Phosphate Induced Arsenic Mobilization as a Potentially Effective In-Situ Remediation Technique—Preliminary Column Tests

Abstract
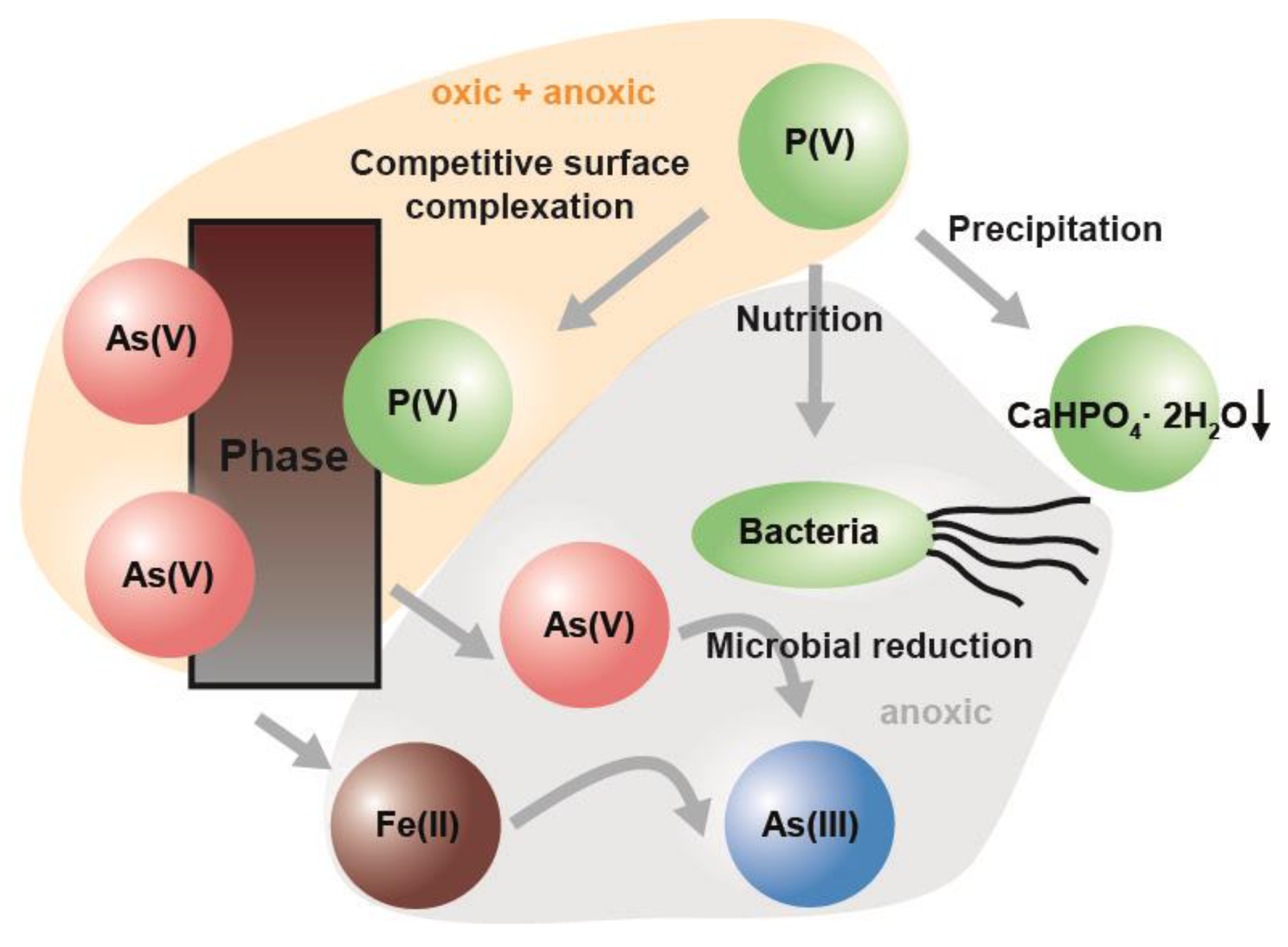
:1. Introduction
2. Materials and Methods
2.1. Aquifer Material, Groundwater Sampling and Analysis
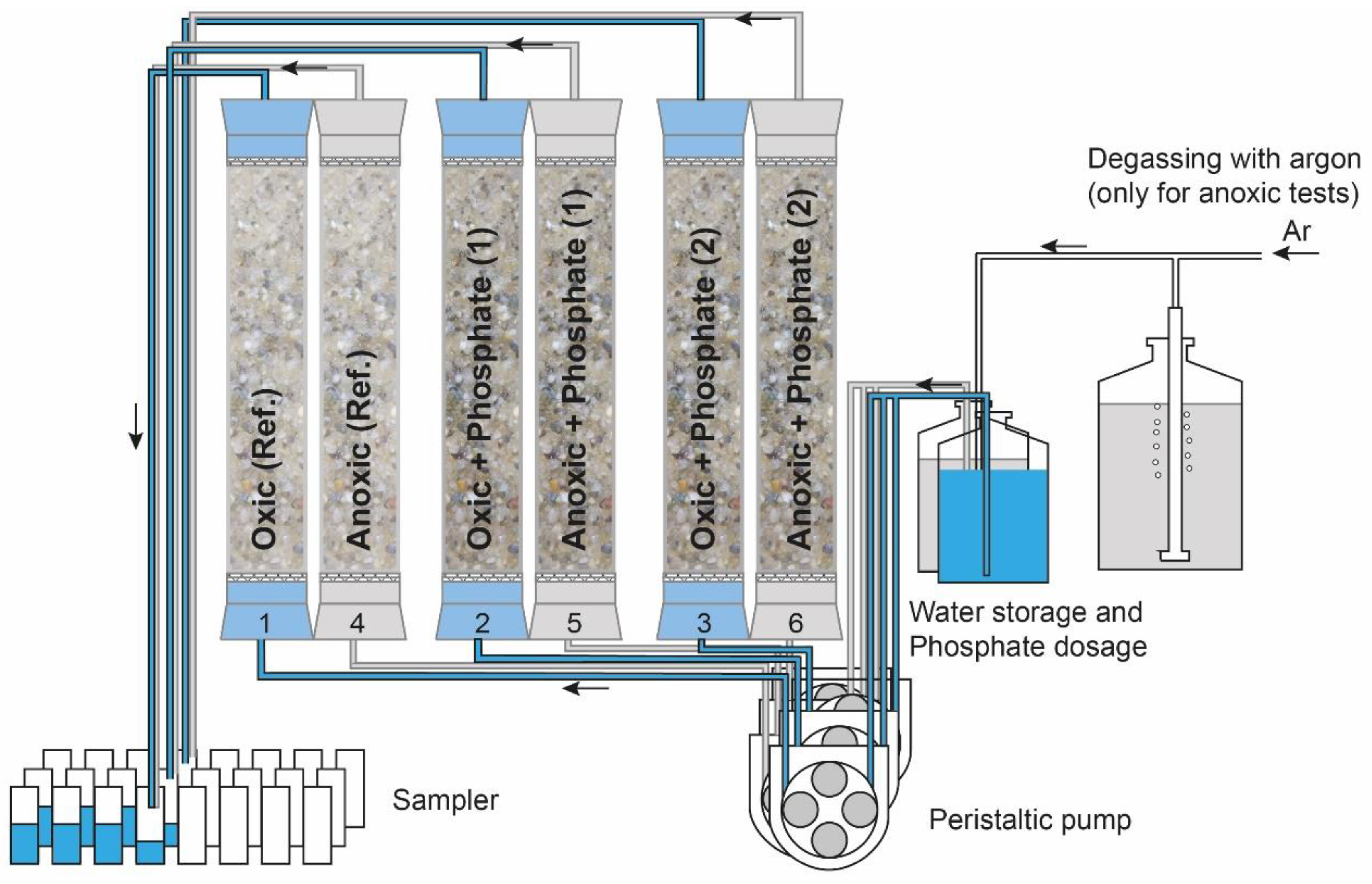
2.2. Column Tests
3. Results
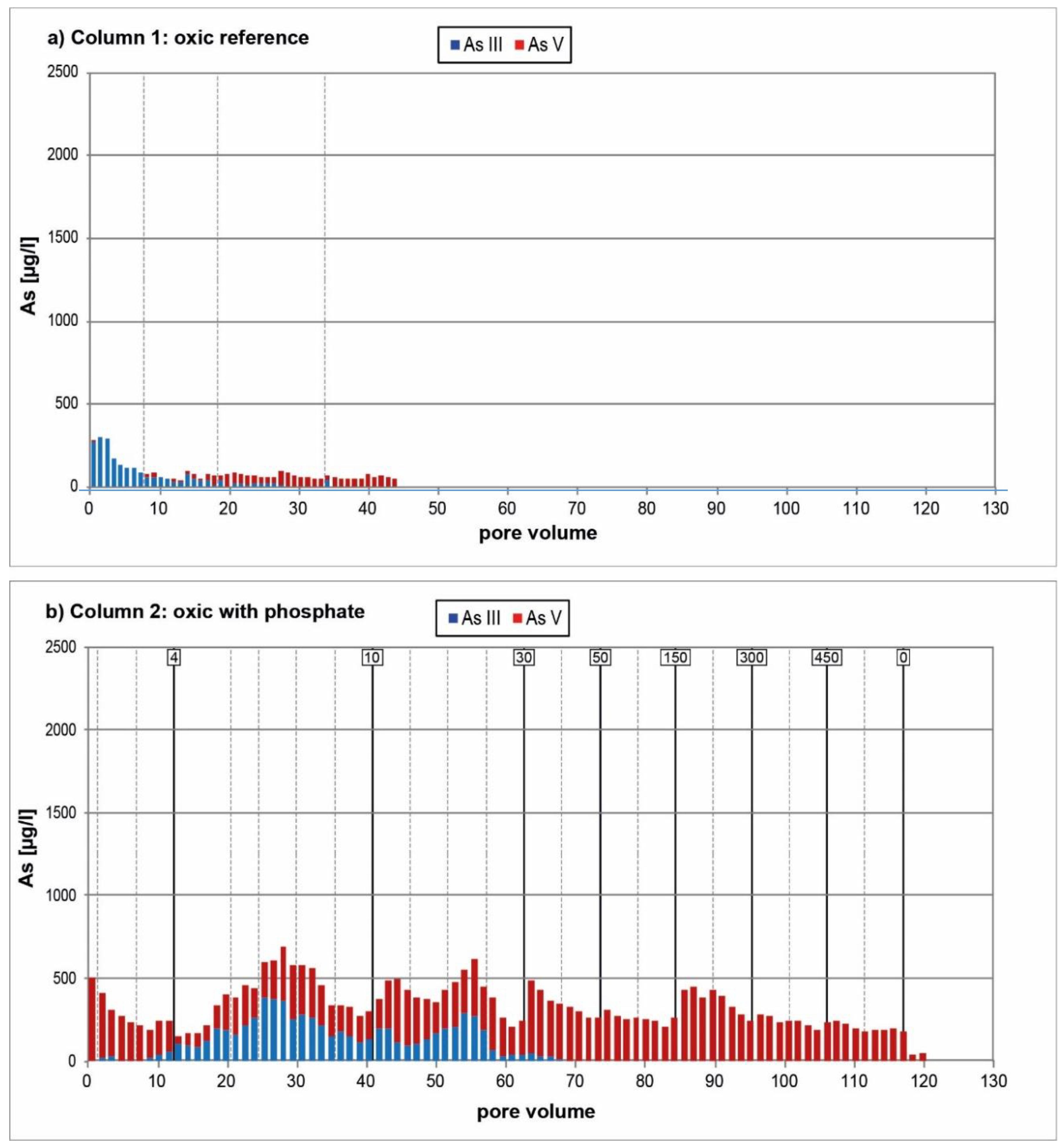
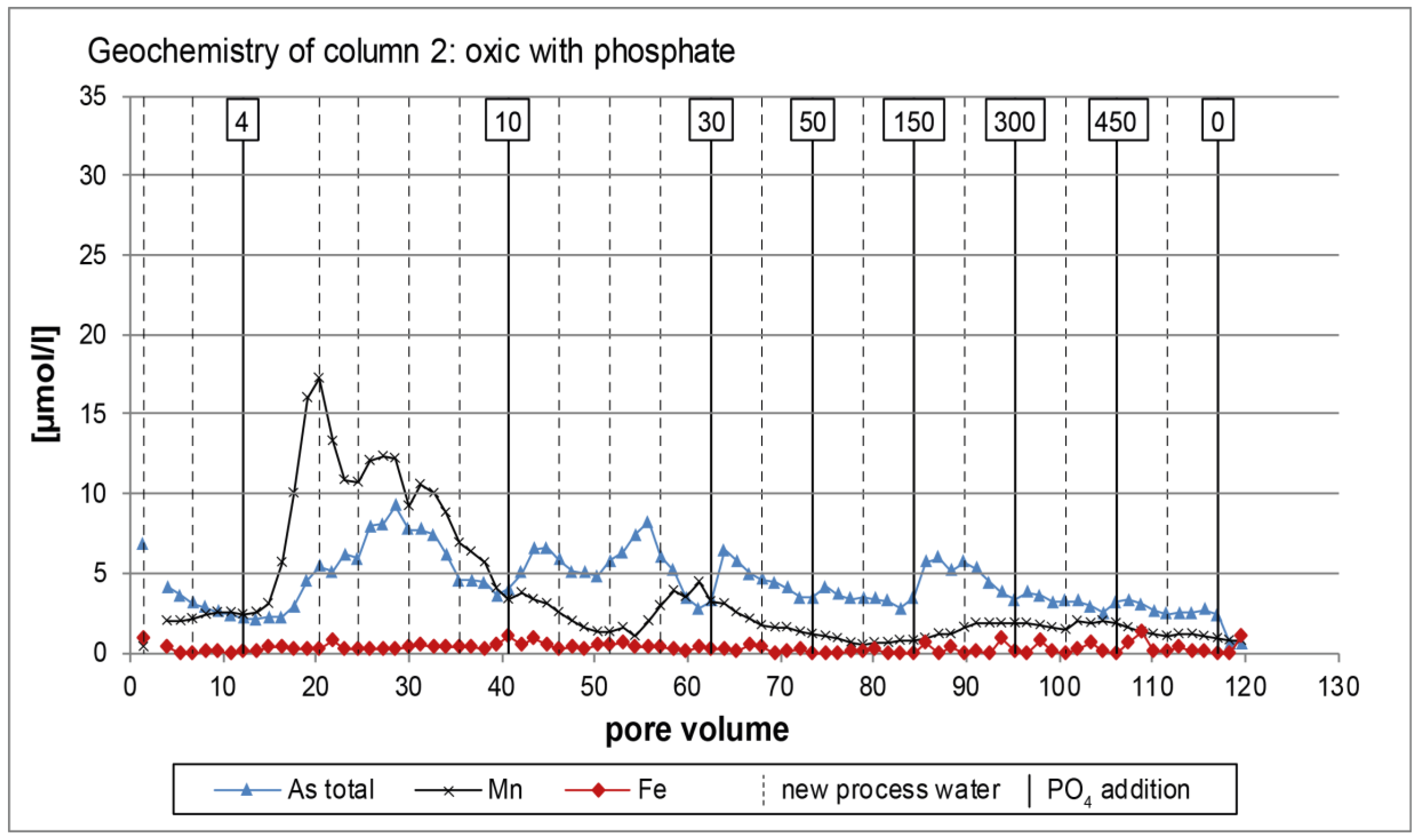
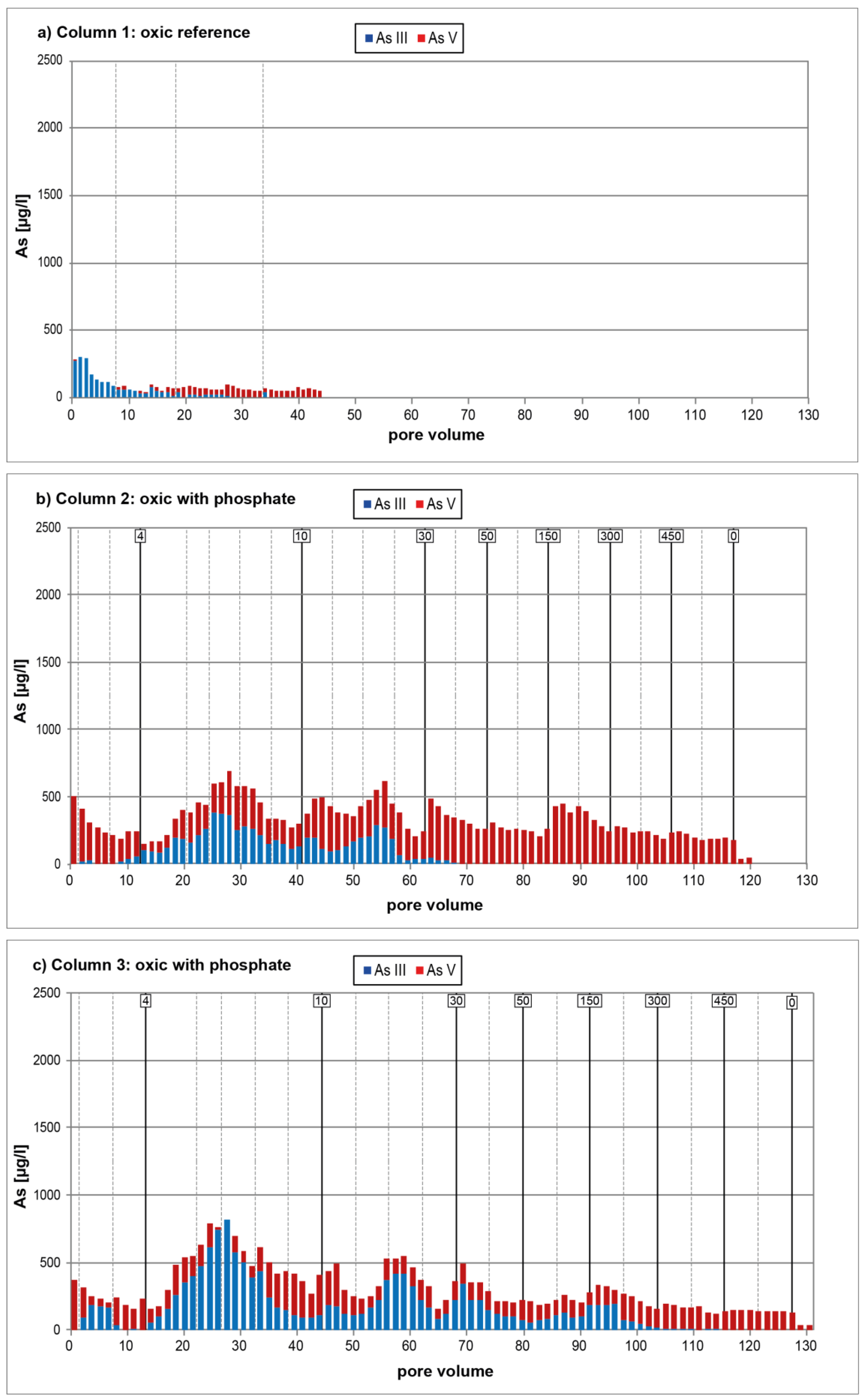
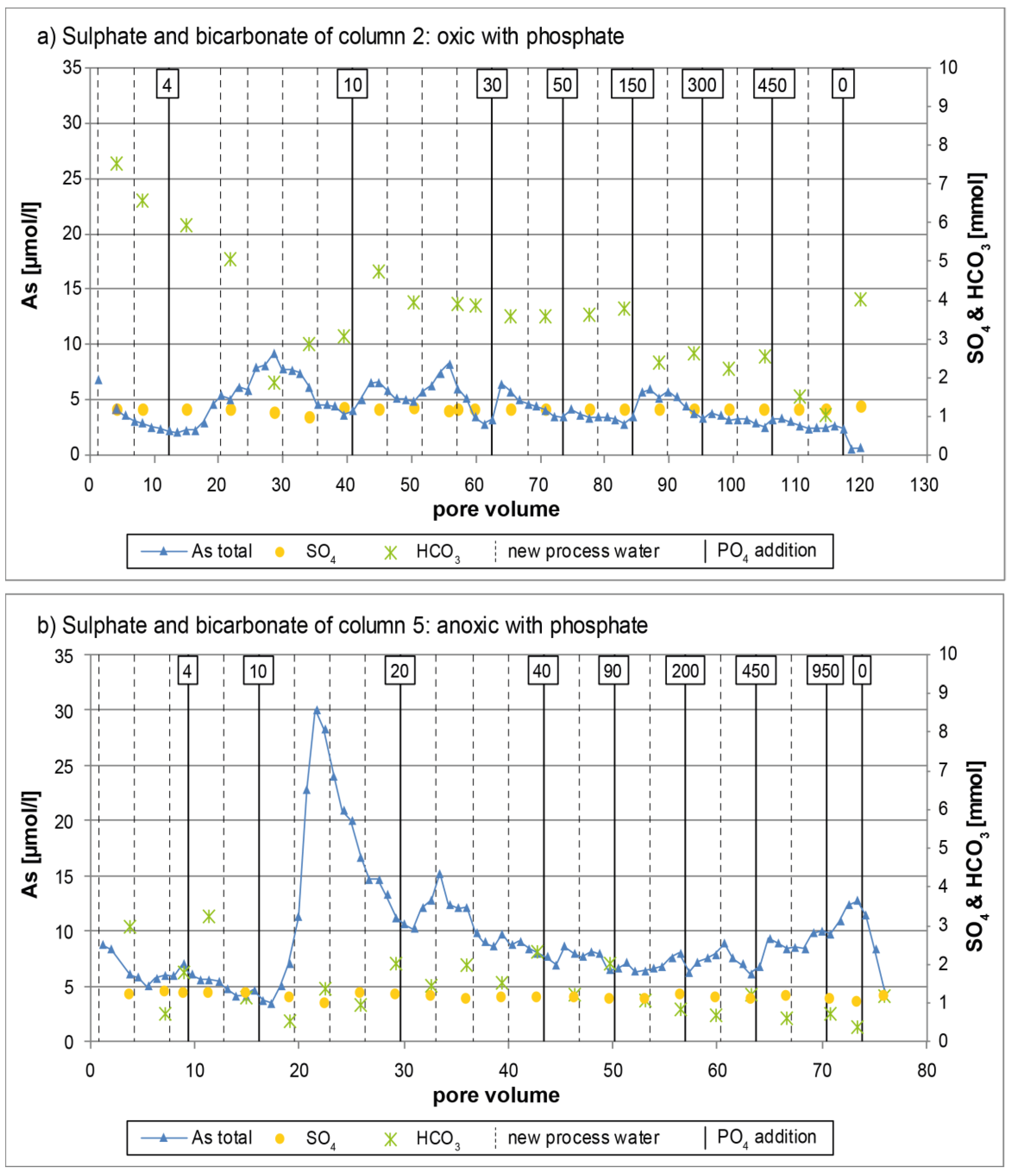
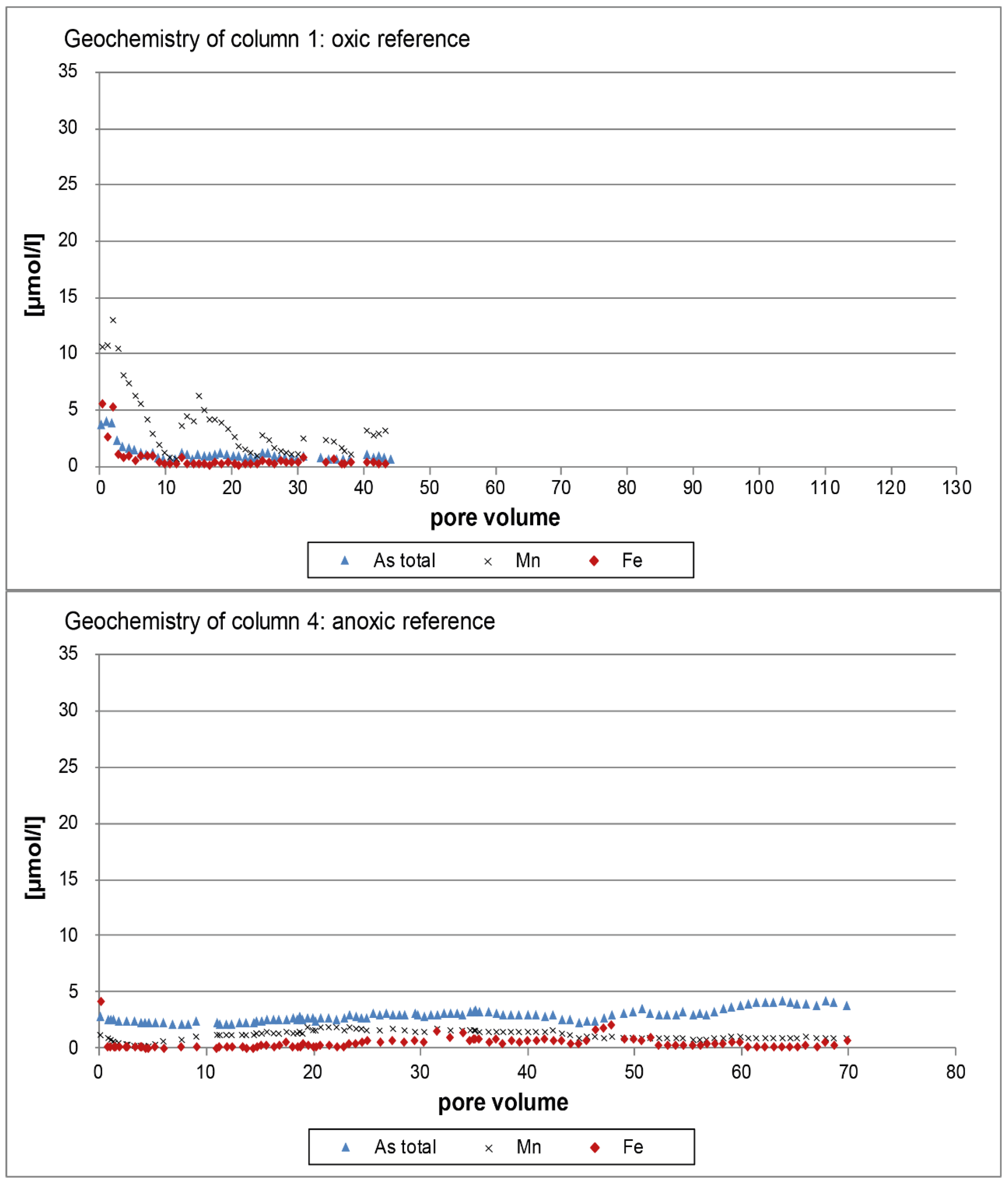
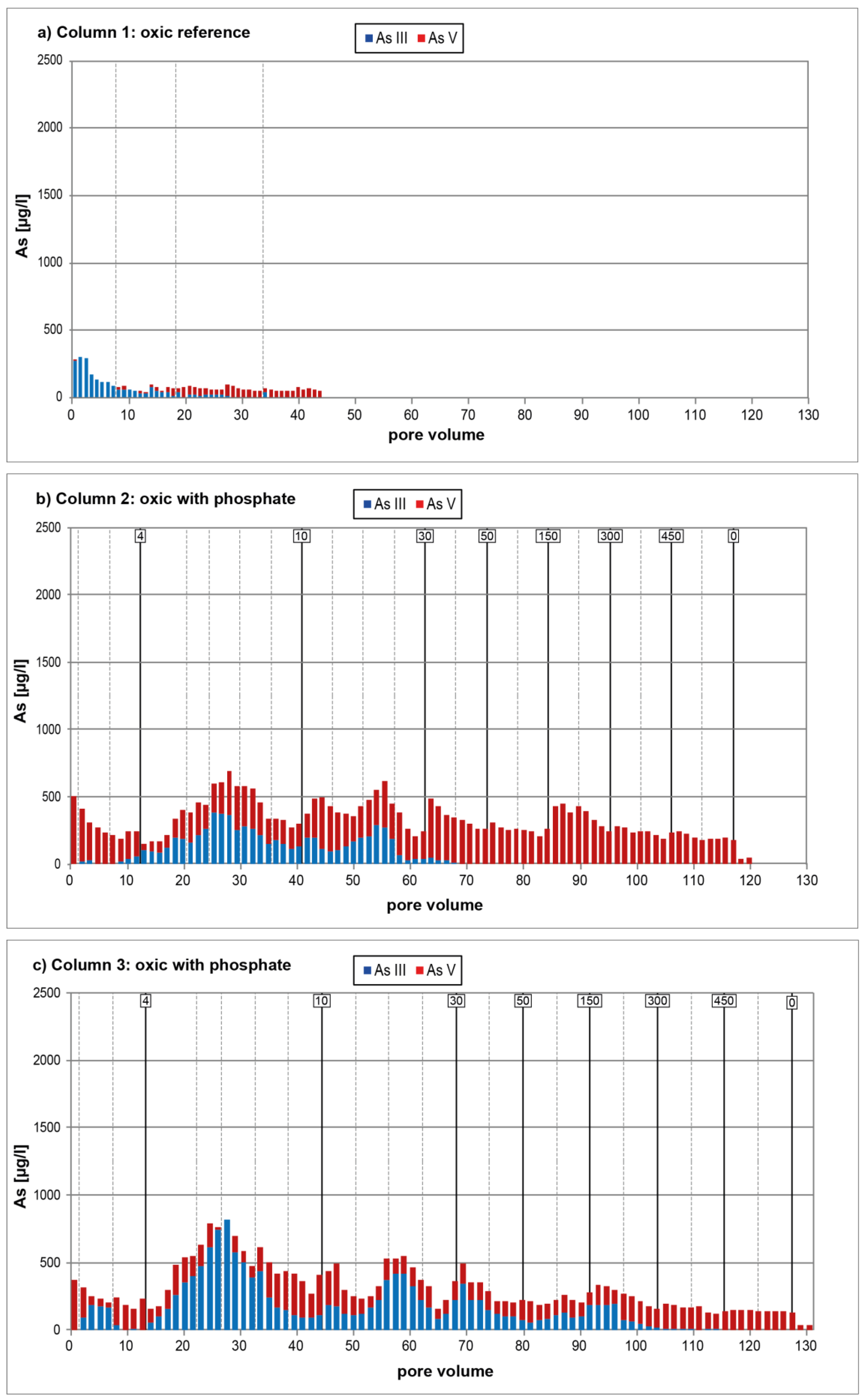
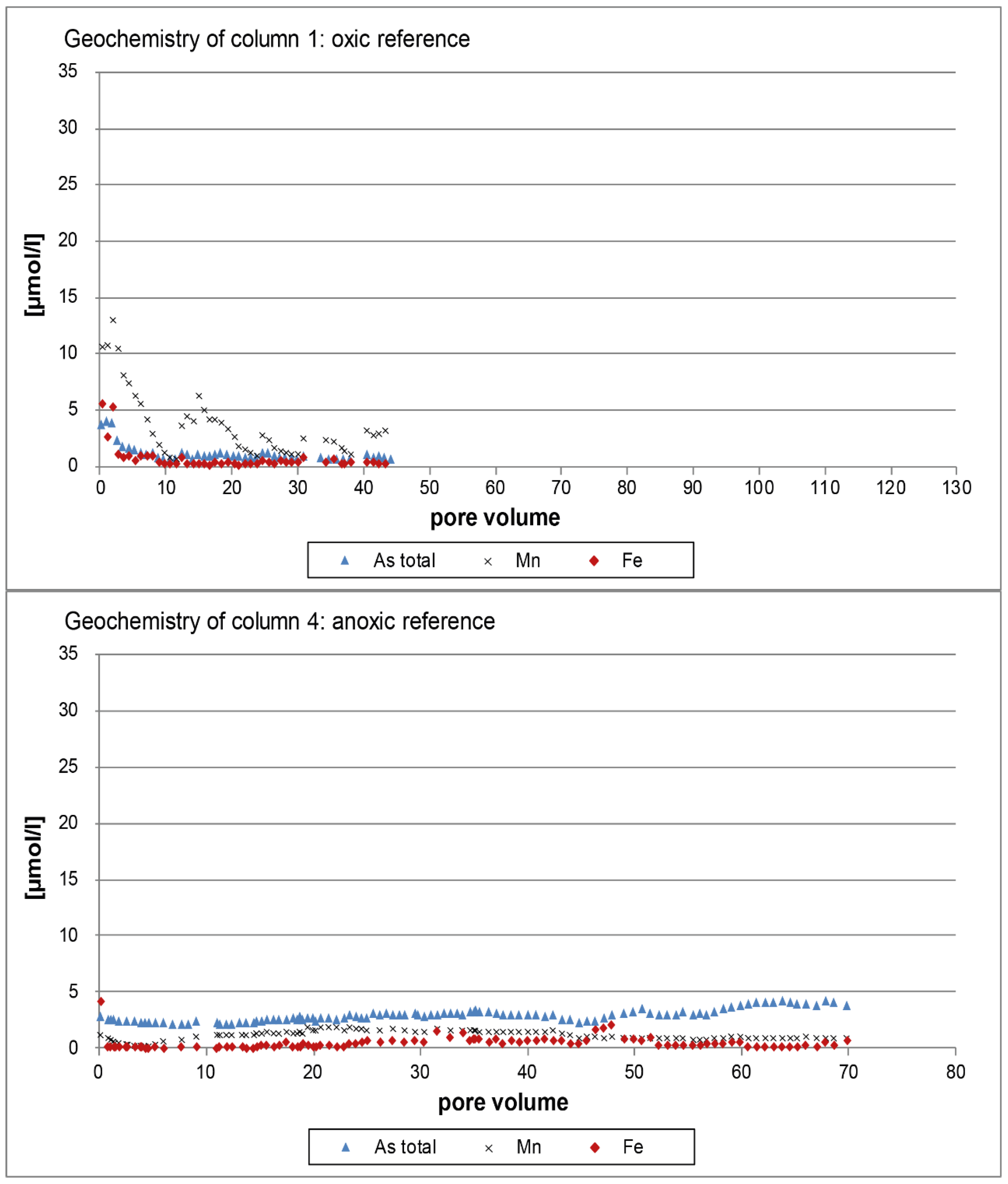
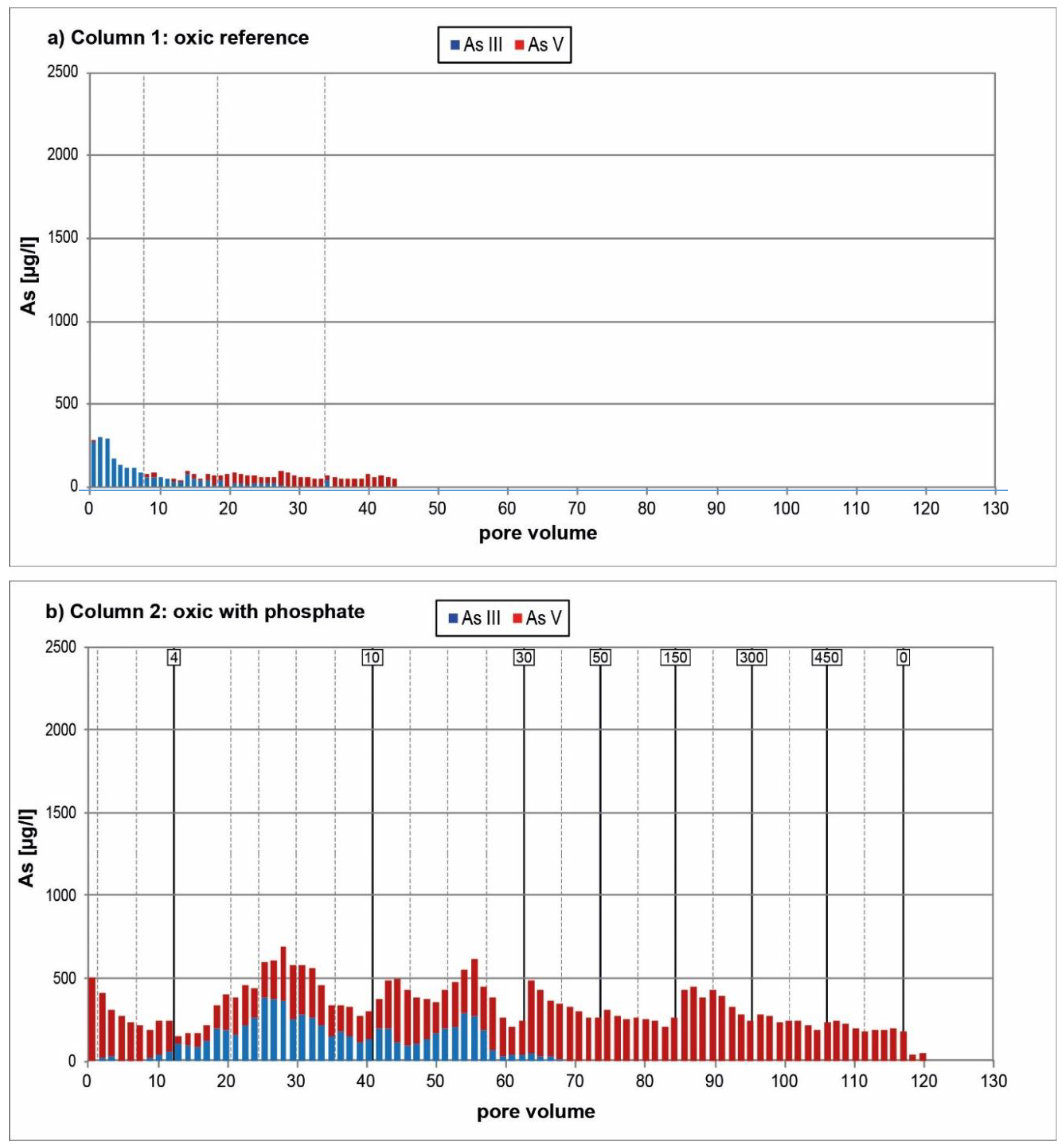
3.1. Oxic Tests
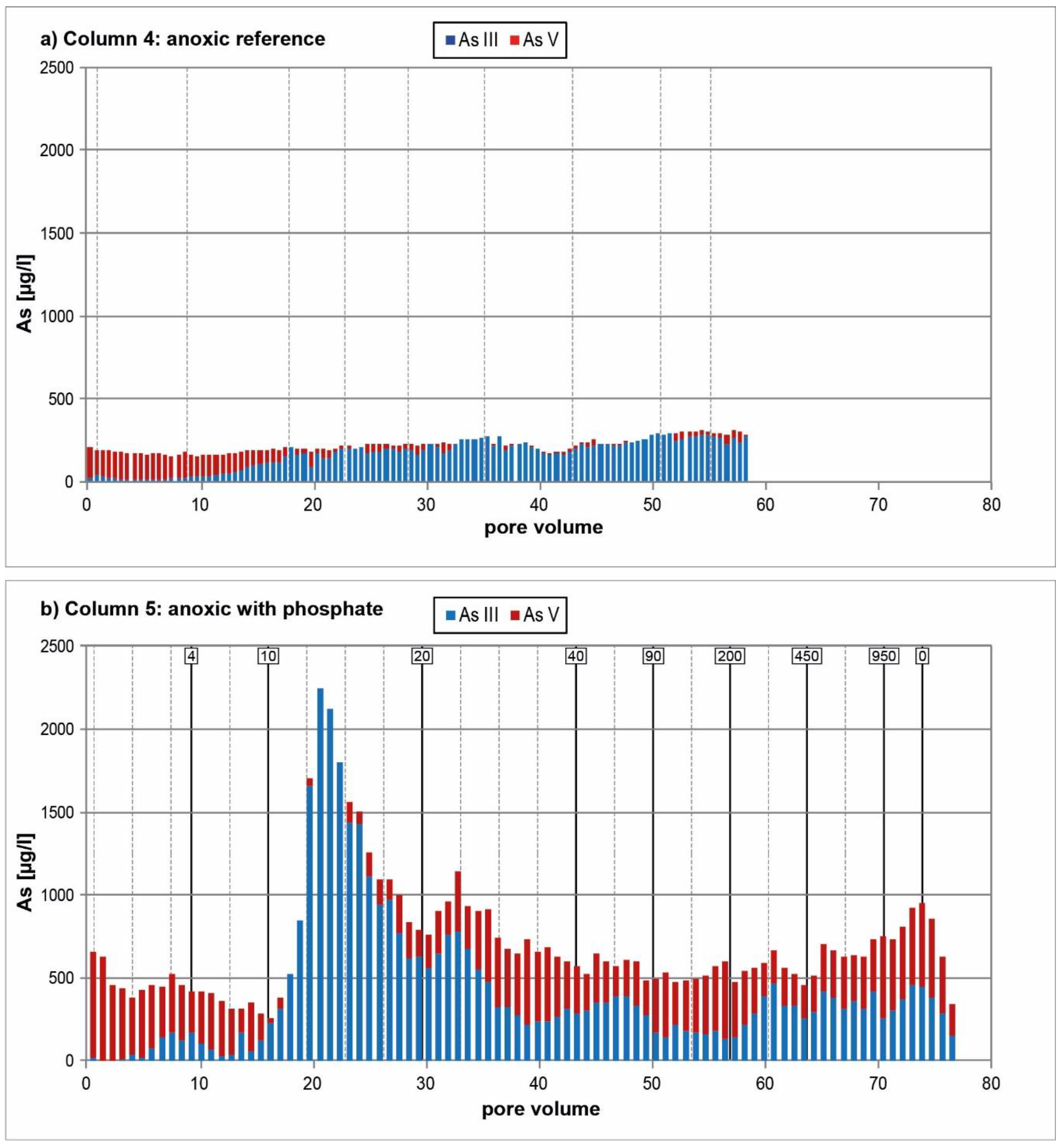
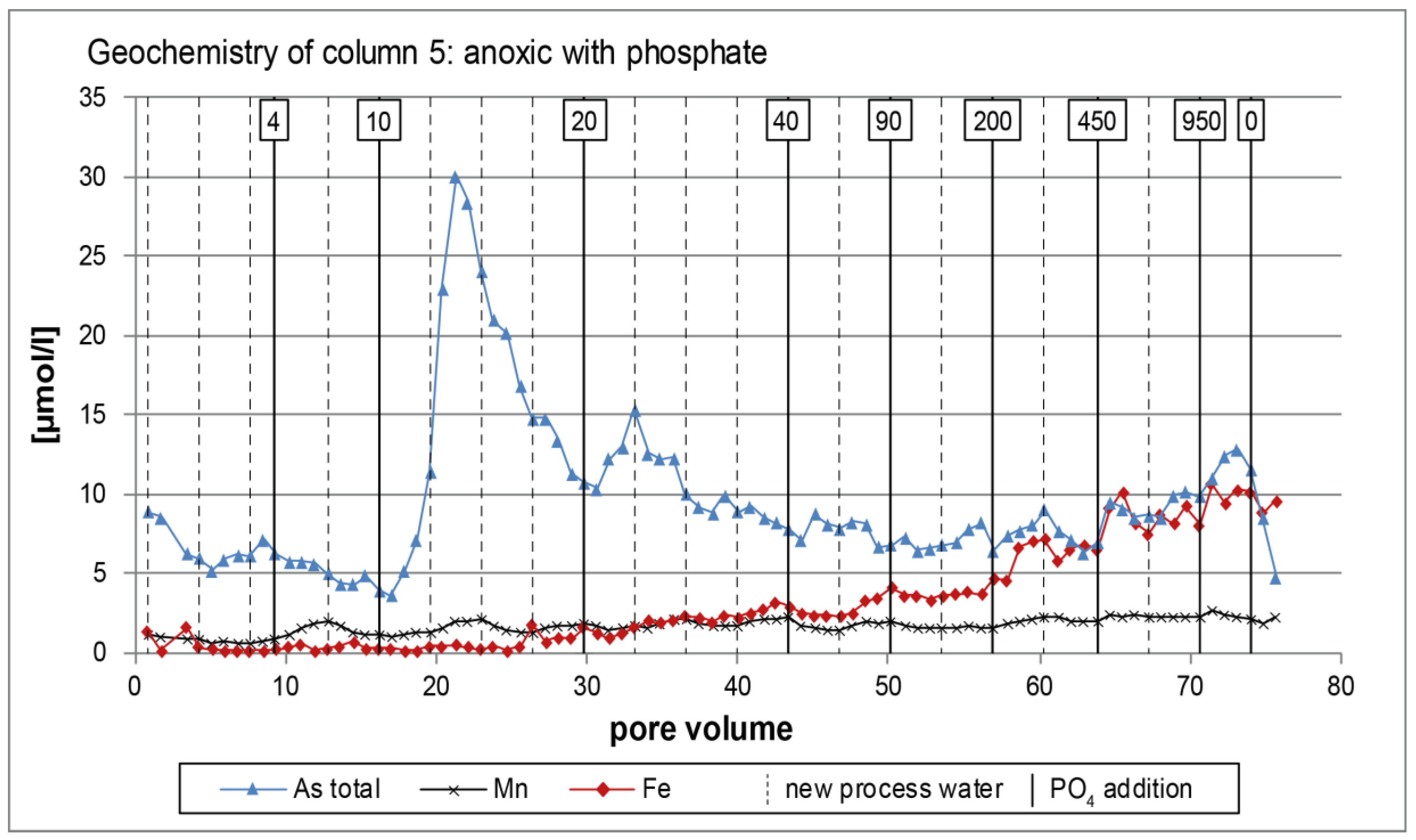
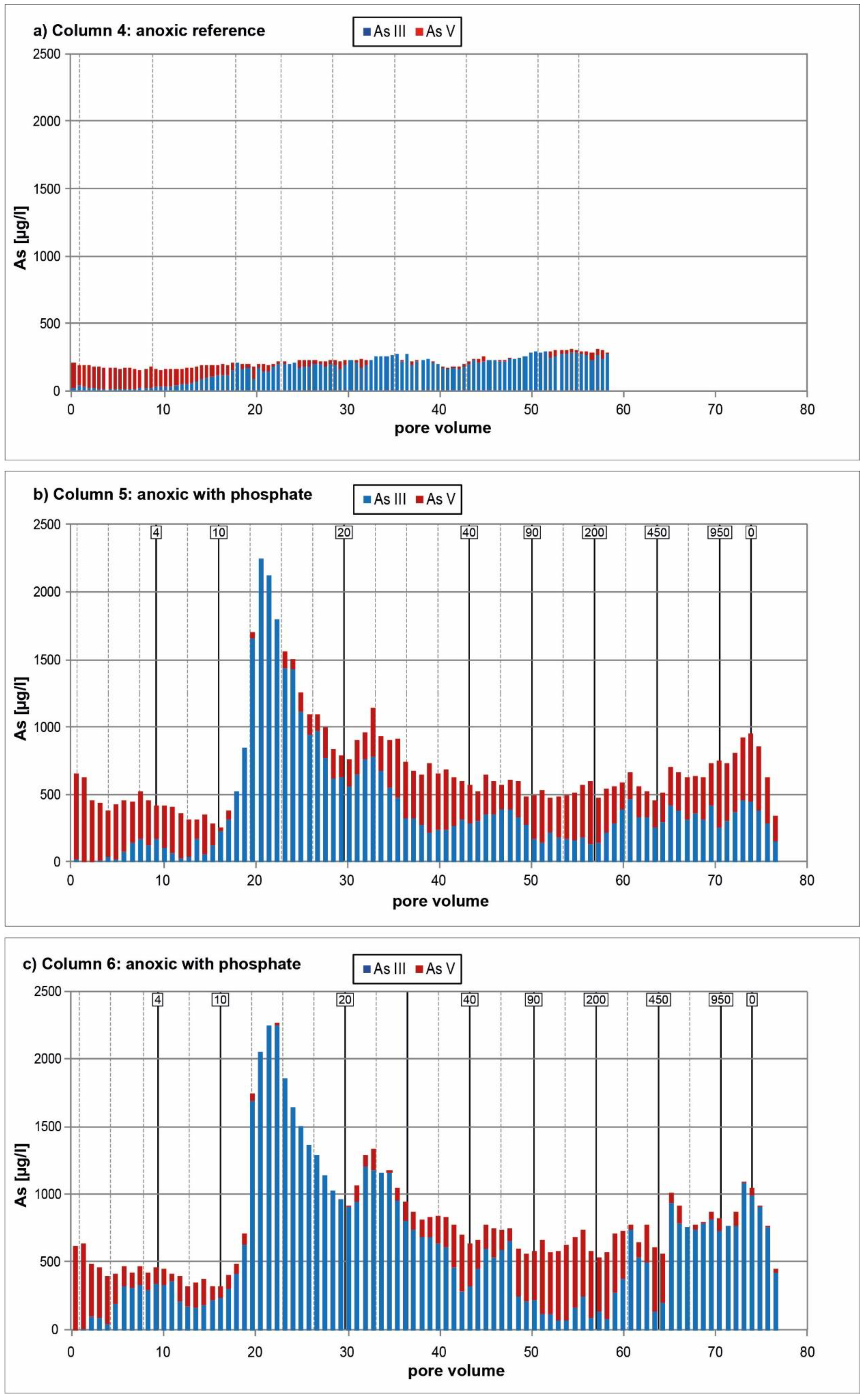
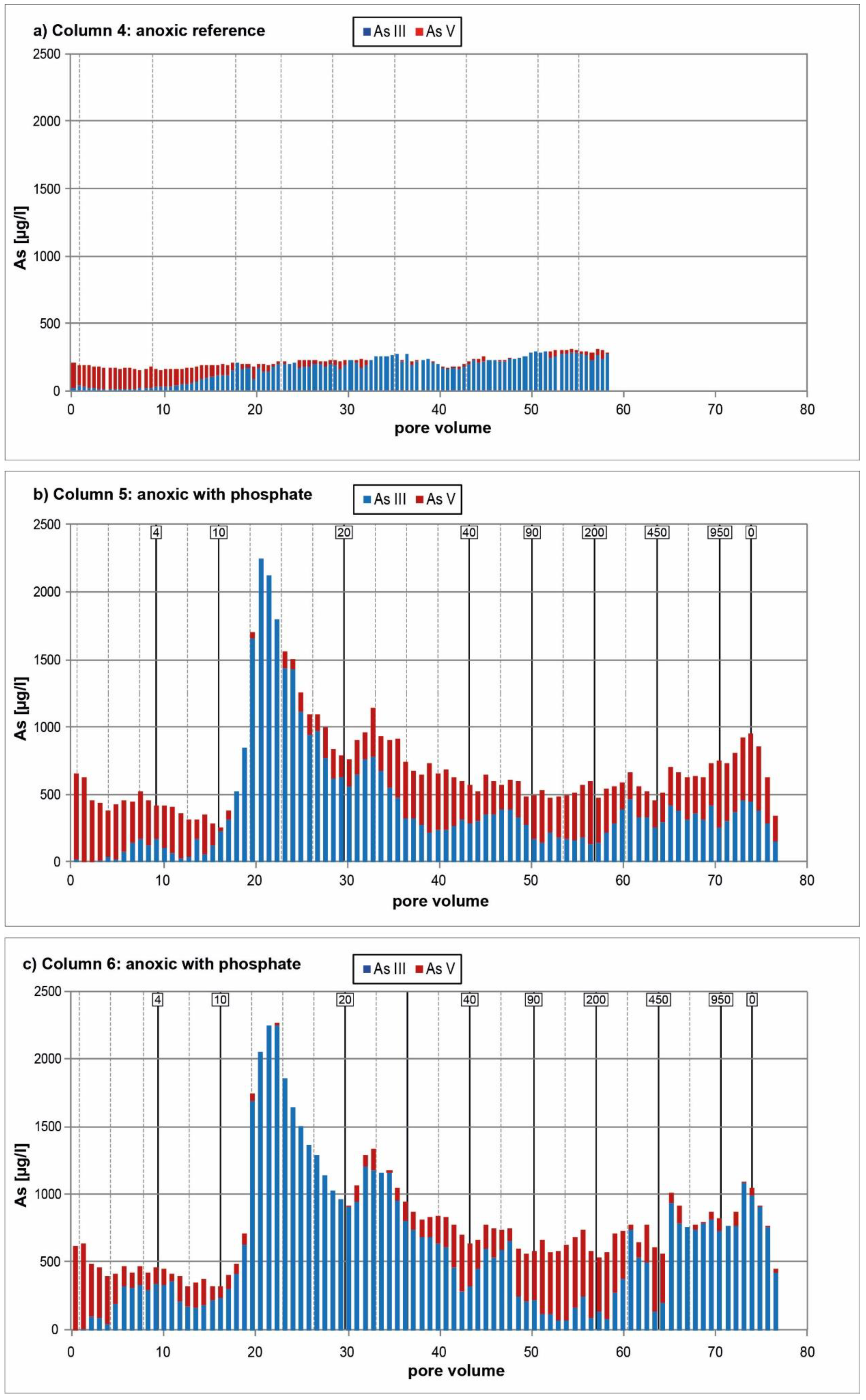
3.2. Anoxic Tests
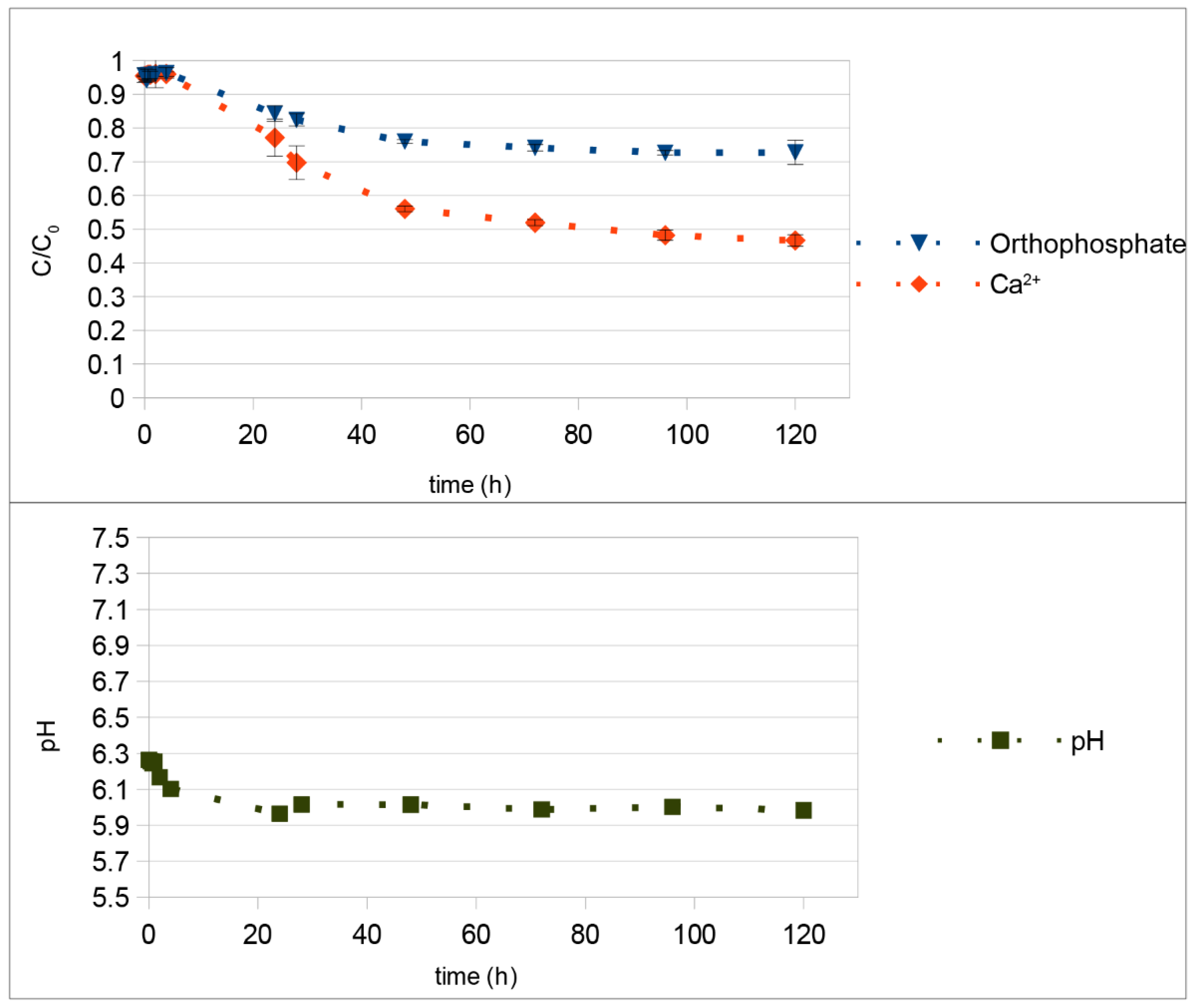
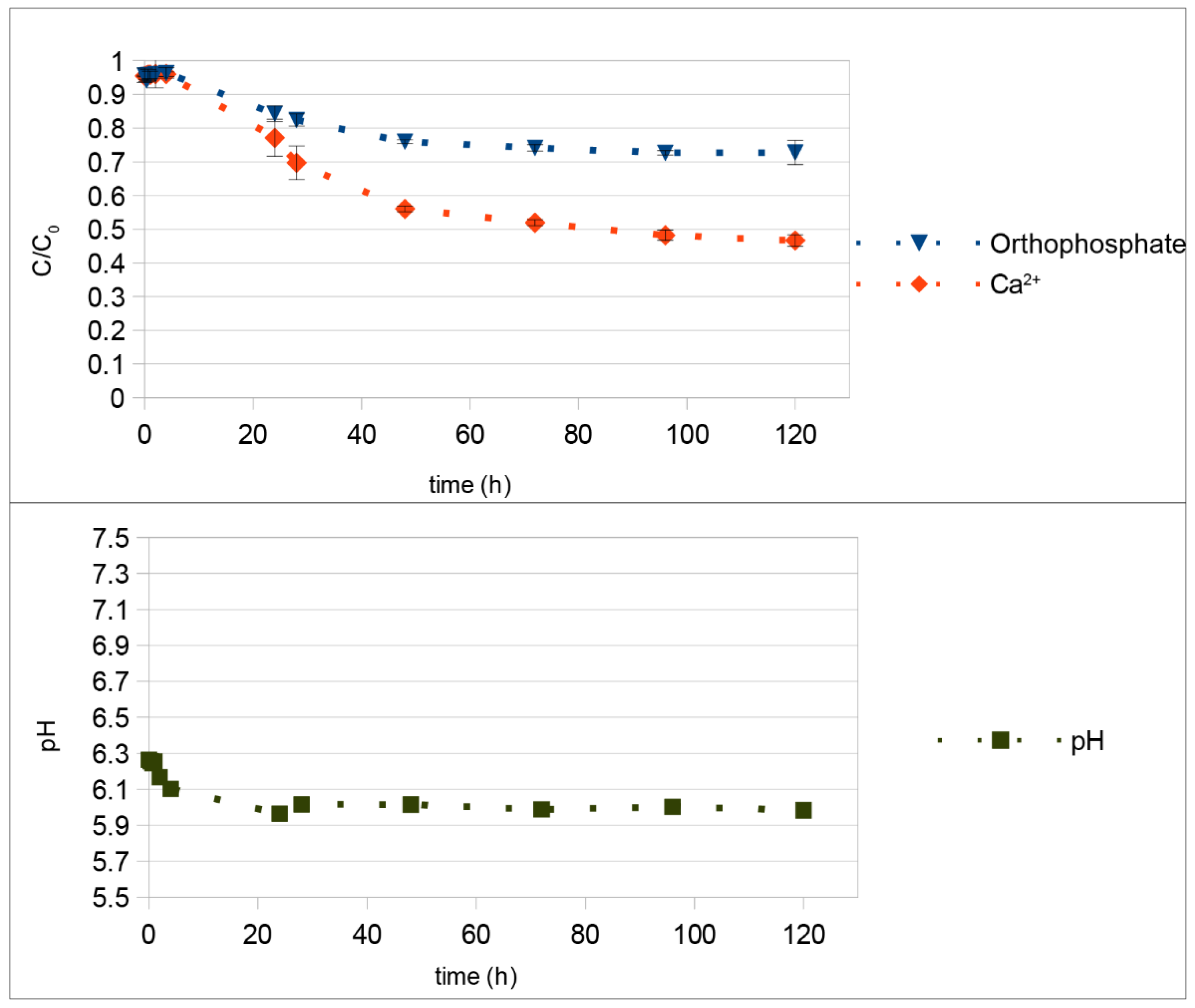
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Field Parameters | |||||||||||
| Sample ID | Sampling Depth | Temperature | Electrical Conductivity | pH Value | Acid Capacity | ORP | Dissolved Oxygen | ||||
| (m bgs) | (°C) | (µS/cm) | (-) | (mmol) | (mV) | ||||||
| GWM 29/1 | 7.80 | 13.3 | 795 | 7.34 | 4.6 | 149 | 1.1 | ||||
| GWM 29/2 | 15.50 | 12.5 | 617 | 7.42 | 4.1 | 49 | 0.2 | ||||
| GWM 29/3 | 22.50 | 13.1 | 646 | 7.39 | 4.1 | 117 | 0.8 | ||||
| Major cations | |||||||||||
| Sample ID | Sampling Depth | Fe | Mn | As | As III | As V | Ca | K | Mg | Na | |
| (m bgs) | (mg/L) | (µg/L) | (µg/L) | (µg/L) | (µg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | ||
| GWM 29/1 | 7.80 | 2.24 | 407 | 667 | 19.0 | 648 | 154 | 9.46 | 9.74 | 18.9 | |
| GWM 29/2 | 15.50 | 1.41 | 462 | 322 | 68.1 | 254 | 120 | 6.29 | 8.51 | 12.1 | |
| GWM 29/3 | 22.50 | 1.21 | 480 | 230 | 26.9 | 203 | 129 | 7.21 | 9.02 | 14.4 | |
| Heavy metals | |||||||||||
| Sample ID | Sampling Depth | Al | Cd | Co | Cr | Cu | Ni | Pb | Sr | Zn | |
| (m bgs) | (µg/L) | (µg/L) | (µg/L) | (µg/L) | (µg/L) | (µg/L) | (µg/L) | (µg/L) | (µg/L) | ||
| GWM 29/1 | 7.80 | <50 | <5 | <5 | <5 | <5 | <5 | <10 | 244 | 60 | |
| GWM 29/2 | 15.50 | <50 | <5 | <5 | <5 | <5 | <5 | <10 | 258 | 29.6 | |
| GWM 29/3 | 22.50 | <50 | <5 | <5 | <5 | <5 | <5 | <10 | 260 | 33.5 | |
| Anions | |||||||||||
| Sample ID | Sampling Depth | NO3 | SO4 | PO4 | F | Cl | Br | ||||
| (m bgs) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | |||||
| GWM 29/1 | 7.80 | 5.4 | 171 | <1 | 0.50 | 20.5 | 0.08 | ||||
| GWM 29/2 | 15.50 | 1.1 | 106 | <1 | 0.20 | 14.9 | <0.01 | ||||
| GWM 29/3 | 22.50 | 1.2 | 126 | <1 | 0.27 | 16.4 | 0.04 | ||||
| Carbon | |||||||||||
| Sample ID | Sampling Depth | HCO3− | TC | TIC | TOC | ||||||
| (m bgs) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | |||||||
| GWM 29/1 | 7.80 | 270 | 58.9 | 53 | 5.87 | ||||||
| GWM 29/2 | 15.50 | 225 | 49.7 | 44.2 | 5.51 | ||||||
| GWM 29/3 | 22.50 | 247 | 52.3 | 48.5 | 3.75 | ||||||
| GWM 29/3 | 22.50 | 247 | 52.3 | 48.5 | 3.75 | ||||||
| Method | Limit of Determination (As) | Reference Material | Standard Deviation |
|---|---|---|---|
| ICP-OES (water) HG-ICP-OES (As(III)) | 50 µg/L 2.50 µg/L | SPW-SW2 TMDA 51.3 3× replication of measurement | 1–15% 1–8% |
| ICP-OES (aqua regia/elutions) | 2.50 mg/kg | BAM U112a, Blank | 2.83–5.35% |
| Photometer (phosphate) | 0.53 mg/L | 6.02% |
| Sample | Depth | As Total | Ca | Mn | Fe | CaCO3 | Carbonate | C Total | Corg (calc.) |
|---|---|---|---|---|---|---|---|---|---|
| (m bgs) | (mg/kg) | (%) | (mg/kg) | (%) | (%) | (as %C) | (%) | (%) | |
| 29-1 + 2 | 1–2 | 35.2 | 0.56 | 151 | 0.63 | n.n. | n.n. | n.n. | n.n. |
| 29-3 | 3 | 12.5 | 1.82 | 105 | 0.45 | 4.30 | 0.52 | 0.64 | 0.12 |
| 29-5 | 5 | 60.5 | 0.72 | 28 | 0.20 | 2.15 | 0.28 | 0.23 | 0 |
| 29-7 | 7 | 25.8 | 1.86 | 56.2 | 0.27 | 6.46 | 0.78 | 0.96 | 0.18 |
| 29-9 | 9 | 28.9 | 3.30 | 68.2 | 0.29 | 7.94 | 0.95 | 1.15 | 0.20 |
| 29-11 | 11 | 33.6 | 2.95 | 85 | 0.41 | 12.2 | 1.47 | 1.36 | 0 |
| 29-13 | 13 | 22.2 | 2.17 | 59.7 | 0.30 | 7.27 | 0.87 | 0.70 | 0 |
| 29-15 | 15 | 25.4 | 2.87 | 66.2 | 0.31 | 6.13 | 0.74 | 0.77 | 0.03 |
| Ion | Concentration (mg/L) | Added as | Source and Quality |
|---|---|---|---|
| Ca2+ | 128 | CaSO42−∙H2O and CaCO3 | MERCK/Grüssing (p.a) |
| Mg2+ | 8 | MgCl22−∙6H2O | AppliChem (p.a) |
| Na+ | 10 | NaHCO3 | Grüssing (p.a) |
| K+ | 5 | KHCO3 | Theoretikum (p.a) |
| NO3− | 5 * | NaNO3 | Grüssing (p.a) |
| SO42− | 120 | CaSO42+ | MERCK (p.a) |
| Cl− | 23 | MgCl22−∙6H2O | AppliChem (p.a) |
| Column | Diameter (m) | Length (m) | Redox Condition | Discharge (L/day) | Phosphate Application |
|---|---|---|---|---|---|
| 1 | 0.005 | 0.3 | Oxic | 0.18 | none |
| 2 | 0.004 | 0.3 | Oxic | 0.22 | via bypass into columns entry |
| 3 | 0.004 | 0.3 | Oxic | 0.22 | |
| 4 | 0.005 | 0.3 | Anoxic | 0.18 | none |
| 5 | 0.005 | 0.3 | Anoxic | 0.18 | added to process water after degassing |
| 6 | 0.005 | 0.3 | Anoxic | 0.18 |
| Sediment Samples | |||
| Aqua regia extraction | As (tot) and cations | 1 g sample with 3 mL 65%, HNO3 and 9 mL 37% HCl, filled up to 50 mL and filtered through folded filter | (HG)-ICP-OES (VISTA-MPX CCD Simultaneous; Varian) |
| Water elution | As (III) and As (tot) | 3.5 g sample add. 40 mL distilled water, filtered by 0.45 µm and stabilized at pH 1 with 6 M HCl | |
| Phosphate elution | As (III) and As (tot) | 3.5 g sample add. 40 mL 1 M NaH2PO4, filtered by 0.45 µm and stabilized at pH 1 with 6 M HCl | |
| Solid sample | TC | SC-Analyzer (SC 144R; Leco) | |
| Water Samples | |||
| Inflow solution and column outflow | As (III), As (tot) and cations | stabilized at pH 1 with 6 M HCl | (HG)-ICP-OES (VISTA-MPX CCD Simultaneous; Varian) |
| Anions | IC (Dionex DX-120; ThermoFisher Scientific) | ||
| TIC/TC | TOC (TOC-VCSN; Shimadzu) | ||
| Phosphate | Molybdenum-blue method | UV-VIS Spectrophotometer; (Specord 50; Analytik Jena) | |
| Tracer test | Bromide | Ion selective electrode (Br 500; WTW); IC (Dionex DX-120; ThermoFisher Scientific) | |
| Average SO4 (mg/L) | |||||
|---|---|---|---|---|---|
| Replicated Water | Column Outflow | Deviation SO4 (%) | |||
| Column | Planned | Measured * | |||
| 1 | reference oxic | 120 | 125 | 129 | 3.25 |
| 2 | oxic + PO4 | 120 | 111 | −7.72 | |
| 3 | oxic + PO4 | 115 | −4.71 | ||
| 4 | reference anoxic | 120 | 121 | 0.17 | |
| 5 | anoxic + PO4 | 121 | 113 | −6.89 | |
| 6 | anoxic + PO4 | 114 | −6.51 | ||


References
- Eljamal, O.; Sasaki, K.; Tsuruyama, S.; Hirajima, T. Kinetic Model of Arsenic Sorption onto Zero-Valent Iron (ZVI). Water Qual. Expo. Health 2011, 2, 125–132. [Google Scholar] [CrossRef]
- Bissen, M.; Frimmel, F.H. Arsenic—A Review. Part II: Oxidation of Arsenic and its Removal in Water Treatment. Acta Hydrochim. Hydrobiol. 2003, 31, 97–107. [Google Scholar] [CrossRef]
- Kuhlmeier, P.D. Sorption and desorption of arsenic from sandy soils: Column studies. J. Soil Contam. 1997, 6, 21–36. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef]
- Guo, H.; Stüben, D.; Berner, Z.; Yu, Q. Characteristics of arsenic adsorption from aqueous solution: Effect of arsenic species and natural adsorbents. Appl. Geochem. 2009, 24, 657–663. [Google Scholar] [CrossRef]
- Bowell, R.J. Sorption of arsenic by iron oxides and oxyhydroxides in soils. Appl. Geochem. 1994, 9, 279–286. [Google Scholar] [CrossRef]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modeling: Hydrous Ferric Oxide; Wiley-Interscience: New York, NY, USA, 1990. [Google Scholar]
- Sø, H.U.; Postma, D.; Jakobsen, R.; Larsen, F. Sorption of phosphate onto calcite; results from batch experiments and surface complexation modeling. Geochim. Cosmochim. Acta 2011, 75, 2911–2923. [Google Scholar] [CrossRef]
- Biswas, A.; Majumder, S.; Neidhardt, H.; Halder, D.; Bhowmick, S.; Mukherjee-Goswami, A.; Kundu, A.; Saha, D.; Berner, Z.; Chatterjee, D. Groundwater chemistry and redox processes: Depth dependent arsenic release mechanism. Appl. Geochem. 2011, 26, 516–525. [Google Scholar] [CrossRef]
- Saunders, J.A.; Lee, M.-K.; Shamsudduha, M.; Dhakal, P.; Uddin, A.; Chowdury, M.T.; Ahmed, K.M. Geochemistry and mineralogy of arsenic in (natural) anaerobic groundwaters. Appl. Geochem. 2008, 23, 3205–3214. [Google Scholar] [CrossRef]
- Rochette, E.A.; Bostick, B.C.; Li, G.; Fendorf, S. Kinetics of Arsenate Reduction by Dissolved Sulfide. Environ. Sci. Technol. 2000, 34, 4714–4720. [Google Scholar] [CrossRef]
- Bostick, B.C.; Fendorf, S. Arsenite sorption on troilite (FeS) and pyrite (FeS2). Geochim. Cosmochim. Acta 2003, 67, 909–921. [Google Scholar] [CrossRef]
- Bundschuh, J.; Hollander, H.; Lena, M. In-Situ Remediation of Arsenic-Contaminated Sites, 1st ed.; Bundschuh, J., Hollander, H., Lena, M., Eds.; CRC Press, Taylor & Francis Group: London, UK, 2014. [Google Scholar]
- Matthess, G. In Situ Treatment of Arsenic Contaminated Groundwater. In Studies in Environmental Science; Elsevier: Amsterdam, The Netherlands, 1981; Volume 17, pp. 291–296. [Google Scholar] [CrossRef]
- Rott, U.; Friedle, M. Eco-friendly and cost-efficient removal of arsenic, iron and manganese by means of subterranean ground-water treatment. Water Supply 2000, 18, 632–636. [Google Scholar]
- Kim, M.-J.; Nriagu, J. Oxidation of arsenite in groundwater using ozone and oxygen. Sci. Total Environ. 2000, 247, 71–79. [Google Scholar] [CrossRef]
- Krüger, T.; Holländer, H.M.; Stummeyer, J.; Harazim, B.; Boochs, P.-W.; Billib, M. In-situ immobilization of arsenic in the subsurface on an anthropogenic contaminated site. In In-Situ Remediation of Arsenic-Contaminated Sites; Bundschuh, J., Holländer, H.M., Ma, L.Q., Eds.; CRC Press, Taylor & Francis Group: London, UK, 2014. [Google Scholar]
- Luong, V.T.; Cañas Kurz, E.E.; Hellriegel, U.; Luu, T.L.; Hoinkis, J.; Bundschuh, J. Iron-based subsurface arsenic removal technologies by aeration: A review of the current state and future prospects. Water Res. 2018, 133, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Onstott, T.C.; Chan, E.; Polizzotto, M.L.; Lanzon, J.; DeFlaun, M.F. Precipitation of arsenic under sulfate reducing conditions and subsequent leaching under aerobic conditions. Appl. Geochem. 2011, 26, 269–285. [Google Scholar] [CrossRef]
- Gemeinhardt, C.; Müller, S.; Weigand, H.; Marb, C. Chemical immobilisation of arsenic in contaminated soils using iron(II)sulphate—advantages and pitfalls. Water Air Soil Pollut. Focus 2006, 6, 281–297. [Google Scholar] [CrossRef]
- Köber, R.; Daus, B.; Ebert, M.; Mattusch, J.; Welter, E.; Dahmke, A. Compost-Based Permeable Reactive Barriers for the Source Treatment of Arsenic Contaminations in Aquifers: Column Studies and Solid-Phase Investigations. Environ. Sci. Technol. 2005, 39, 7650–7655. [Google Scholar] [CrossRef] [PubMed]
- Klaas, N.; Braun, J.; Mackenberg, S. Wissenschaftlicher Bericht Nr. 2007/06 (VEG 23) Entwicklung Eines Immobilisierungsverfahrens für Schwermetalle unter Nutzung des Geogenen Sulfatgehaltes im Grundwasser; VEGAS: Stuttgart, Germany, 2007. [Google Scholar]
- Zeng, H.; Fisher, B.; Giammar, D.E. Individual and Competitive Adsorption of Arsenate and Phosphate to a High-Surface-Area Iron Oxide-Based Sorbent. Environ. Sci. Technol. 2008, 42, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Kent, D.B. The influence of groundwater chemistry on arsenic concentrations and speciation in a quartz sand and gravel aquifer. Geochem. Trans. 2004, 5, 1–12. [Google Scholar] [CrossRef]
- Hongshao, Z.; Stanforth, R. Competitive Adsorption of Phosphate and Arsenate on Goethite. Environ. Sci. Technol. 2001, 35, 4753–4757. [Google Scholar] [CrossRef] [PubMed]
- Luengo, C.; Brigante, M.; Avena, M. Adsorption kinetics of phosphate and arsenate on goethite. A comparative study. J. Colloid Interface Sci. 2007, 311, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.A.; Goldberg, S. Modeling Competitive Adsorption of Arsenate with Phosphate and Molybdate on Oxide Minerals. Soil Sci. Soc. Am. J. 1996, 60, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, F.; Benvenuti, M.; Costagliola, P.; Di Benedetto, F.; Lattanzi, P.; Meneghini, C.; Romanelli, M.; Valenzano, L. Arsenic uptake by natural calcite: An XAS study. Geochim. Cosmochim. Acta 2011, 75, 3011–3023. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Tanaka, K.; Takahashi, Y. Differences in the immobilization of arsenite and arsenate by calcite. Geochim. Cosmochim. Acta 2012, 91, 202–219. [Google Scholar] [CrossRef]
- Sø, H.U.; Postma, D.; Jakobsen, R.; Larsen, F. Sorption and desorption of arsenate and arsenite on calcite. Geochim. Cosmochim. Acta 2008, 72, 5871–5884. [Google Scholar] [CrossRef]
- Pigna, M.; Krishnamurti, G.S.R.; Violante, A. Kinetics of Arsenate Sorption–Desorption from Metal Oxides. Soil Sci. Soc. Am. J. 2006, 70, 2017–2027. [Google Scholar] [CrossRef]
- Alam, M.G.; Tokunaga, S.; Maekawa, T. Extraction of arsenic in a synthetic arsenic-contaminated soil using phosphate. Chemosphere 2001, 43, 1035–1041. [Google Scholar] [CrossRef]
- Woolson, E.A.; Axley, J.H.; Kearney, P.C. The Chemistry and Phytotoxicity of Arsenic in Soils: II. Effects of Time and Phosphorus. Soil Sci. Soc. Am. J. 1973, 37, 254–259. [Google Scholar] [CrossRef]
- Wovkulich, K.; Mailloux, B.J.; Lacko, A.; Keimowitz, A.R.; Stute, M.; Simpson, H.J.; Chillrud, S.N. Chemical treatments for mobilizing arsenic from contaminated aquifer solids to accelerate remediation. Appl. Geochem. 2010, 25, 1500–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, S.E.; Strawn, D.G.; Sparks, D.L. Residence Time Effects on Arsenate Adsorption/Desorption Mechanisms on Goethite. Soil Sci. Soc. Am. J. 2001, 65, 67. [Google Scholar] [CrossRef] [Green Version]
- Maier, M. Untersuchungen zum Reaktiven Transport von Arsen im Grundwasserleiter: Prozessstudie und Entwicklung Einer Neuartigen Sanierungsmethode an Einem Altstandort in Hessen; Ruprecht-Karls-Universität Heidelberg: Heidelberg, Germany, 2014. [Google Scholar] [CrossRef]
- Maier, M.V. Insitu-Mobilisierung von Arsen im Grundwasser. In Handbuch Altlastensanierung und Flächenmanagement (HdA); Franziskus, V., Altenbockum, M., Gerhold, T., Eds.; Hütig Jehle Rehm Verlag: Heidelberg, Germany, 2016; ISBN 978-3-8073-2397-8. [Google Scholar]
- Keon, N.E.; Swartz, C.H.; Brabander, D.J.; Harvey, C.; Hemond, H.F. Validation of an arsenic sequential extraction method for evaluating mobility in sediments. Environ. Sci. Technol. 2001, 35, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Rüde, T.R. Beiträge zur Geochemie des Arsens; Puchelt, H., Ed.; Karlsruher Geochemische Hefte: Karlsruhe, Germany, 1996; Volume 10. [Google Scholar]
- Höhn, R.; Isenbeck-Schröter, M.; Kent, D.B.; Davis, J.A.; Jakobsen, R.; Jann, S.; Niedan, V.; Scholz, C.; Stadler, S.; Tretner, A. Tracer test with As(V) under variable redox conditions controlling arsenic transport in the presence of elevated ferrous iron concentrations. J. Contam. Hydrol. 2006, 88, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution, 2nd ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA; London, UK; New York, NY, USA, 2005. [Google Scholar]
- Kinzelbach, W. Numerische Methoden zur Modellierung des Transports von Schadstoffen im Grundwasser; 2. Aufl.; Oldenbourg: München, Germany; Wien, Austria, 1987. [Google Scholar]
- Isenbeck-Schröter, M. Transportverhalten von Schwermetallkationen und Oxoanionen–Laborversuche in Säulen und ihre Modellierung; Berichte Nr. 67; Fachbereich Geowissenschaften: Bremen, Germany, 1995. [Google Scholar]
- Macur, R.E.; Jackson, C.R.; Botero, L.M.; Mcdermott, T.R.; Inskeep, W.P. Bacterial Populations Associated with the Oxidation and Reduction of Arsenic in an Unsaturated Soil. Environ. Sci. Technol. 2004, 38, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.M.; Malasarn, D.; Saltikov, C.W.; Newman, D.K.; Hering, J.G. Simultaneous Microbial Reduction of Iron(III) and Arsenic(V) in Suspensions of Hydrous Ferric Oxide. Environ. Sci. Technol. 2006, 40, 5950–5955. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-H.; Voegelin, A.; Pombo, S.A.; Lazzaro, A.; Zeyer, J.; Kretzschmar, R. Influence of Arsenate Adsorption to Ferrihydrite, Goethite, and Boehmite on the Kinetics of Arsenate Reduction by Shewanella putrefaciens strain CN-32. Environ. Sci. Technol. 2011, 45, 7701–7709. [Google Scholar] [CrossRef] [PubMed]
- Dhar, R.K.; Zheng, Y.; Saltikov, C.W.; Radloff, K.A.; Mailloux, B.J.; Ahmed, K.M.; van Geen, A. Microbes Enhance Mobility of Arsenic in Pleistocene Aquifer Sand from Bangladesh. Environ. Sci. Technol. 2011, 45, 2648–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Jia, Y.; Wang, S.; Pan, R.; Zhang, X. Bacterial reduction and release of adsorbed arsenate on Fe(III)-, Al- and coprecipitated Fe(III)/Al-hydroxides. J. Environ. Sci. 2012, 24, 440–448. [Google Scholar] [CrossRef]
- Slaughter, D.C.; Macur, R.E.; Inskeep, W.P. Inhibition of microbial arsenate reduction by phosphate. Microbiol. Res. 2012, 167, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.V.; Isenbeck-Schröter, M.; Klose, L.B.; Ritter, S.M.; Scholz, C. In situ-mobilization of arsenic in groundwater–an innovative remediation approach? Procedia Earth Planet. Sci. 2017, 17, 452–455. [Google Scholar] [CrossRef]
- Tretner, A. Sorptions- und Redoxprozesse von Arsen an Oxidischen Oberflächen–Experimentelle Untersuchungen; Ruprecht-Karls-Universität Heidelberg: Heidelberg, Germany, 2002. [Google Scholar] [CrossRef]
- Burger, J. Untersuchungen zur Fällungskinetik von Calciumphosphaten Batchversuche mit Grundwasser; Ruprecht-Karls-University Heidelberg: Heidelberg, Germany, 2014. [Google Scholar]
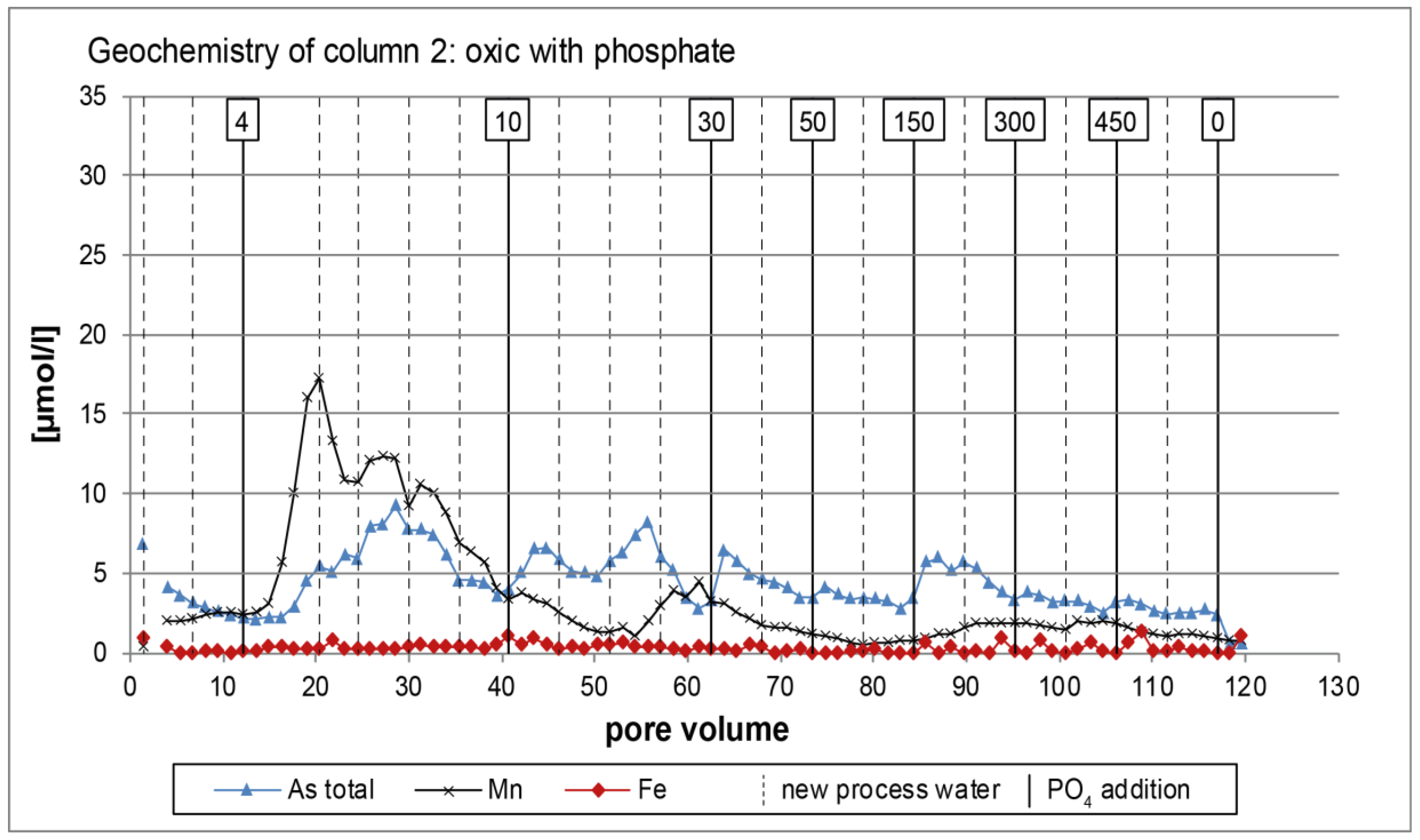
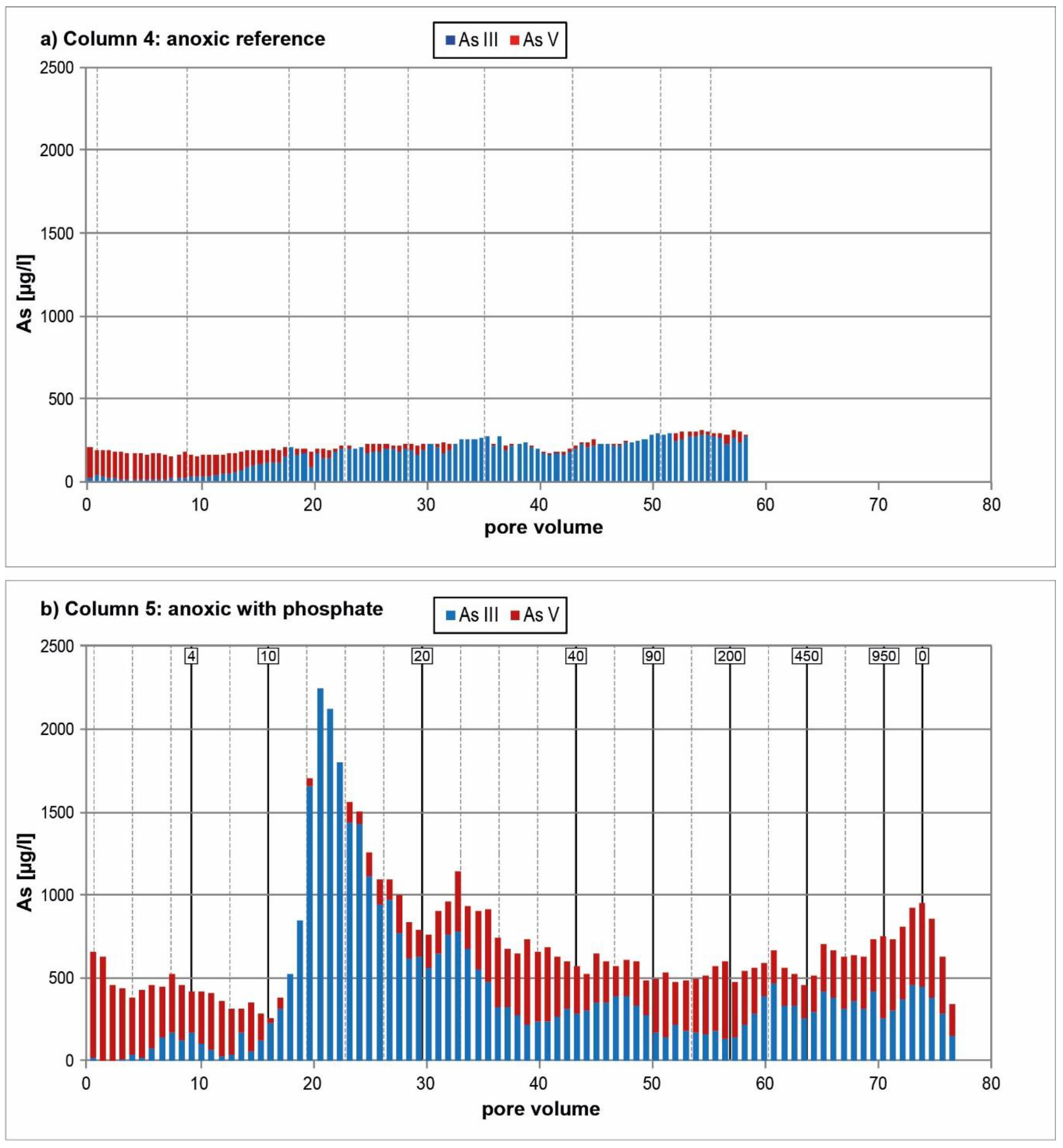
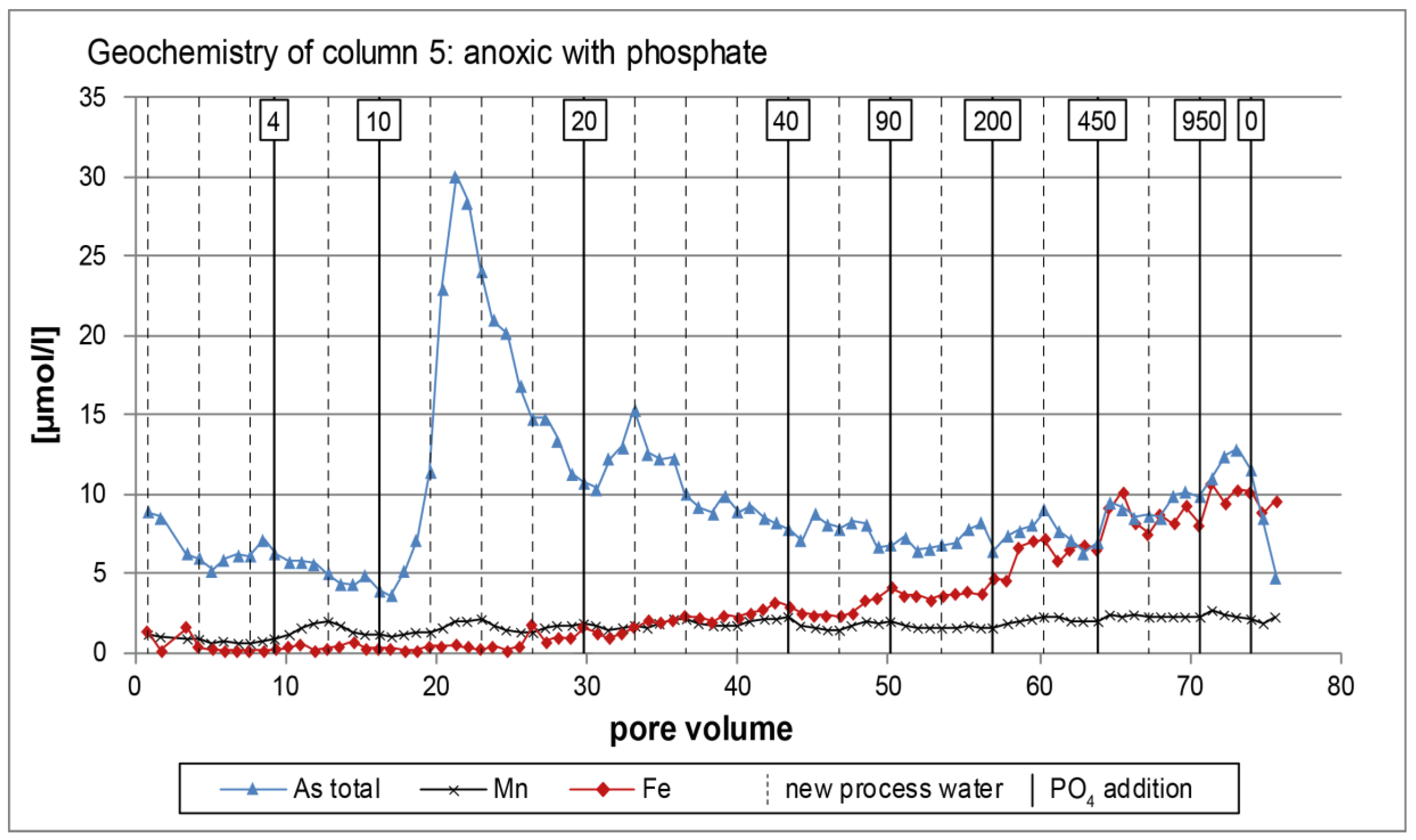

| Column Test | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Cumulative outflow release (mg) | 0.67 * | 4.69 | 4.37 | 3.20 | 11.4 | 13.1 |
| Sedimentary As before test (mg) | 17 ± 5 | 25 ± 0.8 | ||||
| Sedimentary As after test (mg) | 12.1 ± 2.8 | 11.4 ± 3.8 | 15.5 ± 3.3 | 12.9 ± 2.6 | ||
| Sedimentary mass losses (mg) | 4.9 ± 7.8 | 5.6 ± 8.8 | 9.5 ± 4.1 | 12.1 ± 3.4 | ||
| Column | Water Extractable Arsenic (mg) | Phosphate (1 M) Extractable Arsenic (mg) | Arsenic Released from Column (mg) | |||
|---|---|---|---|---|---|---|
| As(III) | As(V) | As(III) | As(V) | As(III) | As(V) | |
| 2 (oxic) | 0.13 | 1.24 | 1.19 | 11.9 | 1.13 | 3.58 |
| 5 (anoxic) | 0.20 | 2.24 | 1.48 | 16.3 | 6.78 | 4.60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maier, M.V.; Wolter, Y.; Zentler, D.; Scholz, C.; Stirn, C.N.; Isenbeck-Schröter, M. Phosphate Induced Arsenic Mobilization as a Potentially Effective In-Situ Remediation Technique—Preliminary Column Tests. Water 2019, 11, 2364. https://doi.org/10.3390/w11112364
Maier MV, Wolter Y, Zentler D, Scholz C, Stirn CN, Isenbeck-Schröter M. Phosphate Induced Arsenic Mobilization as a Potentially Effective In-Situ Remediation Technique—Preliminary Column Tests. Water. 2019; 11(11):2364. https://doi.org/10.3390/w11112364
Chicago/Turabian StyleMaier, Martin V., Yvonne Wolter, Daniel Zentler, Christian Scholz, Charlotte N. Stirn, and Margot Isenbeck-Schröter. 2019. "Phosphate Induced Arsenic Mobilization as a Potentially Effective In-Situ Remediation Technique—Preliminary Column Tests" Water 11, no. 11: 2364. https://doi.org/10.3390/w11112364
APA StyleMaier, M. V., Wolter, Y., Zentler, D., Scholz, C., Stirn, C. N., & Isenbeck-Schröter, M. (2019). Phosphate Induced Arsenic Mobilization as a Potentially Effective In-Situ Remediation Technique—Preliminary Column Tests. Water, 11(11), 2364. https://doi.org/10.3390/w11112364