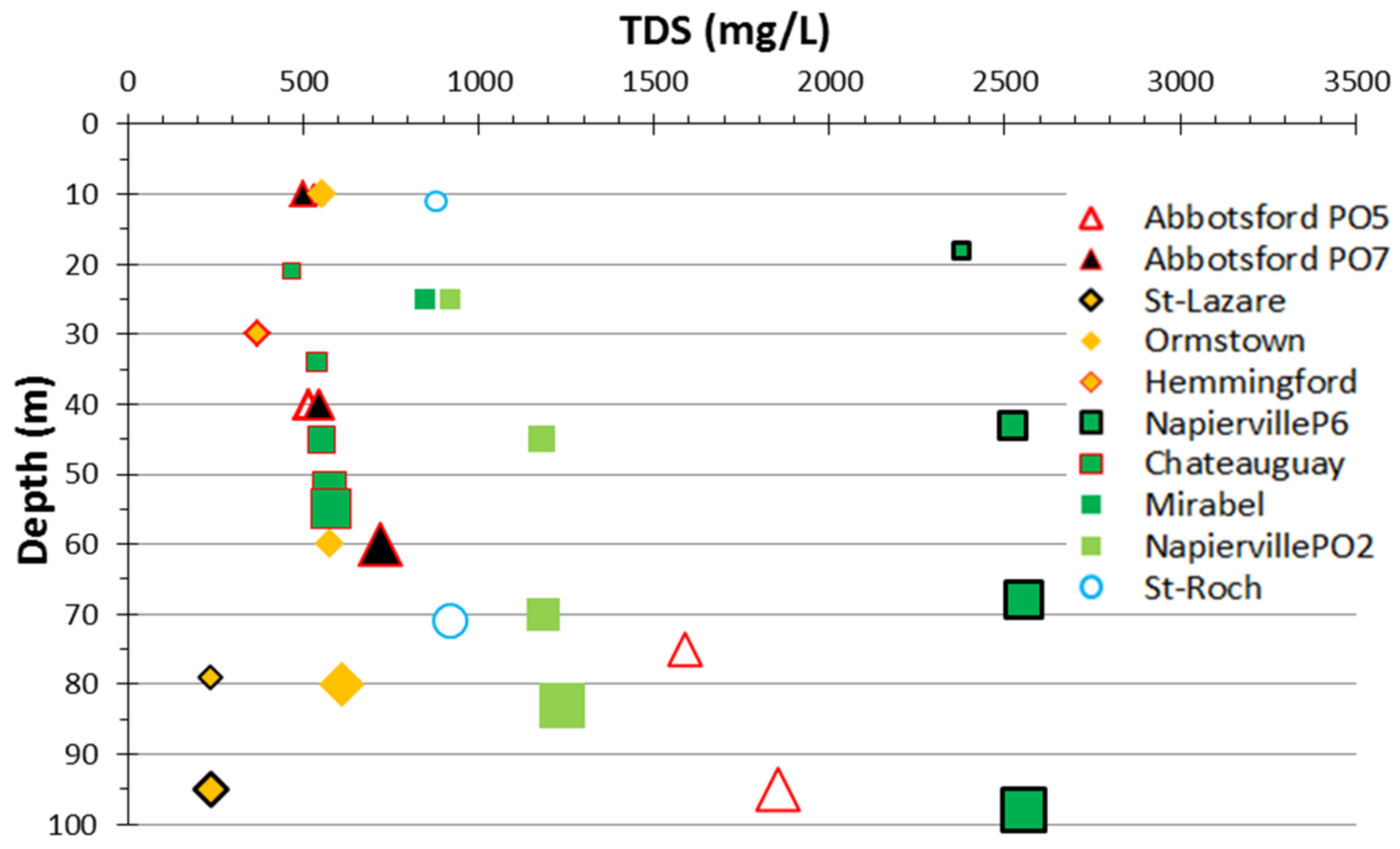
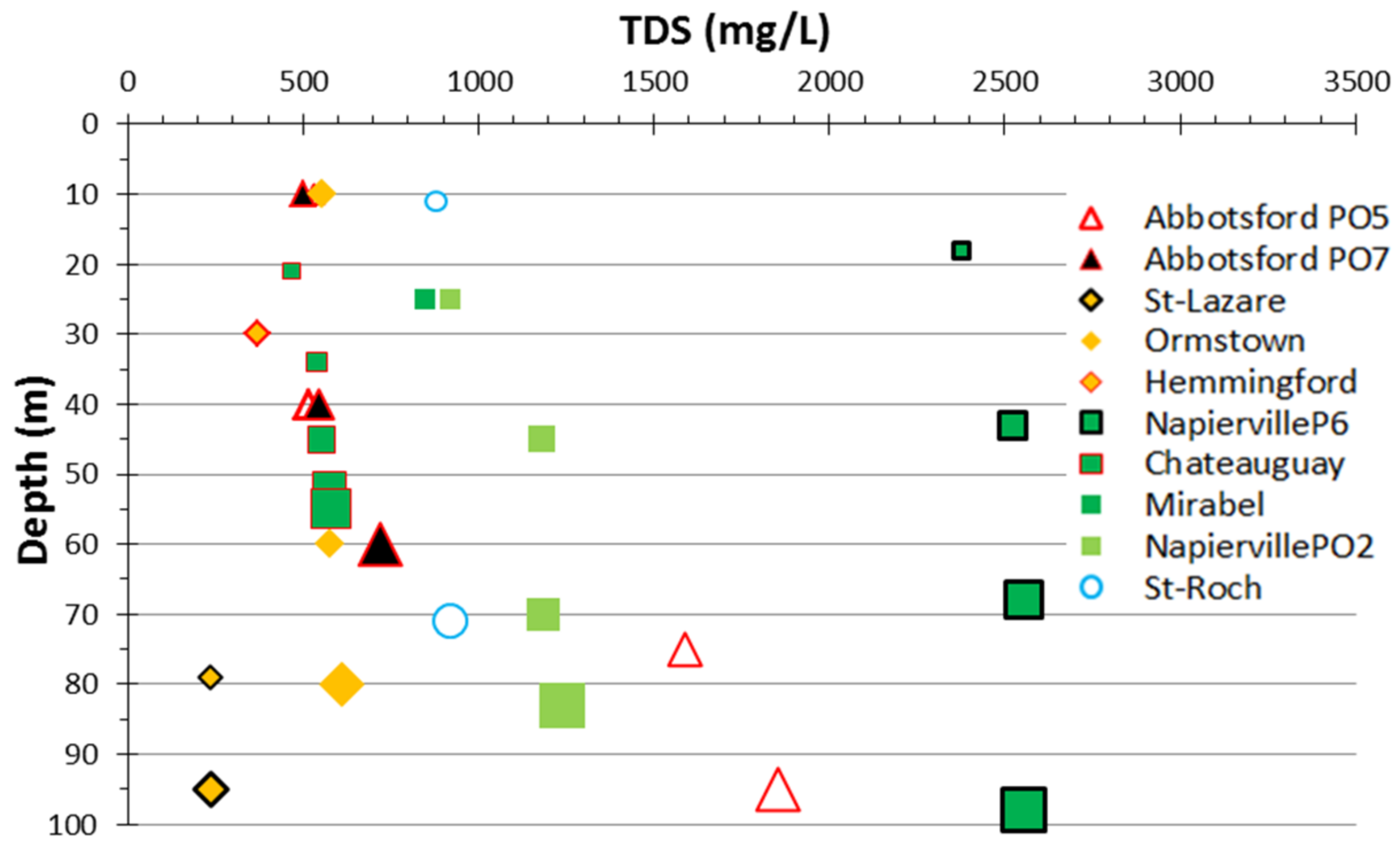
4.3. Hydrogeochemistry Gathered from Passive Sampling into the Wellbores
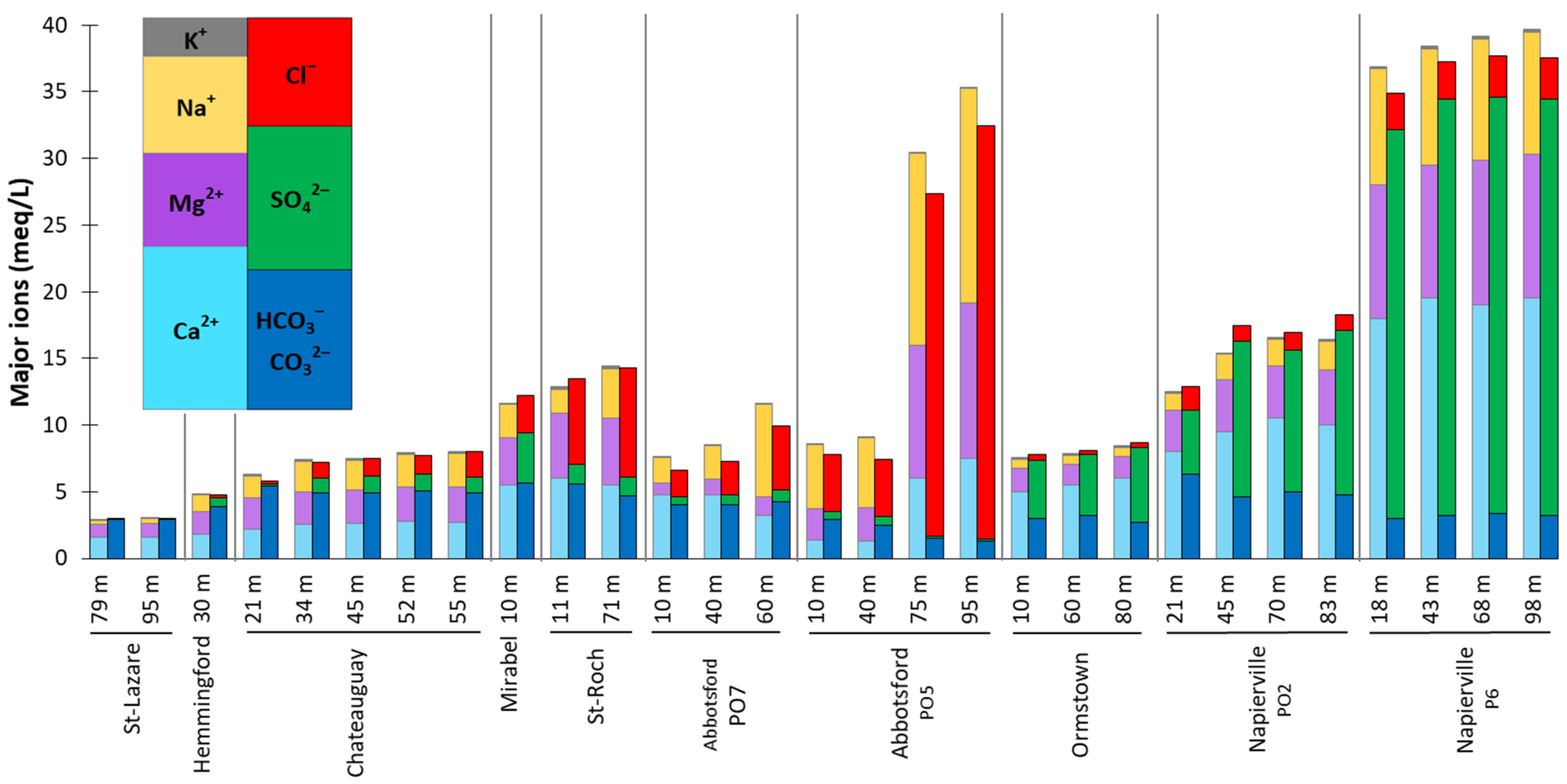
4.3.1. Major Ions Composition
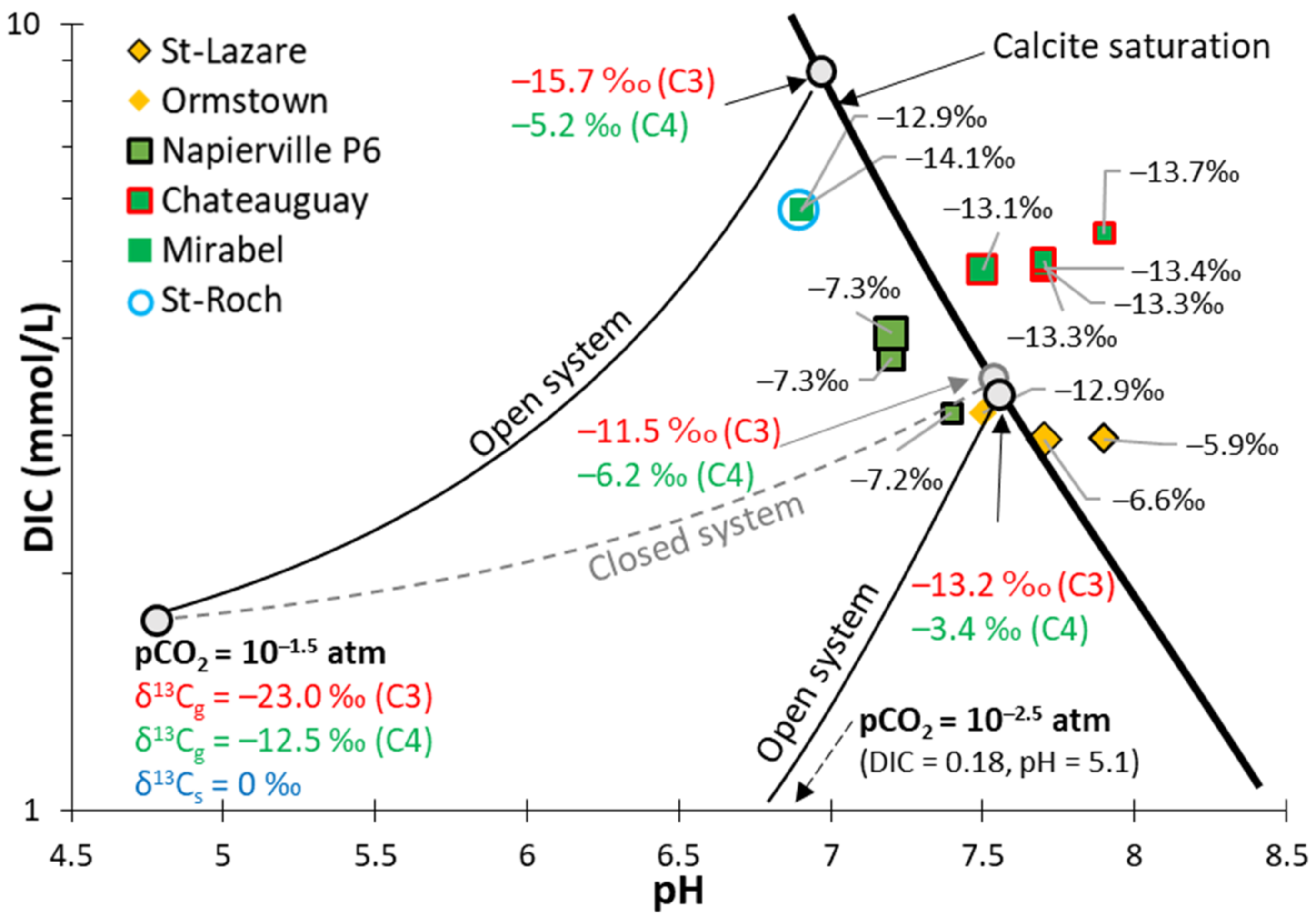
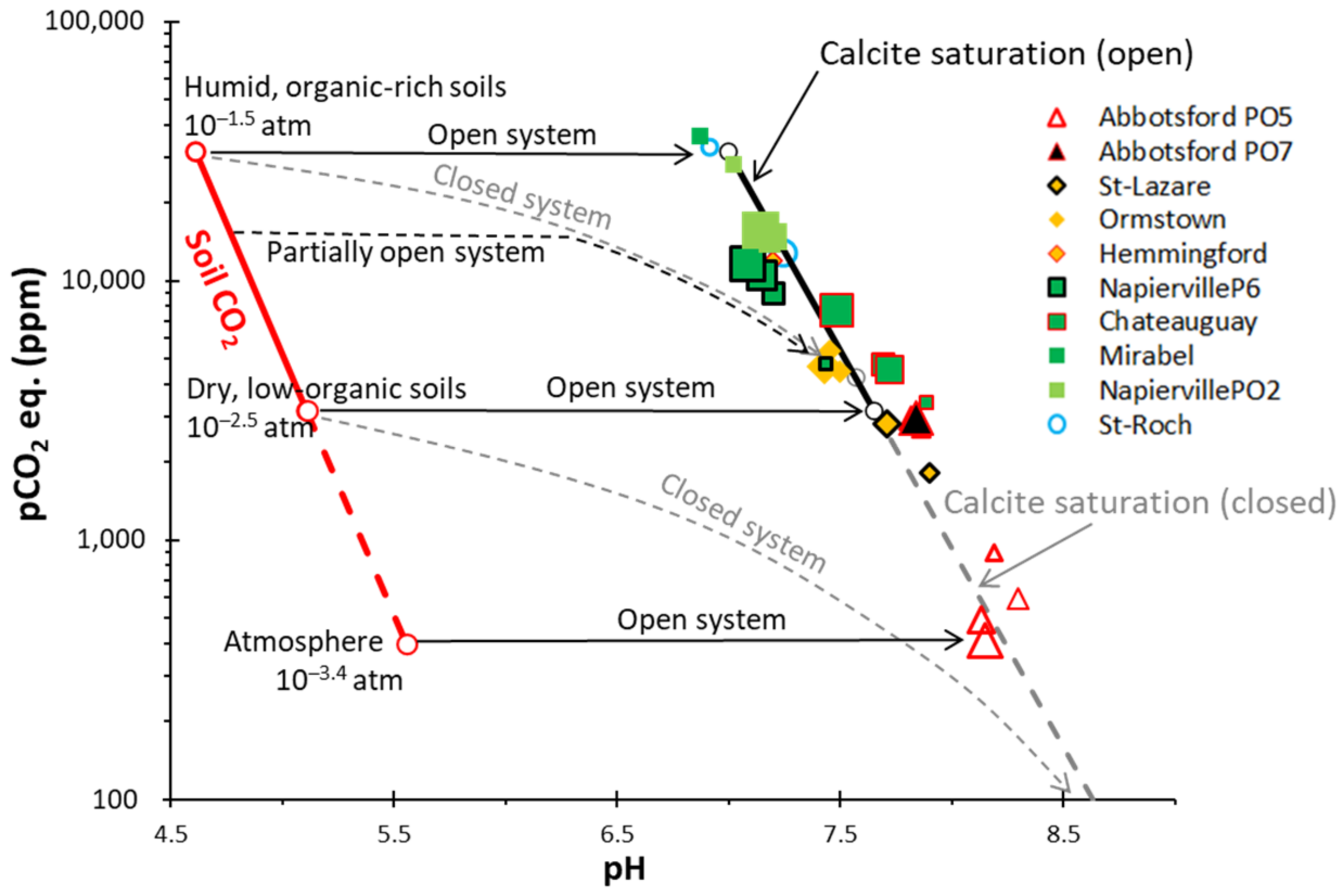
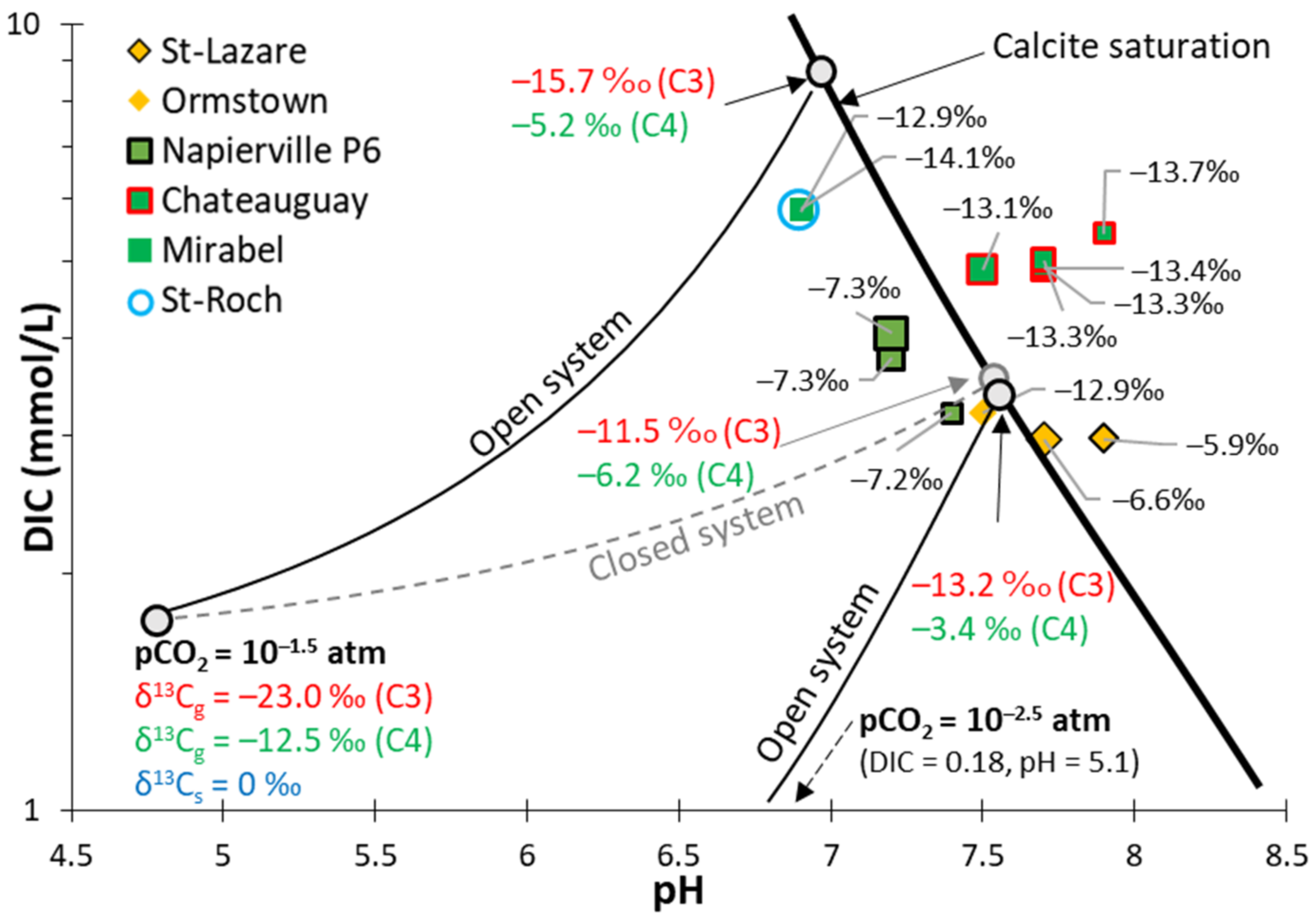
4.3.2. Calco–Carbonic System and δ13C
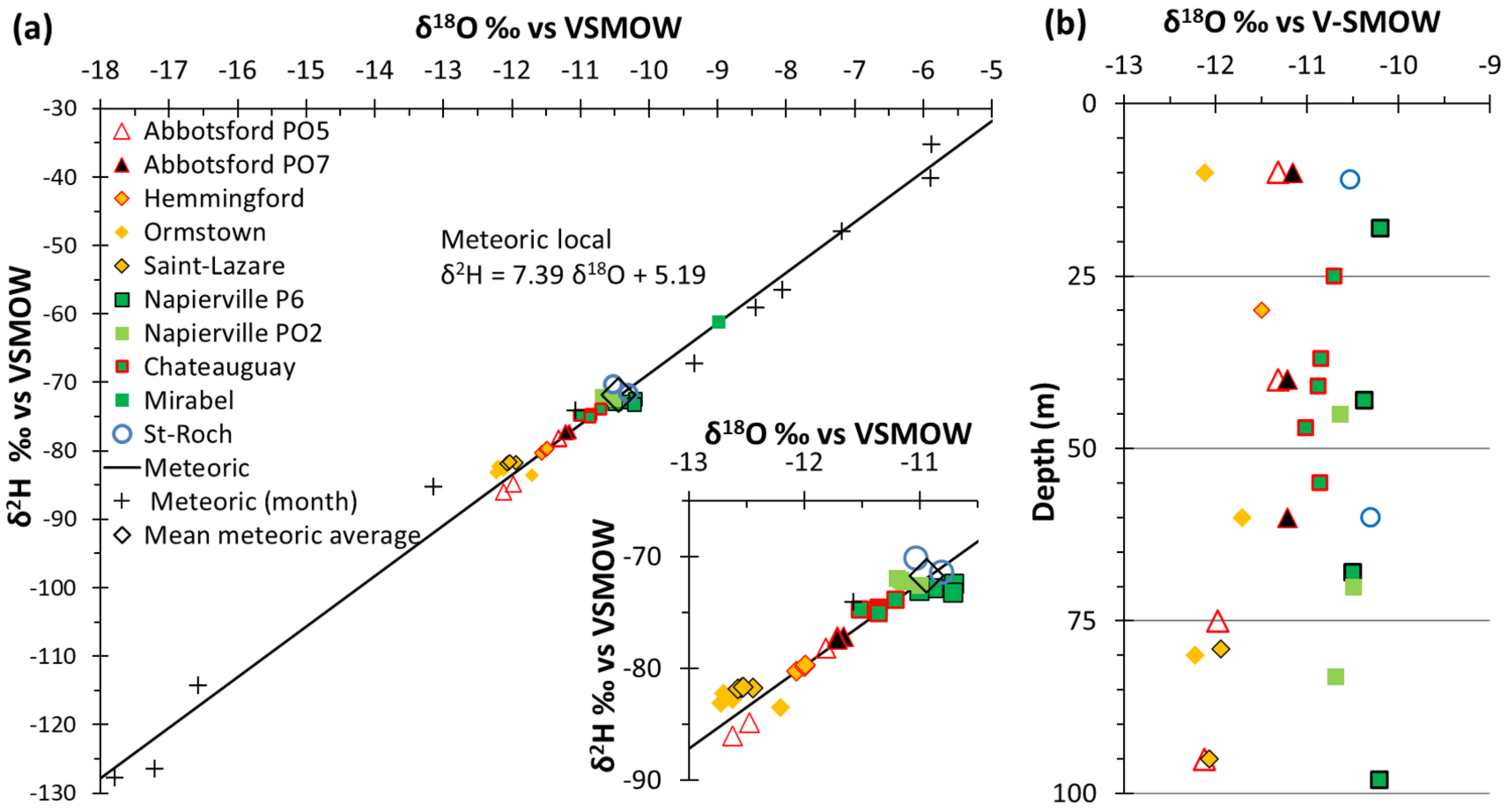
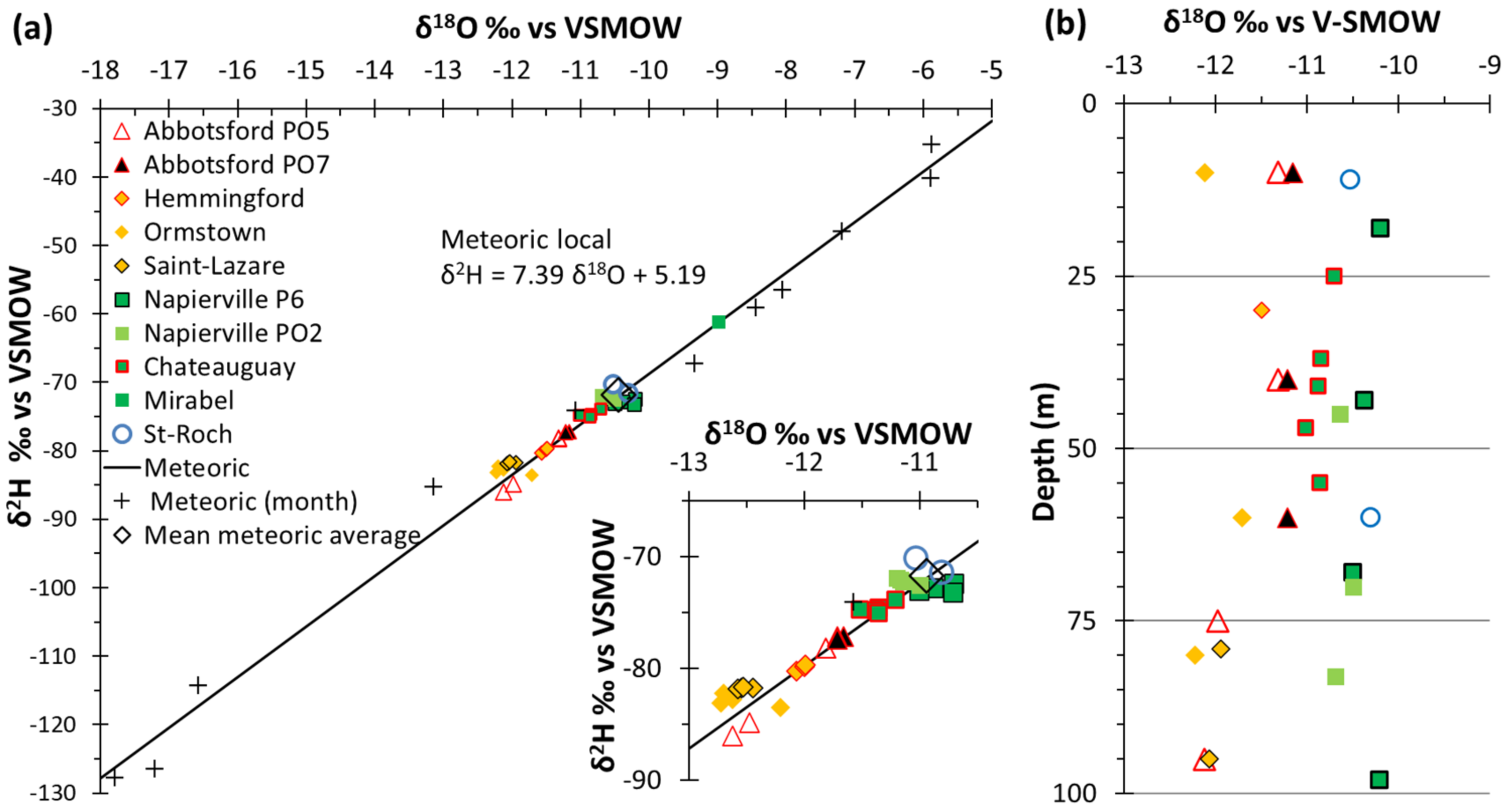
4.3.3. Stable Isotopes of Water
5. Discussions
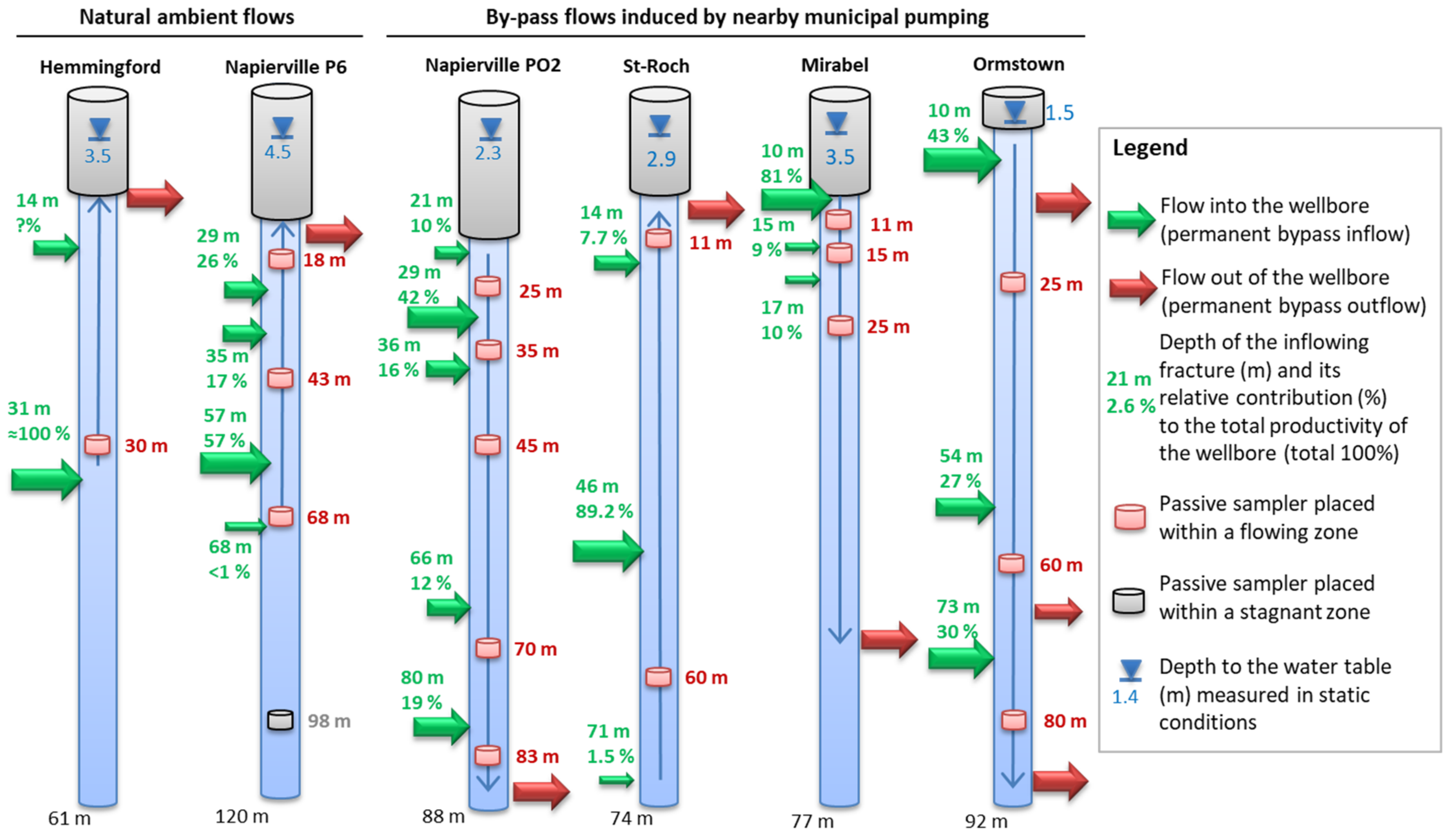
5.1. Representativeness of the Sequential Sampling with Depth in Fractured Aquifers
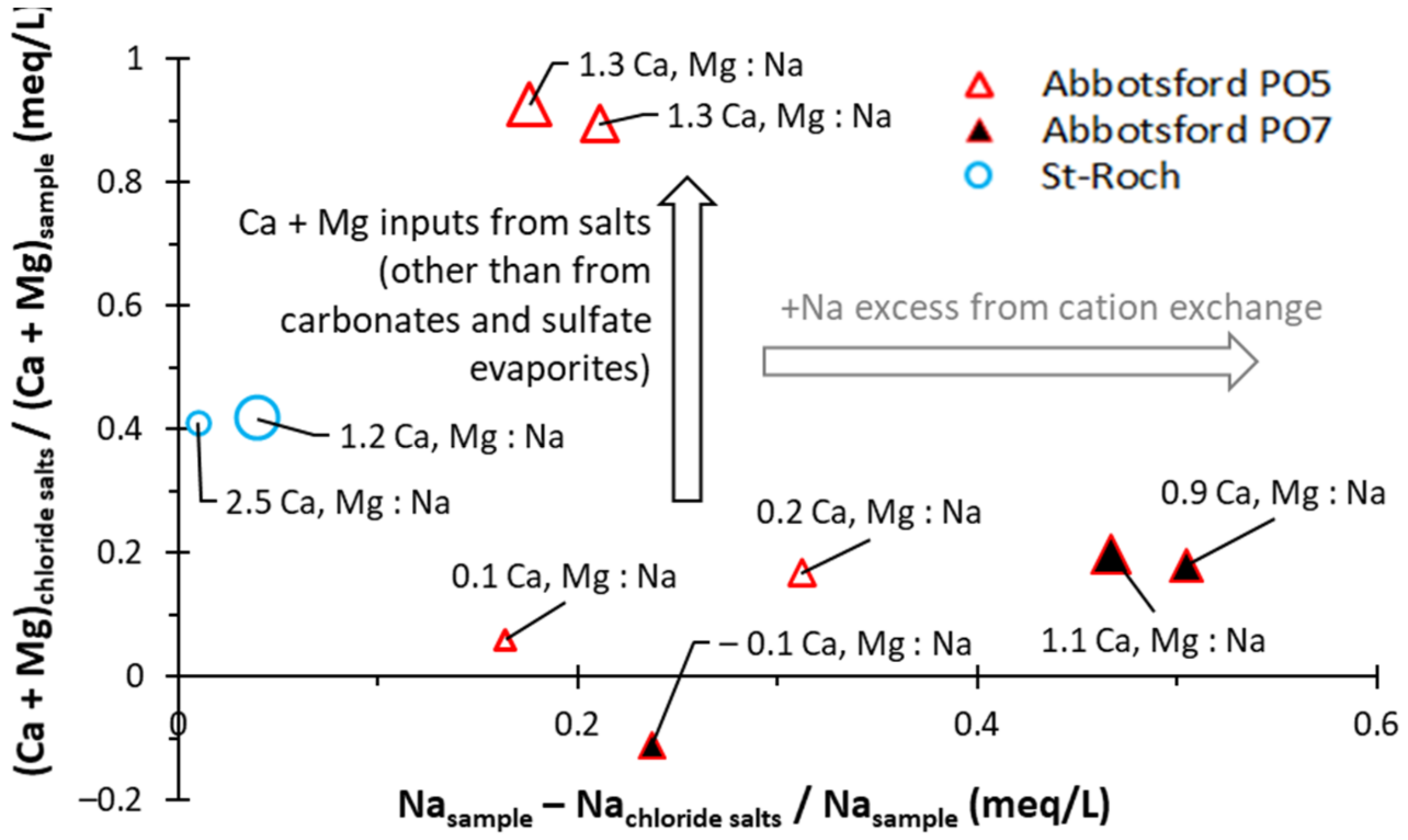
5.2. Sources of Groundwater Salinity and Respective Contributions
5.2.1. Calco–Carbonic Systems
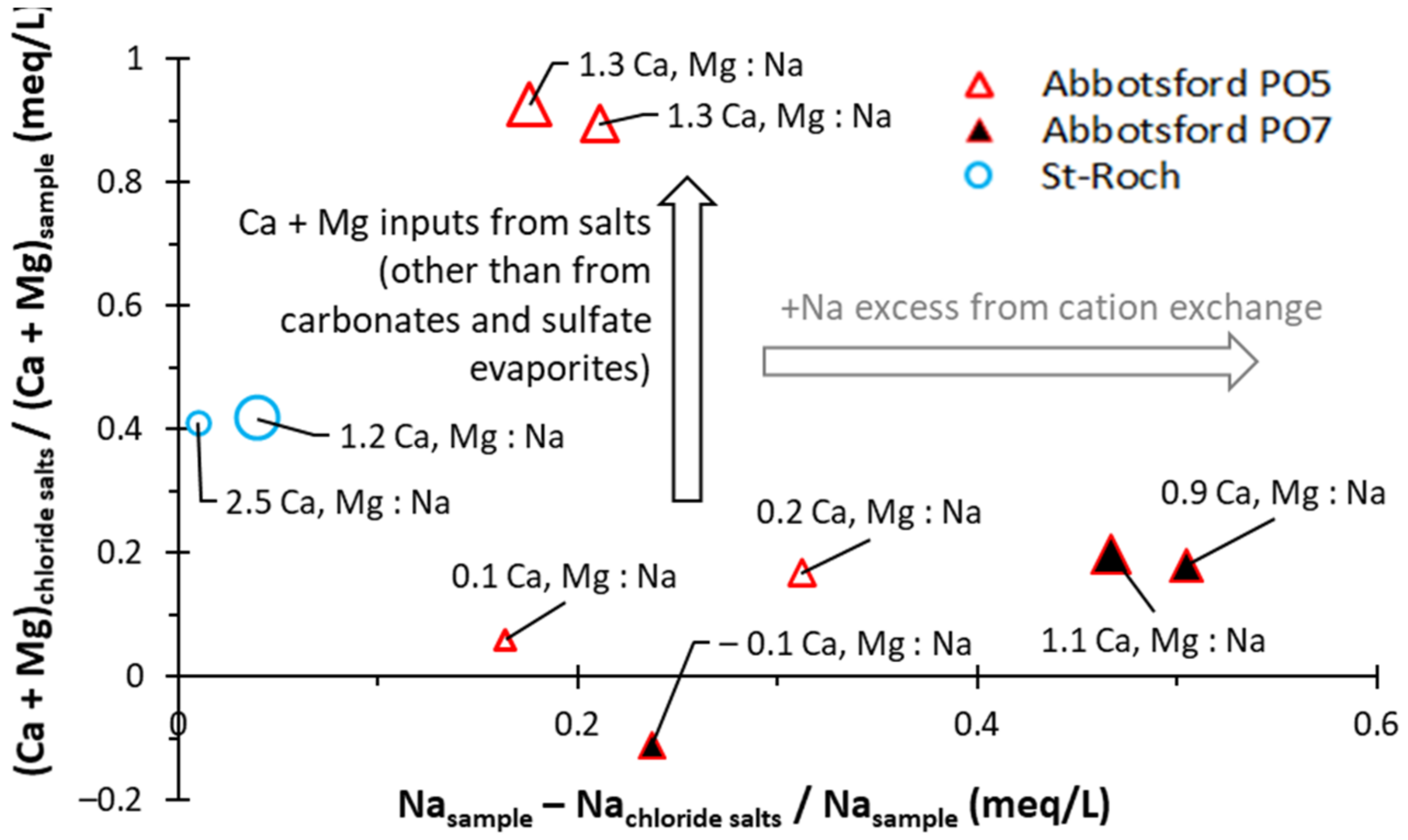
5.2.2. Mineralization from SO42− Inputs
5.2.3. Mineralization from Cl− Inputs
5.2.4. Other Mineralization Related to Na+ Inputs
5.3. Complementary Information Gathered from Isotopic Geochemistry
5.4. Flowing Systems Inferred from Hydrogeochemistry within the Fractured Aquifers
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cloutier, V.; Lefebvre, R.; Savard, M.M.; Bourque, É.; Therrien, R. Hydrogeochemistry and groundwater origin of the Bass-es–Laurentides sedimentary rock aquifer system, St. Lawrence Lowlands, Québec, Canada. Hydrogeol. J. 2006, 14, 573–590. [Google Scholar] [CrossRef]
- Mattei, A.; Barbecot, F.; Goblet, P.; Guillon, S. Pore water isotope fingerprints to understand the spatiotemporal groundwater recharge variability in ungauged watersheds. Vadose Zone J. 2020, 19, 20066. [Google Scholar] [CrossRef]
- Edmunds, W.M.; Cook, J.M.; Darling, W.G.; Kinniburgh, D.G.; Miles, D.L.; Bath, A.H.; Morgan–Jones, M.; Andrews, J.N. Baseline geochemical conditions in the Chalk aquifer, Berkshire, U.K.: A basis for groundwater quality management. Appl. Geochem. 1987, 2, 251–274. [Google Scholar] [CrossRef]
- Mendizabal, I.; Stuyfzand, P.J.; Wiersma, A.P. Hydrochemical system analysis of public supply well fields, to reveal water–quality patterns and define groundwater bodies: The Netherlands. Hydrogeol. J. 2011, 19, 83–100. [Google Scholar] [CrossRef] [Green Version]
- Larocque, M.; Cloutier, V.; Levison, J.; Rosa, E. Results from the Quebec Groundwater Knowledge Acquisition Program. Can. Water Resour. J. Rev. Can. Ressour. Hydr. 2017, 43, 69–74. [Google Scholar] [CrossRef]
- Blanchette, D.; Lefebvre, R.; Nastev, M.; Cloutier, V. Groundwater Quality, Geochemical Processes and Groundwater Evolution in the Chateauguay River Watershed, Quebec, Canada. Can. Water Resour. J. Rev. Can. Res. Hydr. 2010, 35, 503–526. [Google Scholar] [CrossRef] [Green Version]
- Benoit, N.; Nastev, M.; Blanchette, D.; Molson, J. Hydrogeology and hydrogeochemistry of the Chaudière River watershed aquifers, Québec, Canada. Can. Water Resour. J. Rev. Can. Res. Hydr. 2014, 39, 32–48. [Google Scholar] [CrossRef]
- Montcoudiol, N.; Molson, J.; Lemieux, J.M. Groundwater geochemistry of the Outaouais Region (Québec, Canada): A regional–scale study. Hydrogeol. J. 2014, 23, 377–396. [Google Scholar] [CrossRef]
- Boucher, C.; Pinti, D.L.; Roy, M.; Castro, M.C.; Cloutier, V.; Blanchette, D.; Larocque, M.; Hall, C.M.; Wen, T.; Sano, Y. Groundwater age investigation of eskers in the Amos region, Quebec, Canada. J. Hydrol. 2015, 524, 1–14. [Google Scholar] [CrossRef]
- Ghesquière, O.; Walter, J.; Chesnaux, R.; Rouleau, A. Scenarios of groundwater chemical evolution in a region of the Canadian Shield based on multivariate statistical analysis. J. Hydrol. Reg. Stud. 2015, 4, 246–266. [Google Scholar] [CrossRef] [Green Version]
- Vautour, G.; Pinti, D.L.; Méjean, P.; Saby, M.; Meyzonnat, G.; Larocque, M.; Castro, M.C.; Hall, C.M.; Boucher, C.; Roulleau, E.; et al. 3H/3He, 14C and (U–Th)/He groundwater ages in the St. Lawrence Lowlands, Quebec, Eastern Canada. Chem. Geol. 2015, 413, 94–106. [Google Scholar] [CrossRef]
- Saby, M.; Larocque, M.; Pinti, D.L.; Barbecot, F.; Sano, Y.; Castro, M.C. Linking groundwater quality to residence times and regional geology in the St. Lawrence Lowlands, southern Quebec, Canada. Appl. Geochem. 2016, 65, 1–13. [Google Scholar] [CrossRef]
- Walter, J.; Chesnaux, R.; Cloutier, V.; Gaboury, D. The influence of water/rock—Water/clay interactions and mixing in the salinization processes of groundwater. J. Hydrol. Reg. Stud. 2017, 13, 168–188. [Google Scholar] [CrossRef]
- Beaudry, C.; Lefebvre, R.; Rivard, C.; Cloutier, V. Conceptual model of regional groundwater flow based on hydrogeochemistry (Montérégie Est, Québec, Canada). Can. Water Res. J. 2018, 43, 152–172. [Google Scholar] [CrossRef]
- Chaillou, G.; Touchette, M.; Buffin-Bélanger, T.; Cloutier, C.-A.; Hétu, B.; Roy, M.-A. Hydrogeochemical evolution and groundwater mineralization of shallow aquifers in the Bas–Saint–Laurent region, Québec, Canada. Can. Water Res. J. 2018, 43, 136–151. [Google Scholar] [CrossRef]
- Bondu, R.; Cloutier, V.; Rosa, E.; Roy, M. An exploratory data analysis approach for assessing the sources and distribution of naturally occurring contaminants (F, Ba, Mn, As) in groundwater from southern Quebec (Canada). Appl. Geochem. 2020, 114, 104500. [Google Scholar] [CrossRef]
- Vogel, J.C. Investigation of Groundwater Flow with Radiocarbon Isotopes in Hydrology; International Atomic Energy Agency: Vienna, Austria, 1967; pp. 355–369. [Google Scholar]
- Corcho Alvarado, J.A.; Purtschert, R.; Barbecot, F.; Chabault, C.; Rueedi, J.; Schneider, V.; Aeschbach-Hertig, W.; Kipfer, R.; Loosli, H.H. Constraining the age distribution of highly mixed groundwater using 39Ar: A multiple environmental tracer (3H/3He, 85Kr, 39Ar, and 14C) study in the semiconfined Fontainebleau Sands Aquifer (France). Water Res. Res. 2007, 43. [Google Scholar] [CrossRef] [Green Version]
- Visser, A.; Broers, H.P.; Purtschert, R.; Sültenfuß, J.; De Jonge, M. Groundwater age distributions at a public drinking water supply well field derived from multiple age tracers (85Kr,3H/3He, and39Ar). Water Resour. Res. 2013, 49, 7778–7796. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution, 2nd ed.; A.A. Balkema Publishers: Leiden, The Netherlands, 2005; p. 649. [Google Scholar]
- Mayo, A.L. Ambient well-bore mixing, aquifer cross-contamination, pumping stress, and water quality from long-screened wells: What is sampled and what is not? Hydrogeol. J. 2010, 18, 823–837. [Google Scholar] [CrossRef]
- Alvarado, J.A.C.; Barbecot, F.; Purtschert, R. Ambient vertical flow in long-screen wells: A case study in the Fontainebleau Sands Aquifer (France). Hydrogeol. J. 2009, 17, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, D.L.; Cook, P.G.; Simmons, C.T.; Solomon, D.K.; Dogramaci, S. Depth–Resolved Groundwater Chemistry by Longitudinal Sampling of Ambient and Pumped Flows Within Long–Screened and Open Borehole Wells. Water Res. Res. 2019, 55, 9417–9435. [Google Scholar] [CrossRef]
- Tiedeman, C.R.; Hsieh, P.A. Assessing an Open–Well Aquifer Test in Fractured Crystaline Rock. Groundwater 2001, 39, 68–78. [Google Scholar] [CrossRef]
- Clark, T.H. Montreal Area. Department of Natural Resources, General Direction of Mines, Geological Exploration Service. Available online: http://gq.mines.gouv.qc.ca/documents/EXAMINE/RG152/RG152.pdf (accessed on 14 September 2020).
- Tremblay, A.; Pinet, N. Distribution and characteristics of Taconian and Acadian deformation, southern Québec Appalachians. GSA Bull. 1994, 106, 1172–1181. [Google Scholar] [CrossRef]
- Globensky, Y. Géologie des Basses–Terres du Saint–Laurent; Rapport MM85–02; Direction Générale de l’Exploitation Géologique et minérale: Québec, QC, Canada, 1987; p. 70. [Google Scholar]
- Parent, M.; Occhietti, S. Late Wisconsinan Deglaciation and Champlain Sea Invasion in the St. Lawrence Valley, Québec. Géogr. Phys. Quatern. 1988, 42, 215–246. [Google Scholar] [CrossRef] [Green Version]
- Lamothe, M. A New Framework for the Pleistocene Stratigraphy of the Central St. Lawrence Lowland, Southern Québec. Géogr. Phys. Quatern. 1989, 43, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Nadeau, S.; Rosa, E.; Cloutier, V. Stratigraphic sequence map for groundwater assessment and protection of unconsolidated aquifers: A case example in the Abitibi-Témiscamingue region, Québec, Canada. Can. Water Resour. J. Rev. Can. Res. Hydriq. 2017, 43, 113–135. [Google Scholar] [CrossRef]
- Gillon, M.; Barbecot, F.; Gibert, E.; Alvarado, J.A.C.; Marlin, C.; Massault, M. Open to closed system transition traced through the TDIC isotopic signature at the aquifer recharge stage, implications for groundwater 14C dating. Geochim. Cosmochim. Acta 2009, 73, 6488–6501. [Google Scholar] [CrossRef]
- Clark, I.D. Groundwater Geochemistry and Isotopes; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Clark, I.D.; Fritz, P. Environmental Isotopes; Hydrology Lewis Publishers: Boca Raton, FL, USA, 1997. [Google Scholar]
- Cloutier, V.; Lefebvre, R.; Savard, M.M.; Therrien, R. Desalination of a sedimentary rock aquifer system invaded by Pleistocene Champlain Sea water and processes controlling groundwater geochemistry. Environ. Earth Sci. 2010, 59, 977–994. [Google Scholar] [CrossRef]
- Barbecot, F.; Marlin, C.; Gibert, E.; Dever, L. Hydrochemical and isotopic characterisation of the Bathonian and Bajocian coastal aquifer of the Caen area (northern France). Appl. Geochem. 2000, 15, 791–805. [Google Scholar] [CrossRef]
- Han, L.-F.; Plummer, L.N.; Aggarwal, P. A graphical method to evaluate predominant geochemical processes occurring in groundwater systems for radiocarbon dating. Chem. Geol. 2012, 318–319, 88–112. [Google Scholar] [CrossRef]
- Fontes, J.-C.; Garnier, J.-M. Determination of the initial 14C activity of the total dissolved carbon: A review of the existing models and a new approach. Water Resour. Res. 1979, 15, 399–413. [Google Scholar] [CrossRef]
- Cane, G.; Clark, I.D. Tracing Ground Water Recharge in an Agricultural Watershed with Isotopes. Groundwater 1999, 37, 133–139. [Google Scholar] [CrossRef]
- Keith, M.; Weber, J. Carbon and oxygen isotopic composition of selected limestones and fossils. Geochim. Cosmochim. Acta 1964, 28, 1787–1816. [Google Scholar] [CrossRef]
- Dickson, A.G.; Goyet, C. Handbook of Methods for the Analysis of the Various Parameters of the Carbon Dioxide System in Sea Water; Version 2 ORNL/CDIAC–74; Oak Ridge National Lab.: Oak Ridge, TN, USA, 1994. [Google Scholar]
- Meyzonnat, G.; Barbecot, F.; Corcho-Alvarado, J.A.; Tognelli, A.; Zeyen, H.; Mattei, A.; McCormack, R. High–Resolution Wellbore Temperature Logging Combined with a Borehole–Scale Heat Budget: Conceptual and Analytical Approaches to Characterize Hydraulically Active Fractures and Groundwater Origin. Geofluids 2018, 2018, 9461214. [Google Scholar] [CrossRef] [Green Version]
- Meyzonnat, G.; Barbecot, F.; Alvarado, J.A.C.; Lauzon, J.-M.; McCormack, R.; Tognelli, A.; Zeyen, H.; Alazard, M. Borehole Heat Budget Calculator: A New Tool for the Quick Exploitation of High-Resolution Temperature Profiles by Hydrogeologists. J. Water Resour. Prot. 2019, 11, 122–147. [Google Scholar] [CrossRef] [Green Version]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1996; p. 1022. [Google Scholar]
- Zogzas, N.; Maroulis, Z. Effective Moisture Diffusivity Estimation from Drying Data. A Comparison between Various Methods of Analysis. Dry. Technol. 1996, 14, 1543–1573. [Google Scholar] [CrossRef]
- Ogata, A.; Banks, R.B. A Solution of the Differential Equation of Longitudinal Dispersion in Porous Media: Fluid Movement in Earth Materials; US Government Printing Office: Washington, DC, USA, 1961.
- Spitler, J.D.; Javed, S.; Ramstad, R.K. Natural convection in groundwater-filled boreholes used as ground heat exchangers. Appl. Energy 2016, 164, 352–365. [Google Scholar] [CrossRef]
- Larocque, M.; Meyzonnat, G.; Barnetche, D.; Graveline, M.; Ouellet, M. Projet de Connaissance des Eaux Souterraines de la Zone de Vaudreuil–Soulanges—Rapport Scientifique. Rapport Déposé au Ministère du Développement Durable, de l’Environnement et de la Lutte Contre les Changements Climatiques; Université du Québec à Montréal: Montreal, QC, Canada, 2015; p. 202. [Google Scholar]
- Carrier, M.-A.; Lefebvre, R.; Rivard, C.; Parent, M.; Ballard, J.-M.; Benoit, N.; Vigneault, H.; Beaudry, C.; Malet, X.; Lauren-celle, M.; et al. Portrait des ressources en eau souterraine en Montérégie Est, Québec, Canada. In Projet Réalisé Conjointement par l’INRS, la CGC, l’OBV Yamaska et l’IRDA dans le Cadre du Programme d’Acquisition de Connaissances sur les Eaux Souterraines; Rapport Final INRS R–1433; Institut National de la Recherche Scientifique: Québec, QC, Canada, 2013; p. 319. [Google Scholar]
- Gillon, M.; Barbecot, F.; Gibert, E.; Plain, C.; Alvarado, J.A.C.; Massault, M. Controls on 13C and 14C variability in soil C2. Geoderma 2012, 189–190, 431–441. [Google Scholar] [CrossRef]
- Pinti, D.L.; Béland-Otis, C.; Tremblay, A.; Castro, M.C.; Hall, C.M.; Marcil, J.S.; Lapointe, R. Fossil brines preserved in the St. Lawrence Lowlands, Québec, Canada as revealed by their chemistry and noble gas isotopes. Geochim. Cosmochim. Acta 2011, 75, 4228–4243. [Google Scholar] [CrossRef]
- Garett, D.E. Handbook of Lithium and Natural Calcium Chloride; Elsevier Academic Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Garrett, D.E. Potash: Deposits, Processing, Properties and Uses; Chapman & Hall: London, UK, 1996; p. 734. [Google Scholar]
- Charbonneau, P. Sels de voirie: Une utilisation nécessaire mais lourde de conséquences. Nat. Can. 2006, 130, 75–81. [Google Scholar]
- Warr, O.; Giunta, T.; Onstott, T.C.; Kieft, T.L.; Harris, R.L.; Nisson, D.M.; Lollar, B.S. The role of low-temperature 18O exchange in the isotopic evolution of deep subsurface fluids. Chem. Geol. 2021, 561, 120027. [Google Scholar] [CrossRef]
- Grenier, J.-F. Caractérisation Pétrographique et Pétrophysique du Groupe de Potsdam dans le Forage A203, Basses–Terres du Saint–Laurent; Mémoire de Maitrise INRS; Institut National de la Recherche Scientifique: Québec, QC, Canada, 2014; p. 162. [Google Scholar]
- Barbecot, F.; Guillon, S.; Pili, E.; Larocque, M.; Gibert-Brunet, E.; Hélie, J.-F.; Noret, A.; Plain, C.; Schneider, V.; Mattei, A.; et al. Using Water Stable Isotopes in the Unsaturated Zone to Quantify Recharge in Two Contrasted Infiltration Regimes. Vadose Zone J. 2018, 17, 170170. [Google Scholar] [CrossRef] [Green Version]
- Caley, T.; Roche, D.M.; Waelbroeck, C.; Michel, E. Oxygen stable isotopes during the Last Glacial Maximum climate: Perspectives from data–model (LOVECLIM) comparison. Clim. Past 2014, 10, 1939–1955. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lithology | Localization | Wellbore | Depth * (m) | T (°C) | pH | TDS mg/L | Water Type | Ca2+ mg/L | Mg2+ mg/L | Na+ mg/L | K+ mg/L | Alk. as HCO3− mg/L | Cl− mg/L | SO42− mg/L | Na/Cl (molar) | δ2H ‰VSMOW | δ18O ‰VSMOW | δ13C ‰VPDB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Red schist | Abbotsford | PO5 | 10 | 8.5 | 8.2 | 531 | Na,Mg—Cl,HCO3 | 27 | 29 | 110 | 3.1 | 181 | 150 | 31 | 1.1 | −78.18 | −11.31 | |
| 40 | 8.4 | 8.3 | 515 | Na,Mg—Cl,HCO3 | 26 | 30 | 120 | 0.3 | 157 | 150 | 32 | 1.2 | −78.16 | −11.32 | ||||
| 75 | 8.4 | 8.1 | 1589 | Na,Mg—Cl | 120 | 120 | 330 | 0.6 | 99 | 910 | 9 | 0.6 | −84.85 | −11.98 | ||||
| 95 | 8.5 | 8.2 | 1854 | Na,Mg—Cl | 150 | 140 | 370 | 0.6 | 87 | 1100 | 6 | 0.5 | −86.00 | −12.12 | ||||
| PO7 | 10 | 9.0 | 7.9 | 502 | Ca—HCO3,Cl | 95 | 11 | 43 | 1.5 | 254 | 69 | 29 | 1.0 | −77.14 | −11.16 | |||
| 40 | 8.8 | 7.8 | 545 | Ca,Na—HCO3,Cl | 95 | 14 | 58 | 1.3 | 254 | 89 | 34 | 1.0 | −77.18 | −11.22 | ||||
| 60 | 8.7 | 7.8 | 720 | Na,Ca—Cl,HCO3 | 64 | 17 | 160 | 1.2 | 266 | 170 | 42 | 1.5 | −77.37 | −11.22 | ||||
| Sandstone | Hemmingford | P–Cl | 30 | 9.0 | 7.2 | 370 | Ca,Mg—HCO3 | 37 | 20 | 28 | 3.2 | 242 | 8.9 | 31 | 4.9 | −79.72 | −11.49 | |
| St. Lazare | P11 | 79 | 8.3 | 7.9 | 236 | Ca,Mg—HCO3 | 32 | 11 | 8.2 | 2.2 | 181 | 0.6 | 0.5 | 21.1 | −81.79 | −11.95 | −5.9 | |
| 95 | 8.4 | 7.7 | 237 | Ca,Mg—HCO3 | 32 | 12 | 8.9 | 2.3 | 181 | 0.6 | 0.5 | 23.3 | −81.80 | −12.07 | −6.6 | |||
| Ormstown | Dumas 10 | 10 | 8.2 | 7.5 | 553 | Ca—SO4,HCO3 | 100 | 21 | 16 | 4.9 | 187 | 14 | 210 | 1.8 | −82.83 | −12.12 | −12.9 | |
| 60 | 8.2 | 7.5 | 580 | Ca—SO4,HCO3 | 110 | 19 | 15 | 4.7 | 199 | 12 | 220 | 1.9 | −83.49 | −11.71 | ||||
| 80 | 8.2 | 7.4 | 610 | Ca—SO4,HCO3 | 120 | 20 | 15 | 4.6 | 168 | 13 | 270 | 1.8 | −83.14 | −12.23 | ||||
| Dolomite | Napierville | P6 | 18 | 8.3 | 7.4 | 2378 | Ca,Mg—SO4 | 360 | 120 | 200 | 8.5 | 193 | 96 | 1400 | 3.2 | −72.43 | −10.21 | −7.2 |
| 43 | 8.2 | 7.2 | 2523 | Ca,Mg—SO4 | 390 | 120 | 200 | 8.3 | 205 | 99 | 1500 | 3.1 | −72.81 | −10.38 | −7.3 | |||
| 68 | 8.2 | 7.2 | 2556 | Ca,Mg—SO4 | 380 | 130 | 210 | 8.3 | 217 | 110 | 1500 | 2.9 | −73.01 | −10.50 | −7.3 | |||
| 98 | 8.2 | 7.1 | 2554 | Ca,Mg—SO4 | 390 | 130 | 210 | 8.6 | 205 | 110 | 1500 | 2.9 | −73.23 | −10.21 | ||||
| PO2 | 21 | 9.1 | 7.0 | 921 | Ca—HCO3,SO4 | 160 | 37 | 30 | 4.8 | 395 | 64 | 230 | 0.7 | −72.05 | −10.67 | |||
| 45 | 8.7 | 7.2 | 1179 | Ca,Mg—SO4,HCO3 | 190 | 47 | 43 | 5.1 | 291 | 43 | 560 | 1.5 | −72.25 | −10.64 | ||||
| 70 | 8.6 | 7.2 | 1183 | Ca—SO4,HCO3 | 210 | 47 | 47 | 5.3 | 317 | 47 | 510 | 1.5 | −72.54 | −10.50 | ||||
| 83 | 8.6 | 7.2 | 1239 | Ca,Mg—SO4,HCO3 | 200 | 50 | 49 | 5.4 | 301 | 44 | 590 | 1.7 | −71.89 | −10.70 | ||||
| Chateauguay | MELCC 03090013 | 21 | 8.3 | 7.9 | 460 | Mg,Ca,Na—HCO3 | 43 | 29 | 37 | 5.1 | 339 | 8.9 | 6 | 6.4 | −73.92 | −10.72 | −13.7 | |
| 33–35 | 8.2 | 7.7 | 532 | Ca,Mg,Na—HCO3 | 51 | 30 | 53 | 5.1 | 304 | 44 | 55 | 1.9 | −74.96 | −10.89 | −13.4 | |||
| 45 | 8.1 | 7.7 | 546 | Ca,Mg,Na—HCO3 | 52 | 29 | 51 | 4.9 | 305 | 44 | 57 | 1.7 | −74.92 | −10.85 | −13.3 | |||
| 52 | 8.1 | 7.7 | 566 | Ca,Mg,Na—HCO3 | 55 | 32 | 55 | 5.5 | 314 | 50 | 63 | 1.7 | −74.74 | −11.02 | −13.3 | |||
| 55 | 8.0 | 7.5 | 575 | Ca,Mg,Na—HCO3 | 54 | 32 | 59 | 5.5 | 304 | 67 | 60 | 1.4 | −75.06 | −10.93 | −13.1 | |||
| Mirabel | Charles | 10 | 9.9 | 6.9 | 849 | Ca,Mg—HCO3,SO4 | 110 | 43 | 57 | 4.1 | 355 | 100 | 180 | 0.9 | −61.09 | −8.99 | −14.1 | |
| Limestone | St. Roch | 2017–04 | 11 | 9.8 | 6.9 | 879 | Ca,Mg—Cl,HCO3 | 120 | 59 | 41 | 8.1 | 353 | 230 | 68 | 0.3 | −70.19 | −10.53 | −11.9 |
| 71 | 9.2 | 7.2 | 919 | Ca,Mg—Cl,HCO3 | 110 | 60 | 85 | 9.1 | 297 | 290 | 68 | 0.5 | −71.45 | −10.31 | ||||
| Detection threshold/analytical error (±) | 0.3 | 0.1 | 0.1 | 0.1 | 1 | 0.05 | 5.0 | – | ±1.5 | ±0.05 | ±0.1 | |||||||
| Sampling Mode | Depth (m) | pH | EC μS/cm | Ca2+ mg/L | Mg2+ mg/L | Na+ mg/L | K+ mg/L | HCO3− mg/L | Cl− mg/L | SO42− mg/L | δ18O ‰ VSMOW | δ 2H ‰ VSMOW |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Passive Sampler | 25 | 7.89 | 507 | 43 | 29 | 37 | 5 | 339 | 8.7 | 6.4 | −10.71 | −73.89 |
| 37 | 7.7 | 667 | 51 | 30 | 52 | 5.1 | 305 | 43 | 53 | −10.85 | −74.62 | |
| 41 | 7.69 | 679 | 53 | 30 | 52 | 5 | 308 | 46 | 59 | −10.88 | −74.83 | |
| Low flow pumping | 25 | 7.91 | 534 | 43 | 28 | 36 | 4.9 | 344 | 9.7 | 7.1 | −10.75 | −73.95 |
| 37 | 7.81 | 621 | 47 | 27 | 48 | 4.9 | 300 | 38 | 39 | −10.90 | −75.07 | |
| 41 | 7.67 | 680 | 53 | 31 | 54 | 5.3 | 307 | 46 | 58 | −10.91 | −74.89 | |
| Detection threshold/± error % | 0.1 | 5 | 0.3 | 0.1 | 0.1 | 0.1 | 1 | 0.05 | 0.5 | ±0.05‰ | ±1.5‰ | |
| Passive Sampler | Depth (m) | pH | EC μS/cm | Ca2+ mg/L | Mg2+ mg/L | Na+ mg/L | K+ mg/L | HCO3− mg/L | Cl− mg/L | SO42− mg/L | δ18O‰ VSMOW | δ2H‰ VSMOW |
| 25 | 7.89 | 507 | 43 | 29 | 37 | 5 | 339 | 8.7 | 6.4 | −10.71 | −73.89 | |
| 37 | 7.7 | 667 | 51 | 30 | 52 | 5.1 | 305 | 43 | 53 | −10.85 | −74.62 | |
| 41 | 7.69 | 679 | 53 | 30 | 52 | 5 | 308 | 46 | 59 | −10.88 | −74.83 | |
| 47 | 7.72 | 715 | 55 | 32 | 55 | 5.3 | 314 | 50 | 63 | −11.02 | −74.74 | |
| Mixing (calculated from samplers) | 7.70 | 672 | 52 | 30 | 52 | 5.1 | 308 | 44 | 54 | −10.89 | −74.74 | |
| Pumping’s discharge (sampled) | 7.65 | 612 | 50 | 29 | 51 | 4.9 | 307 | 42 | 48 | −10.89 | −74.95 | |
| % error | −1% | −10% | −4% | −4% | −2% | −4% | 0% | −4% | −14% | 0.004‰ | 0.21‰ | |
| Detection threshold/±analytical error | ±0.1 | ±5 | 0.3 | 0.1 | 0.1 | 0.1 | 1 | 0.05 | 0.5 | ±0.05‰ | ±1.5‰ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyzonnat, G.; Barbecot, F.; Corcho Alvarado, J.; Pinti, D.L.; Lauzon, J.-M.; McCormack, R. Depth–Sequential Investigation of Major Ions, δ18O, δ2H and δ13C in Fractured Aquifers of the St. Lawrence Lowlands (Quebec, Canada) Using Passive Samplers. Water 2021, 13, 1806. https://doi.org/10.3390/w13131806
Meyzonnat G, Barbecot F, Corcho Alvarado J, Pinti DL, Lauzon J-M, McCormack R. Depth–Sequential Investigation of Major Ions, δ18O, δ2H and δ13C in Fractured Aquifers of the St. Lawrence Lowlands (Quebec, Canada) Using Passive Samplers. Water. 2021; 13(13):1806. https://doi.org/10.3390/w13131806
Chicago/Turabian StyleMeyzonnat, Guillaume, Florent Barbecot, José Corcho Alvarado, Daniele Luigi Pinti, Jean-Marc Lauzon, and Renald McCormack. 2021. "Depth–Sequential Investigation of Major Ions, δ18O, δ2H and δ13C in Fractured Aquifers of the St. Lawrence Lowlands (Quebec, Canada) Using Passive Samplers" Water 13, no. 13: 1806. https://doi.org/10.3390/w13131806
APA StyleMeyzonnat, G., Barbecot, F., Corcho Alvarado, J., Pinti, D. L., Lauzon, J.-M., & McCormack, R. (2021). Depth–Sequential Investigation of Major Ions, δ18O, δ2H and δ13C in Fractured Aquifers of the St. Lawrence Lowlands (Quebec, Canada) Using Passive Samplers. Water, 13(13), 1806. https://doi.org/10.3390/w13131806