Reductive/Oxidative Sequential Bioelectrochemical Process for Perchloroethylene Removal





Abstract
1. Introduction
2. Materials and Methods
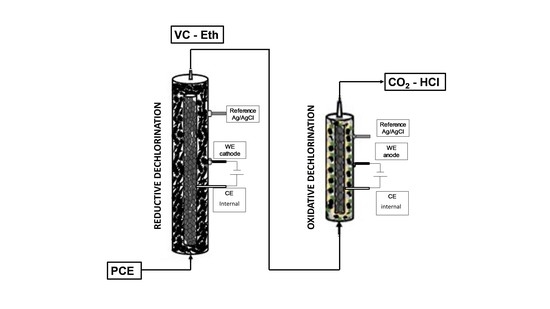
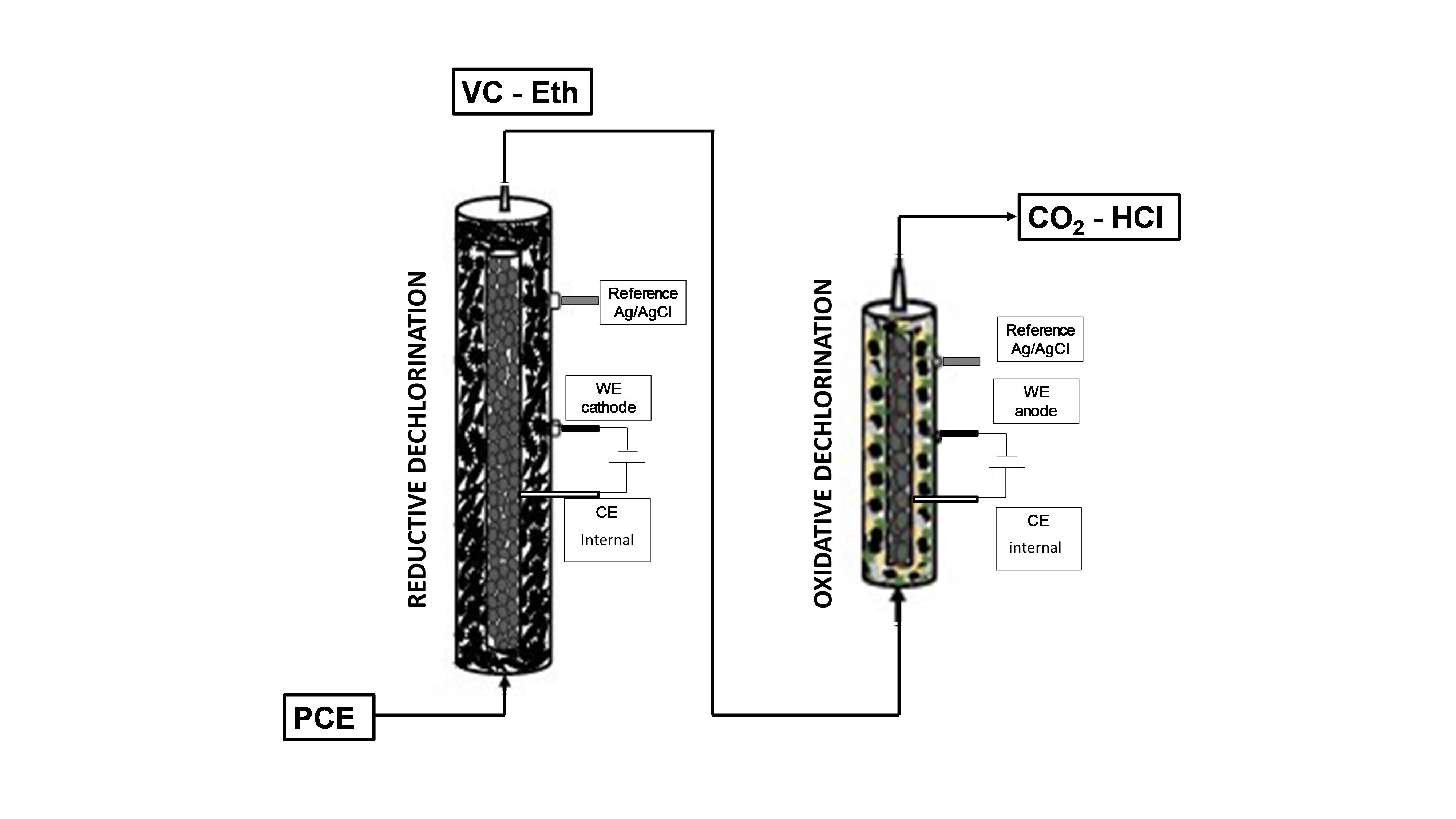
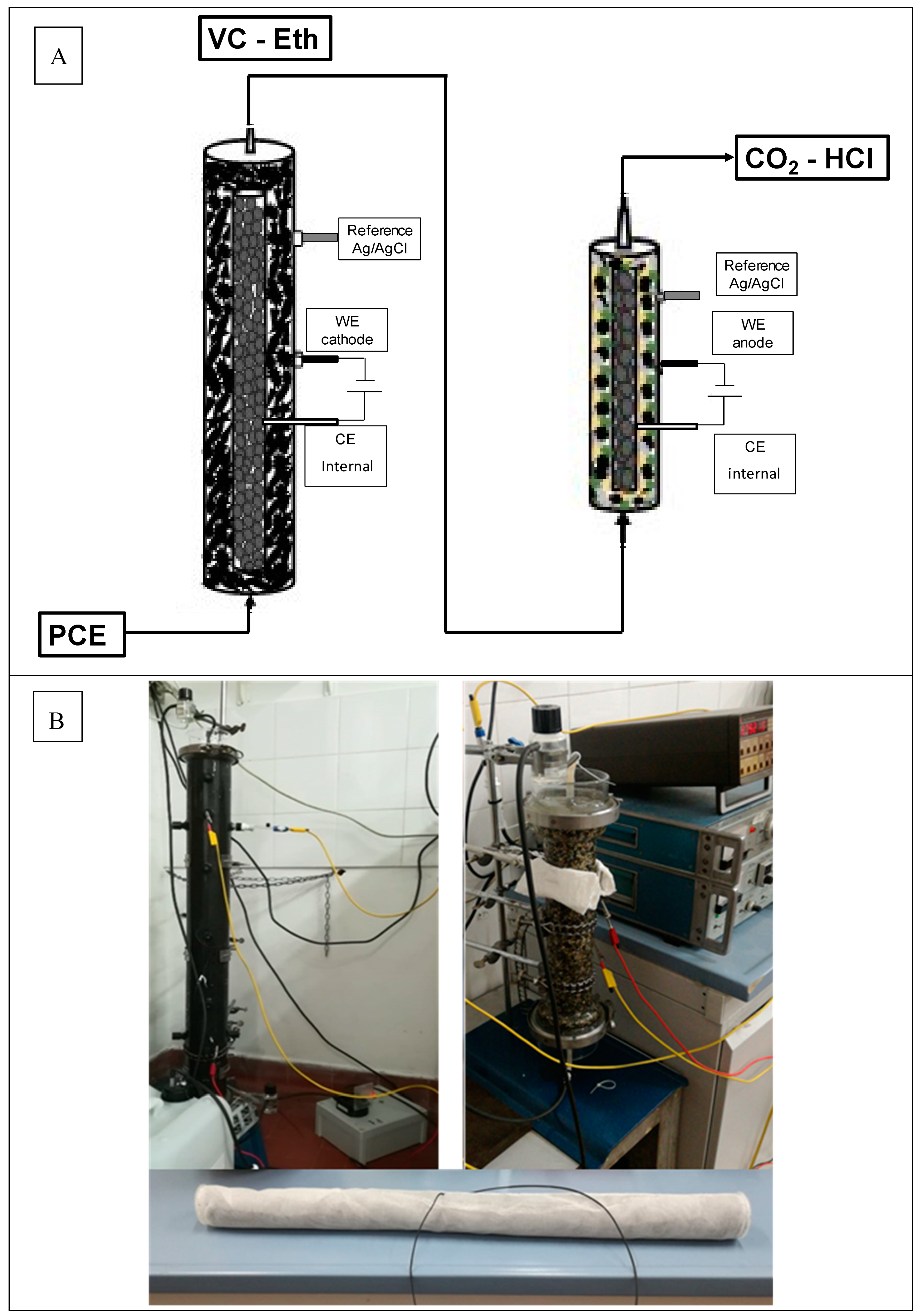
2.1. Sequential Process Setup
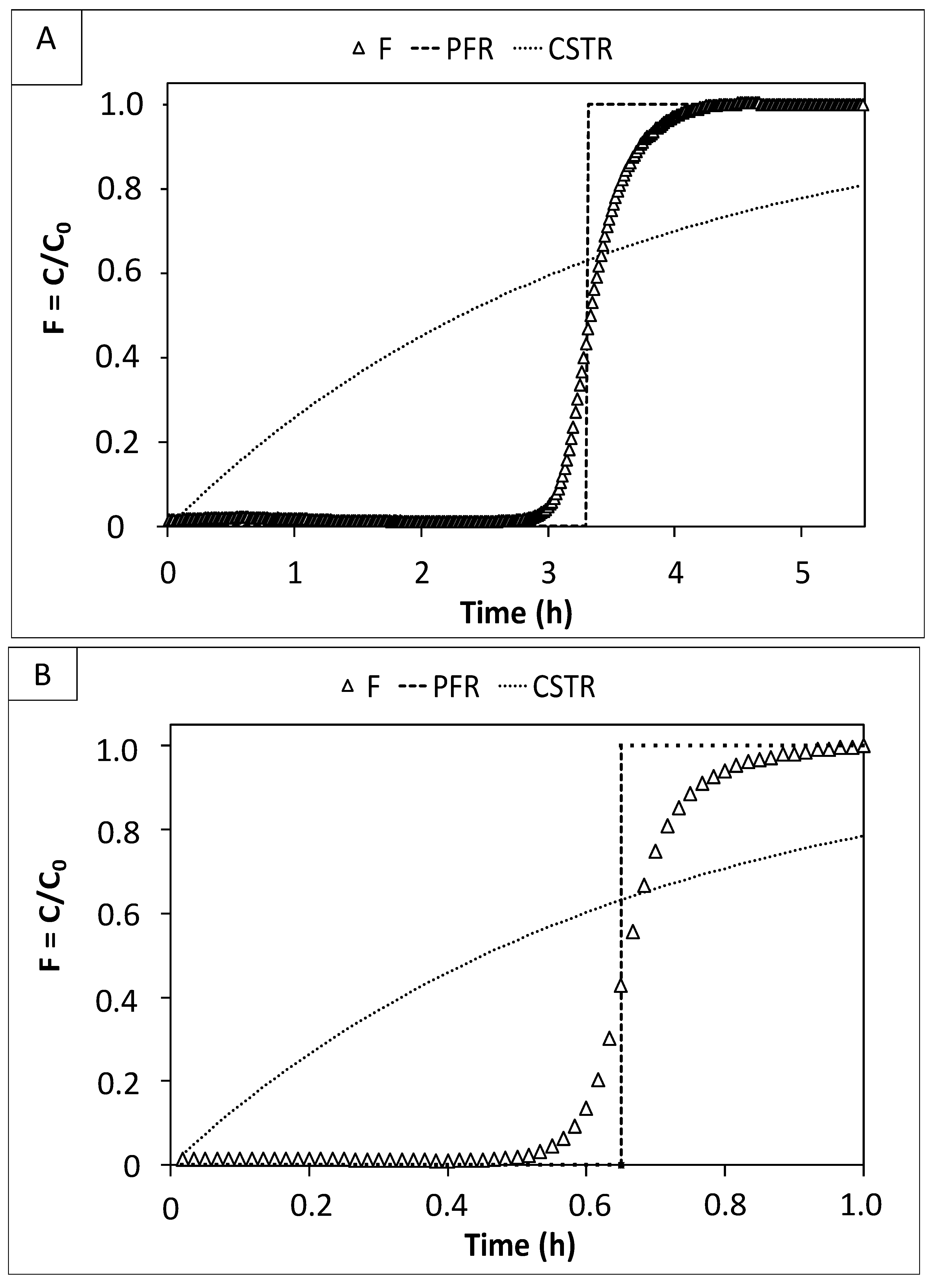
2.2. Tracer Test
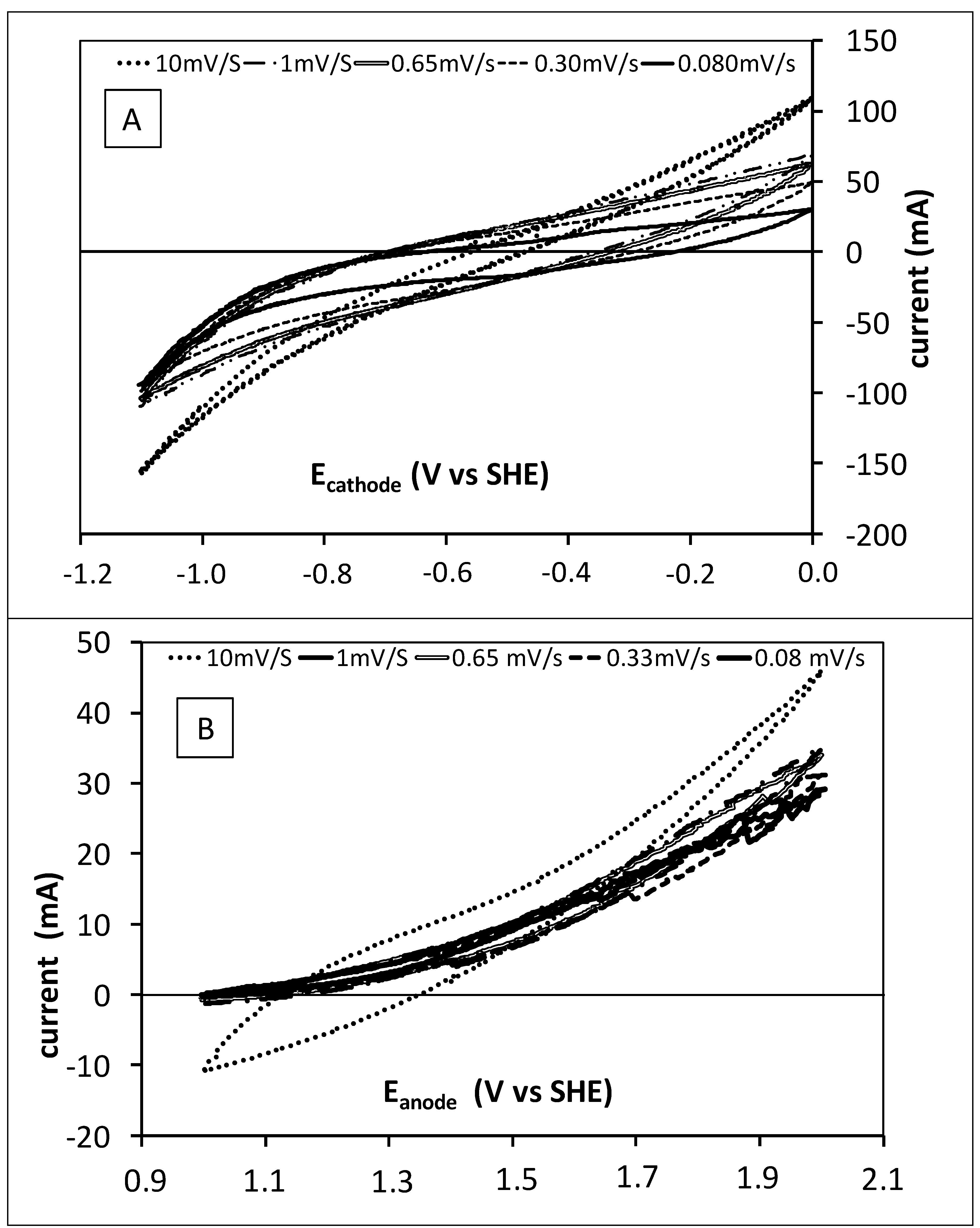
2.3. Cyclic Voltammetry
2.4. Analytical Methods
2.5. Calculations
2.6. Chemicals
3. Results
3.1. Preliminary Fluid Dynamics and Electrochemical Characterization of the Tubular Reactors
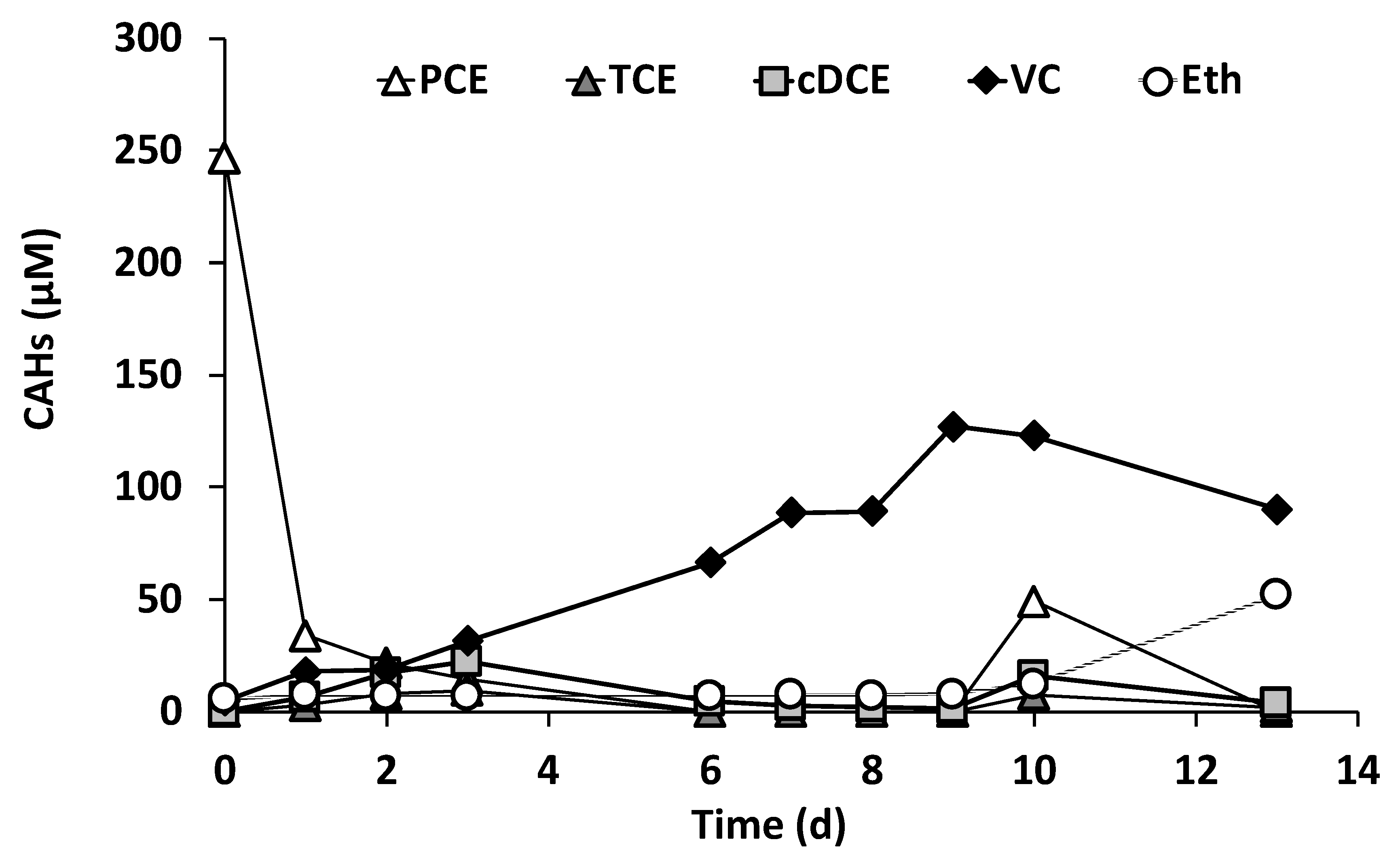
3.2. Reactor Inoculation
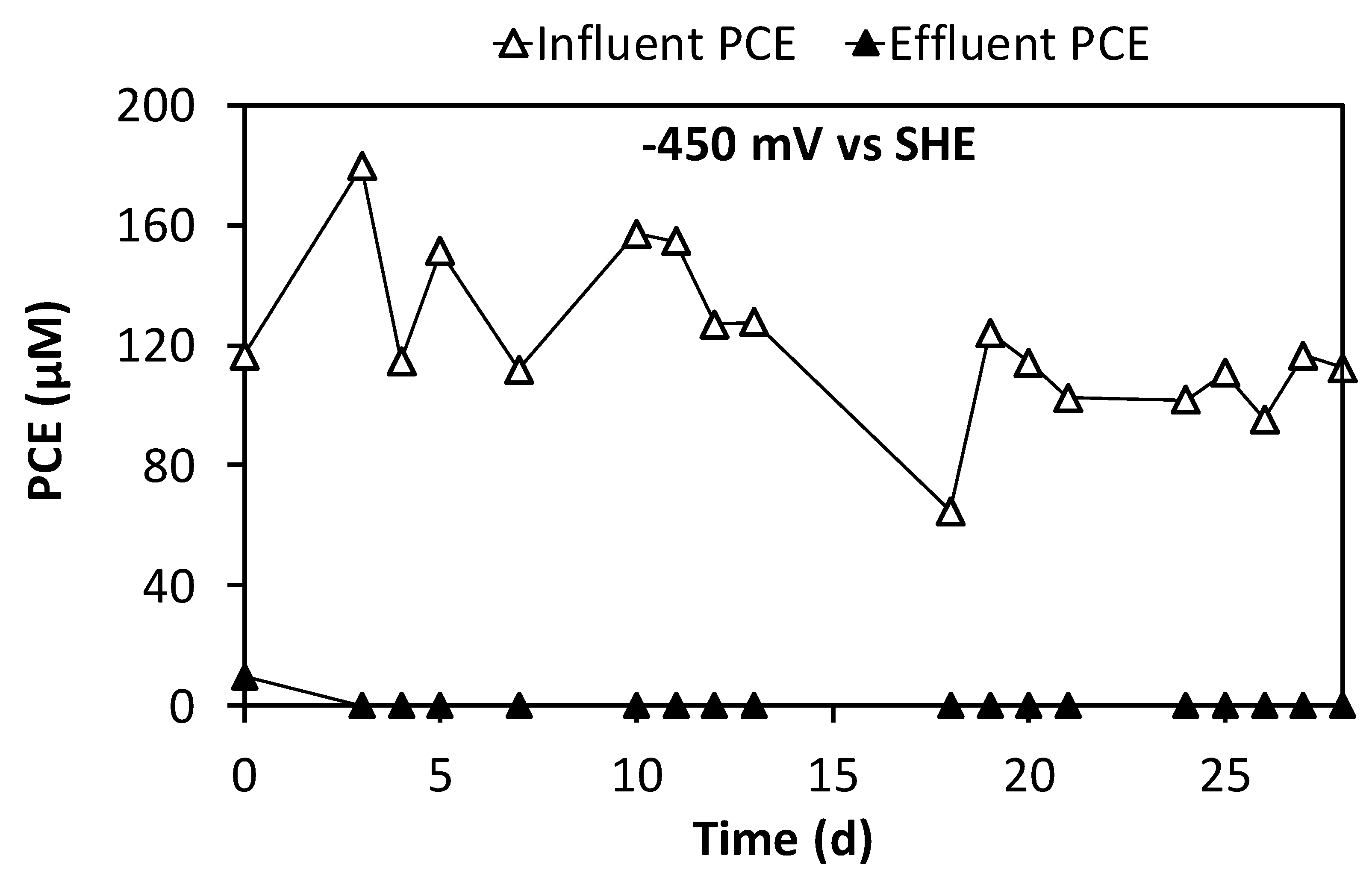
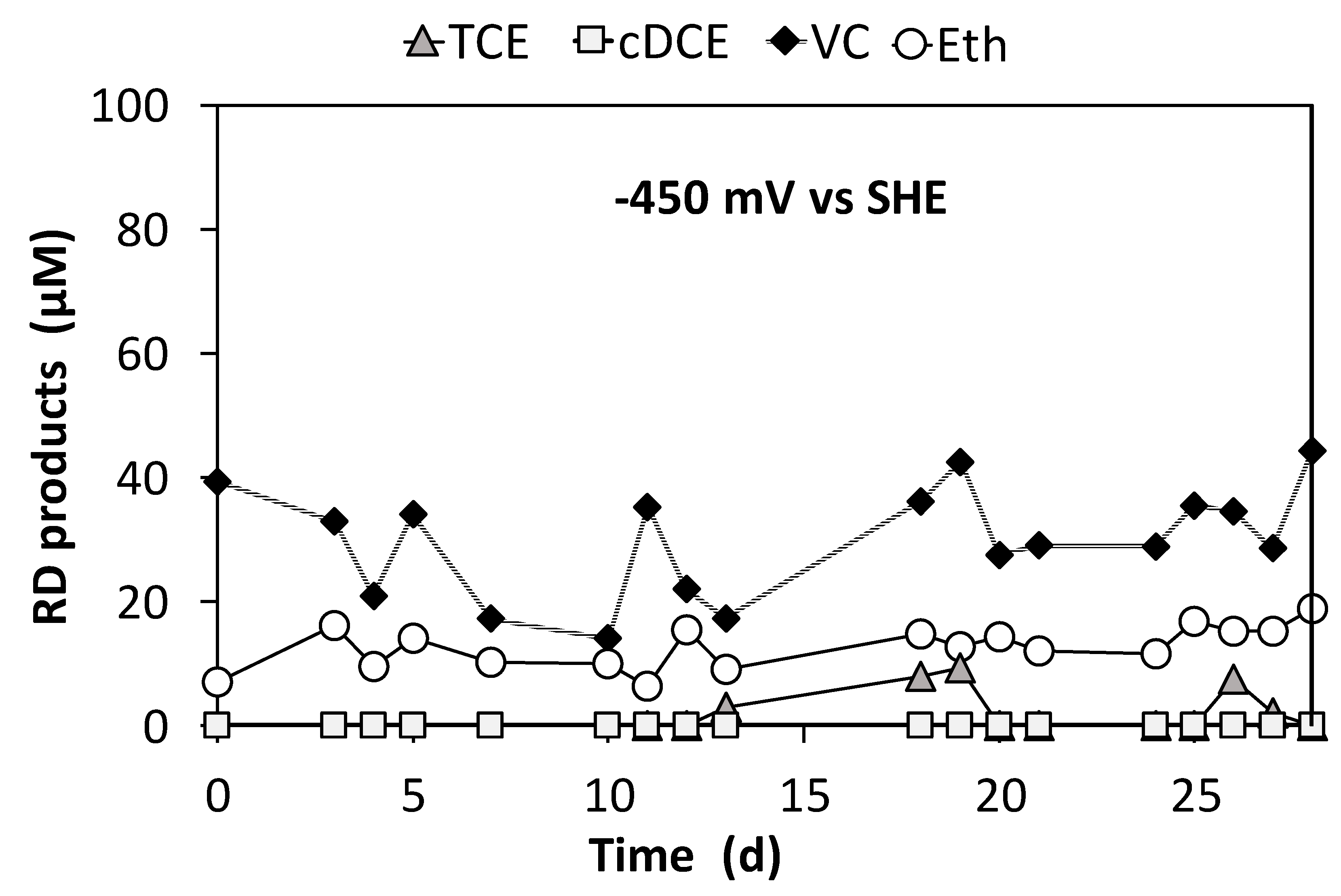
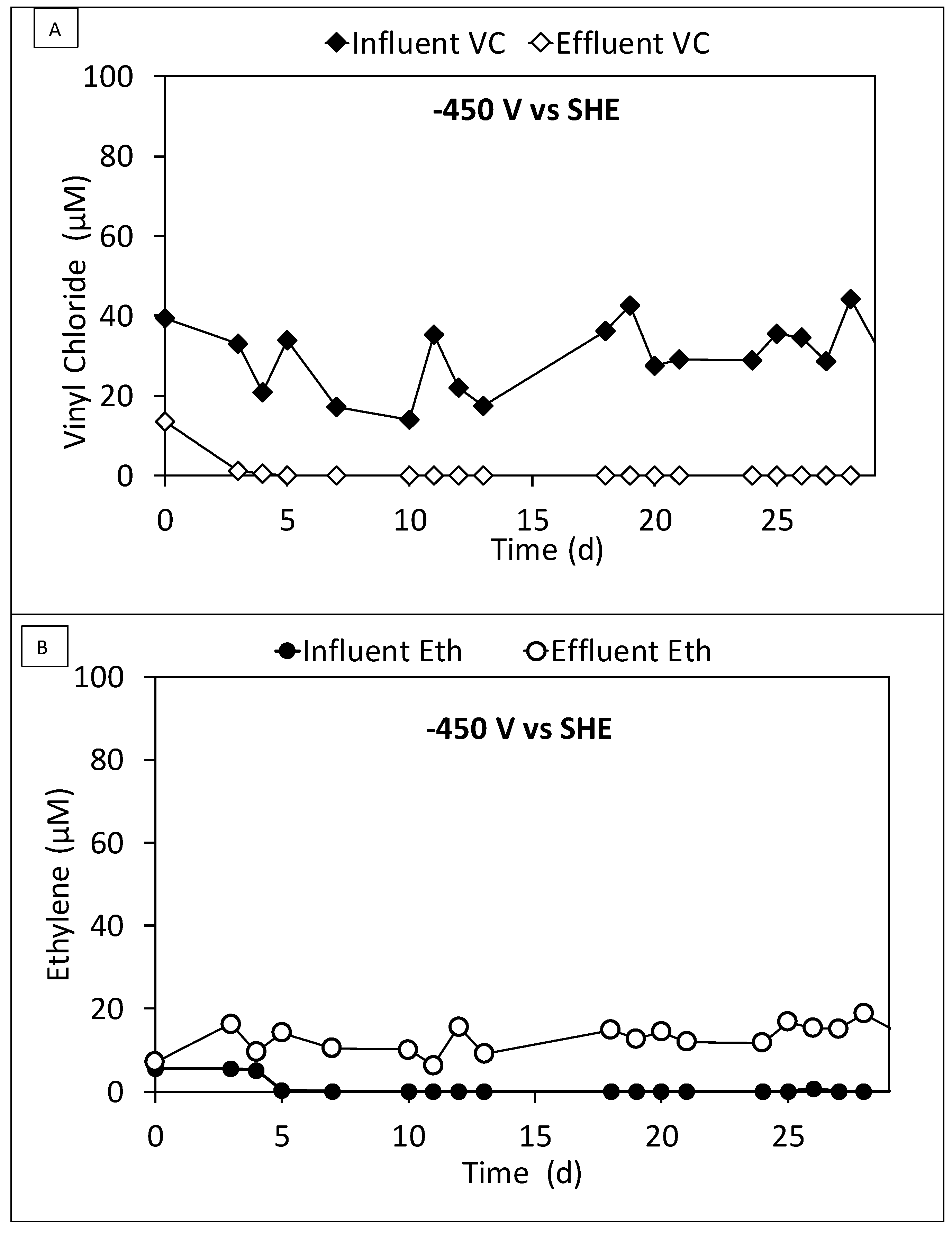
3.3. Continuous Flow Operation of the Sequential Oxidative/Reductive Process
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bradley, P.M. History and ecology of chloroethene biodegradation: A Review. Biorem. J. 2003, 7, 81–109. [Google Scholar] [CrossRef]
- Moran, M.J.; Zogorski, J.S.; Squillace, P.J. Chlorinated solvents in groundwater of the United States. Environ. Sci. Technol. 2007, 41, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Pierro, L.; Matturro, B.; Rossetti, S.; Sagliaschi, M.; Sucato, S.; Alesi, E.; Bartsch, E.; Arjmand, F.; Papini, M.P. Polyhydroxyalkanoate as a slow-release carbon source for in situ bioremediation of contaminated aquifers: From laboratory investigation to pilot-scale testing in the field. New Biotechnol. 2017, 37, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Aulenta, F.; Gossett, J.M.; Papini, M.P.; Rossetti, S.; Majone, M. Comparative study of methanol, butyrate, and hydrogen as electron donors for long-term dechlorination of tetrachloroethene in mixed anerobic cultures. Biotechnol. Bioeng. 2005, 91, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Vainberg, S.; Condee, C.W.; Steffan, R.J. Large-scale production of bacterial consortia for remediation of chlorinated solvent-contaminated groundwater. J. Ind. Microbiol. Biotechnol. 2009, 36, 1189–1197. [Google Scholar] [CrossRef]
- Matturro, B.; Presta, E.; Rossetti, S. Reductive dechlorination of tetrachloroethene in marine sediments: Biodiversity and dehalorespiring capabilities of the indigenous microbes. Sci. Total Environ. 2016, 545, 445–452. [Google Scholar] [CrossRef]
- Seshadri, R.; Adrian, L.; Fouts, D.E.; Eisen, J.A.; Phillippy, A.M.; Methe, B.A.; Ward, N.L.; Nelson, W.C.; Deboy, R.T.; Khouri, H.M.; et al. Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes. Science 2005, 307, 105–108. [Google Scholar] [CrossRef]
- Luijten, M.L.G.C.; Roelofsen, W.; Langenhoff, A.A.M.; Schraa, G.; Stams, A.J.M. Hydrogen threshold concentrations in pure cultures of halorespiring bacteria and at a site polluted with chlorinated ethenes. Environ. Microbiol. 2004, 6, 646–650. [Google Scholar] [CrossRef]
- Majone, M.; Verdini, R.; Aulenta, F.; Rossetti, S.; Tandoi, V.; Kalogerakis, N.; Agathos, S.; Puig, S.; Zanaroli, G.; Fava, F. In situ groundwater and sediment bioremediation: barriers and perspectives at European contaminated sites. New Biotechnol. 2015, 32, 133–146. [Google Scholar] [CrossRef]
- Nam, S.H.; An, Y.J. Assessing the ecotoxicity of vinyl chloride using green alga P. subcapitata, nematode C. elegans, and the SOS chromotest in a closed system without headspace. Sci. Total Environ. 2010, 408, 3148–3152. [Google Scholar] [CrossRef]
- Gossett, J.M. Sustained aerobic oxidation of vinyl chloride at low oxygen concentrations. Environ. Sci. Technol. 2010, 44, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Wendlandt, K.D.; Stottmeister, U.; Helm, J.; Soltmann, B.; Jechorek, M.; Beck, M. The potential of methane-oxidizing bacteria for applications in environmental biotechnology. Eng. Life Sci. 2010, 10, 87–102. [Google Scholar] [CrossRef]
- Mattes, T.E.; Alexander, A.K.; Coleman, N.V. Aerobic biodegradation of the chloroethenes: pathways, enzymes, ecology, and evolution. FEMS Microbiol. Rev. 2010, 34, 445–475. [Google Scholar] [CrossRef] [PubMed]
- Noell, A.L. Estimation of sequential degradation rate coefficients for chlorinated ethenes. Pract. Period. Hazard. Toxic Radioact. Waste Manag. 2009, 13, 35–44. [Google Scholar] [CrossRef]
- Devlin, J.F.; Katic, D.; Barker, J.F. In situ sequenced bioremediation of mixed contaminants in groundwater. J. Contam. Hydrol. 2004, 69, 233–261. [Google Scholar] [CrossRef]
- Tiehm, A.; Müller, A.; Alt, S.; Jacob, H.; Schad, H.; Weingran, C. Development of a groundwater biobarrier for the removal of polycyclic aromatic hydrocarbons, BTEX, and heterocyclic hydrocarbons. Water Sci. Technol. 2008, 58, 1349–1355. [Google Scholar] [CrossRef]
- Logan, B.E.; Rabaey, K. Conversion of wastes into bioelectricity and chemicals by using microbial electrochemical technologies. Science 2012, 337, 686–690. [Google Scholar] [CrossRef]
- Rabaey, K.; Angenent, L.; Schröder, U.; Keller, J. Bioelectrochemical systems: from extracellular electron transfer to biotechnological application. Water Intell. Online 2009, 8, 9781780401621. [Google Scholar] [CrossRef]
- Rozendal, R.A.; Hamelers, H.V.M.; Rabaey, K.; Keller, J.; Buisman, C.J.N. Towards practical implementation of bioelectrochemical wastewater treatment. Trends Biotechnol. 2008, 26, 450–459. [Google Scholar] [CrossRef]
- Aulenta, F.; Canosa, A.; Reale, P.; Rossetti, S.; Panero, S.; Majone, M. Microbial reductive dechlorination of trichloroethene to ethene with electrodes serving as electron donors without the external addition of redox mediators. Biotechnol. Bioeng. 2009, 103, 85–91. [Google Scholar] [CrossRef]
- Aulenta, F.; Catervi, A.; Majone, M.; Panero, S.; Reale, P.; Rossetti, S. Electron transfer from a solid-state electrode assisted by methyl viologen sustains efficient microbial reductive dechlorination of TCE. Environ. Sci. Technol. 2007, 41, 2554–2559. [Google Scholar] [CrossRef] [PubMed]
- Verdini, R.; Aulenta, F.; de Tora, F.; Lai, A.; Majone, M. Relative contribution of set cathode potential and external mass transport on TCE dechlorination in a continuous-flow bioelectrochemical reactor. Chemosphere 2015, 136, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Aulenta, F.; Tocca, L.; Verdini, R.; Reale, P.; Majone, M. Dechlorination of trichloroethene in a continuous-flow bioelectrochemical reactor: Effect of cathode potential on rate, selectivity, and electron transfer mechanisms. Environ. Sci. Technol. 2011, 45, 8444–8451. [Google Scholar] [CrossRef] [PubMed]
- Aulenta, F.; Verdini, R.; Zeppilli, M.; Zanaroli, G.; Fava, F.; Rossetti, S.; Majone, M. Electrochemical stimulation of microbial cis-dichloroethene (cis-DCE) oxidation by an ethene-assimilating culture. New Biotechnol. 2013, 30, 749–755. [Google Scholar] [CrossRef]
- Lohner, S.T.; Becker, D.; Mangold, K.M.; Tiehm, A. Sequential reductive and oxidative biodegradation of chloroethenes stimulated in a coupled bioelectro-process. Environ. Sci. Technol. 2011, 45, 6491–6497. [Google Scholar] [CrossRef]
- Lai, A.; Aulenta, F.; Mingazzini, M.; Palumbo, M.T.; Papini, M.P.; Verdini, R.; Majone, M. Bioelectrochemical approach for reductive and oxidative dechlorination of chlorinated aliphatic hydrocarbons (CAHs). Chemosphere 2017, 169, 351–360. [Google Scholar] [CrossRef]
- Kinoshita, K.; Bett, J. Electrochemical oxidation of carbon black in concentrated phosphoric acid at 135°C. Carbon 1973, 11, 237–247. [Google Scholar] [CrossRef]
- Chouler, J.; Bentley, I.; Vaz, F.; O’Fee, A.; Cameron, P.J.; Di Lorenzo, M. Exploring the use of cost-effective membrane materials for Microbial Fuel Cell based sensors. Electrochim. Acta 2017, 231, 319–326. [Google Scholar] [CrossRef]
- Rozendal, R.A.; Sleutels, T.H.J.A.; Hamelers, H.V.M.; Buisman, C.J.N. Effect of the type of ion exchange membrane on performance, ion transport, and pH in biocatalyzed electrolysis of wastewater. Water Sci. Technol. 2008, 57, 1757–1762. [Google Scholar] [CrossRef]
- Zeppilli, M.; Paiano, P.; Villano, M.; Majone, M. Anodic vs. cathodic potentiostatic control of a methane producing microbial electrolysis cell aimed at biogas upgrading. Biochem. Eng. J. 2019, 152, 107393. [Google Scholar] [CrossRef]
- Matturro, B.; Pierro, L.; Frascadore, E.; Petrangeli Papini, M.; Rossetti, S. Microbial community changes in a chlorinated solvents polluted aquifer over the field scale treatment with poly-3-Hydroxybutyrate as amendment. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Fox, G.E.; Magrum, L.J.; Woese, C.R.; Wolfe, R.S. Methanogens: reevaluation of a unique biological group. Microbiol. Rev. 1979, 43, 260–296. [Google Scholar] [PubMed]
- Zeikus, J.G. The biology of methanogenic bacteria. Bacteriol. Rev. 1977, 41, 514–541. [Google Scholar] [PubMed]
- Gossett, J.M. Measurement of Henry’s law constants for C1 and C2 chlorinated hydrocarbons. Environ. Sci. Technol. 1987, 21, 202–208. [Google Scholar] [CrossRef]
- Zeppilli, M.; Lai, A.; Villano, M.; Majone, M. Anion vs. cation exchange membrane strongly affect mechanisms and yield of CO2 fixation in a microbial electrolysis cell. Chem. Eng. J. 2016, 304, 10–19. [Google Scholar] [CrossRef]
- Villano, M.; Ralo, C.; Zeppilli, M.; Aulenta, F.; Majone, M. Influence of the set anode potential on the performance and internal energy losses of a methane-producing microbial electrolysis cell. Bioelectrochemistry 2016, 107, 1–6. [Google Scholar] [CrossRef]
- Bard, A.; Faulkner, J. Electrochemical Methods Fundamentals and Applications; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Savéant, J.M. Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A practical beginner’s guide to cyclic voltammetry. J. Chem. Educ. 2017, 95, 197–206. [Google Scholar] [CrossRef]
- Villano, M.; Aulenta, F.; Giuliano, A.; Ciucci, C.; Ferri, T.; Majone, M. Bioelectrochemical reduction of CO2 to CH4 via direct and indirect extracellular electron transfer by a hydrogenophilic methanogenic culture. Bioresour. Technol. 2010, 101, 3085–3090. [Google Scholar] [CrossRef]
- Dutta, P.K.; Rabaey, K.; Yuan, Z.; Rozendal, R.A.; Keller, J. Electrochemical sulfide removal and recovery from paper mill anaerobic treatment effluent. Water Res. 2010, 44, 2563–2571. [Google Scholar] [CrossRef]
- Watanabe, T.; Hashimoto, S.; Kuroda, M. Simultaneous nitrification and denitrification in a single reactor using bio-electrochemical process. Water Sci. Technol. 2002, 46, 163–169. [Google Scholar] [CrossRef]
- Ter Heijne, A.; de Rink, R.; Liu, D.; Klok, J.B.M.; Buisman, C.J.N. Bacteria as an Electron Shuttle for Sulfide Oxidation. Environ. Sci. Technol. Lett. 2018, 5, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Zeppilli, M.; Simoni, M.; Paiano, P.; Majone, M. Two-Side cathode microbial electrolysis cell for nutrients recovery and biogas upgrading. Chem. Eng. J. 2019, 370, 466–476. [Google Scholar] [CrossRef]
- Guiot, S.R.; Cimpoia, R.; Kuhn, R.; Alaplantive, A. Electrolytic methanogenic−methanotrophic coupling for tetrachloroethylene bioremediation: Proof of concept. Environ. Sci. Technol. 2008, 42, 3011–3017. [Google Scholar] [CrossRef] [PubMed]
- Knauss, K.G.; Dibley, M.J.; Leif, R.N.; Mew, D.A.; Aines, R.D. The aqueous solubility of trichloroethene (TCE) and tetrachloroethene (PCE) as a function of temperature. Appl. Geochem. 2000, 15, 501–512. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HRT | Reductive Reactor | Oxidative Reactor |
|---|---|---|
| Working electrode (h) | 2.6 | 0.8 |
| Overall reactor (h) | 3.3 | 1 |
| Experimental (h) | 3.3 | 0.7 |
| Reductive Reactor Potential | −450 mV versus SHE |
| PCE removal rate (μmol/d) | 191 ± 13 |
| PCE removal efficiency (%) | 100 ± 1 |
| Coulombic efficiency of reductive dechlorination (RD) (%) | 5 ± 1 |
| Coulombic efficiency CH4 (%) | 22 ± 4 |
| Oxidative Reactor Potential | +1.4 V versus SHE |
| Vinyl chloride (VC) removal efficiency (%) | 100 ± 2 |
| Ethylene removal efficiency (%) | 92 ± 5 |
| Coulombic efficiency of oxidative VC-ethylene (%) | 7 ± 2 |
| Coulombic efficiency of oxidative CH4 (%) | 8 ± 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeppilli, M.; Dell’Armi, E.; Cristiani, L.; Petrangeli Papini, M.; Majone, M. Reductive/Oxidative Sequential Bioelectrochemical Process for Perchloroethylene Removal. Water 2019, 11, 2579. https://doi.org/10.3390/w11122579
Zeppilli M, Dell’Armi E, Cristiani L, Petrangeli Papini M, Majone M. Reductive/Oxidative Sequential Bioelectrochemical Process for Perchloroethylene Removal. Water. 2019; 11(12):2579. https://doi.org/10.3390/w11122579
Chicago/Turabian StyleZeppilli, Marco, Edoardo Dell’Armi, Lorenzo Cristiani, Marco Petrangeli Papini, and Mauro Majone. 2019. "Reductive/Oxidative Sequential Bioelectrochemical Process for Perchloroethylene Removal" Water 11, no. 12: 2579. https://doi.org/10.3390/w11122579
APA StyleZeppilli, M., Dell’Armi, E., Cristiani, L., Petrangeli Papini, M., & Majone, M. (2019). Reductive/Oxidative Sequential Bioelectrochemical Process for Perchloroethylene Removal. Water, 11(12), 2579. https://doi.org/10.3390/w11122579