Performance Evaluation of Small Sized Powdered Ferric Hydroxide as Arsenic Adsorbent

 ,
, 
 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Experimental Methods
2.1. Reagents
2.2. Material Characterization
2.3. Water Matrix
2.4. Batch Adsorption Procedure
2.5. Chemical Analytics
3. Results and Discussion
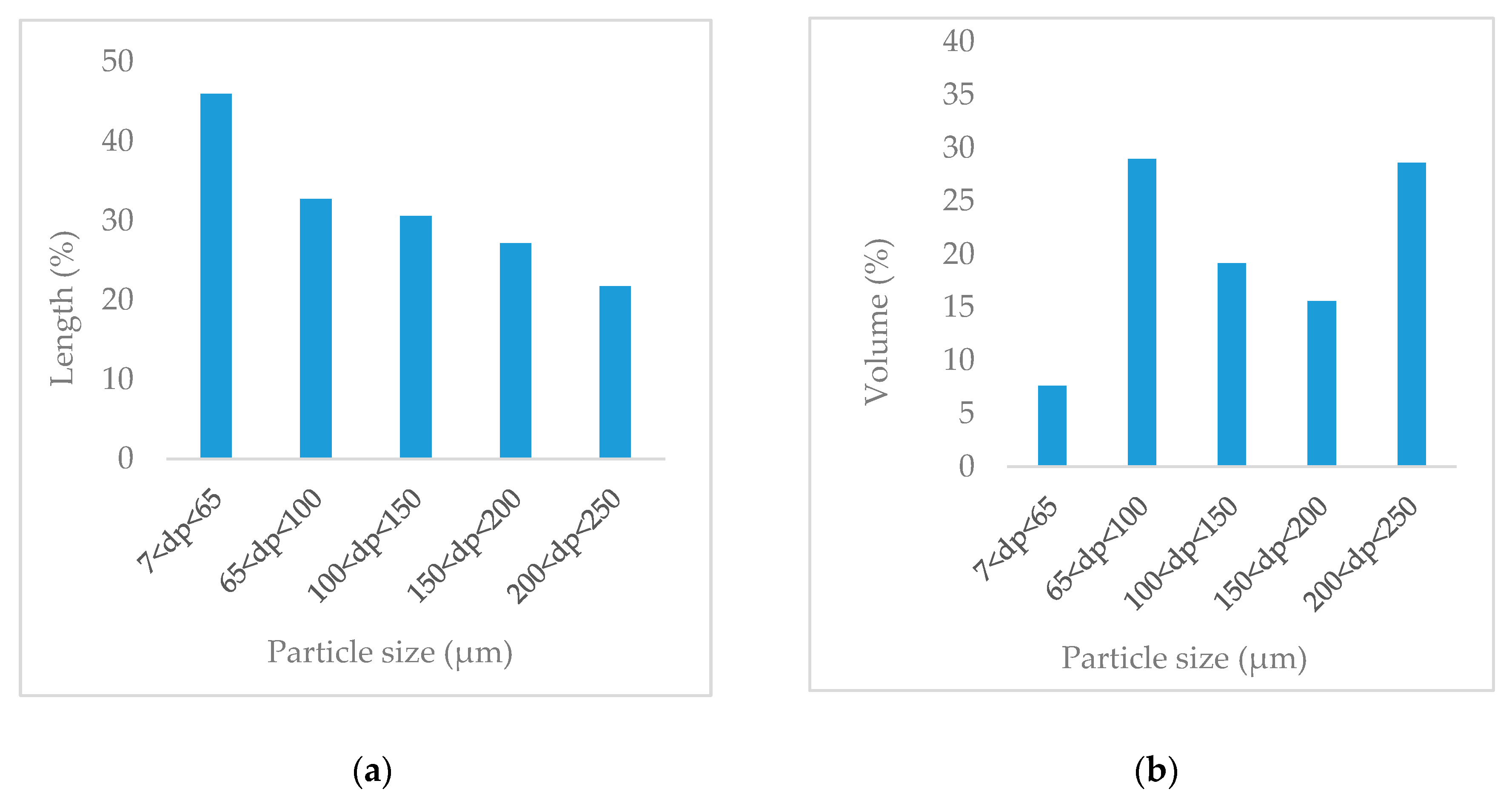
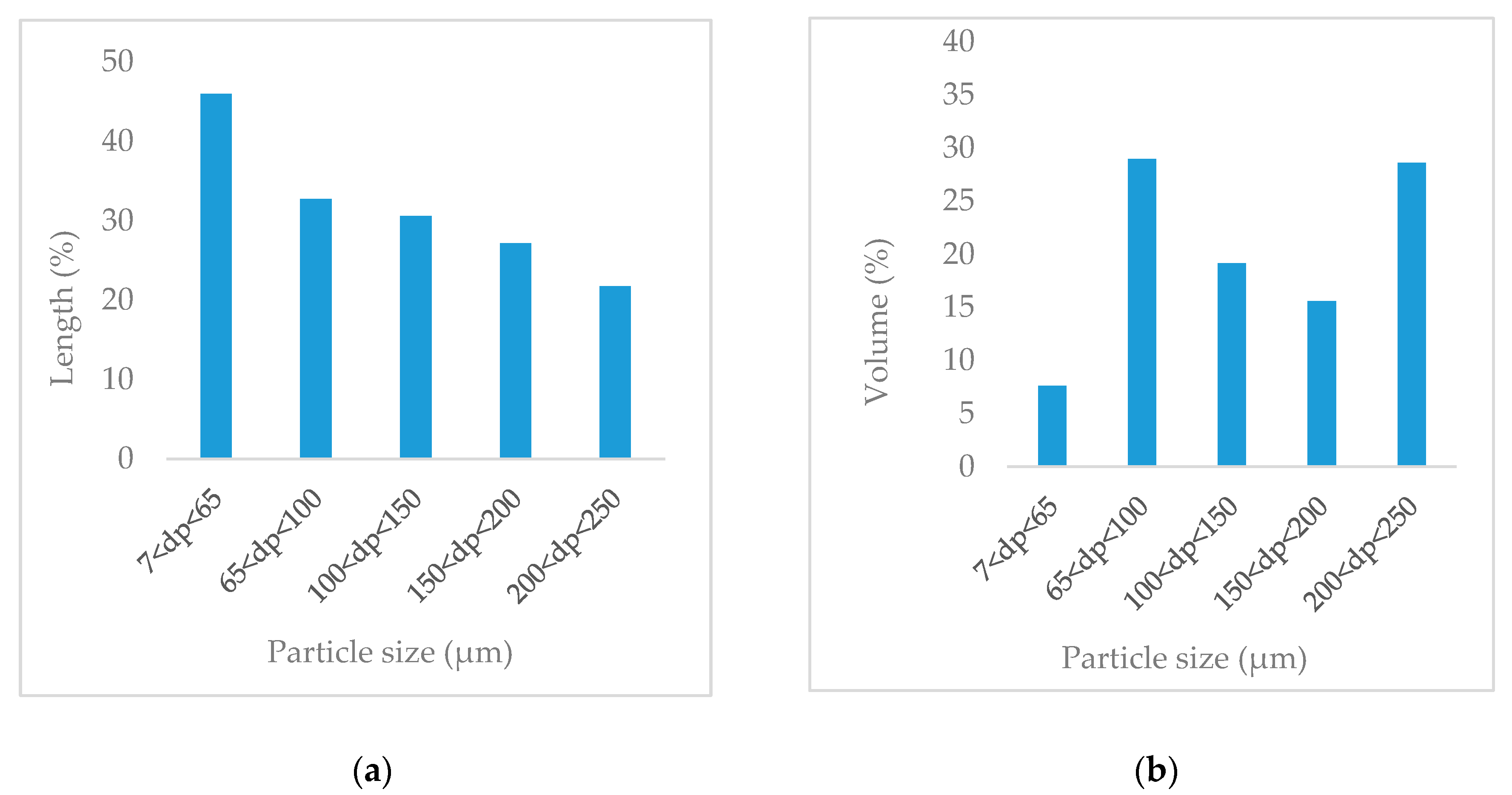
3.1. Particle Size Distribution
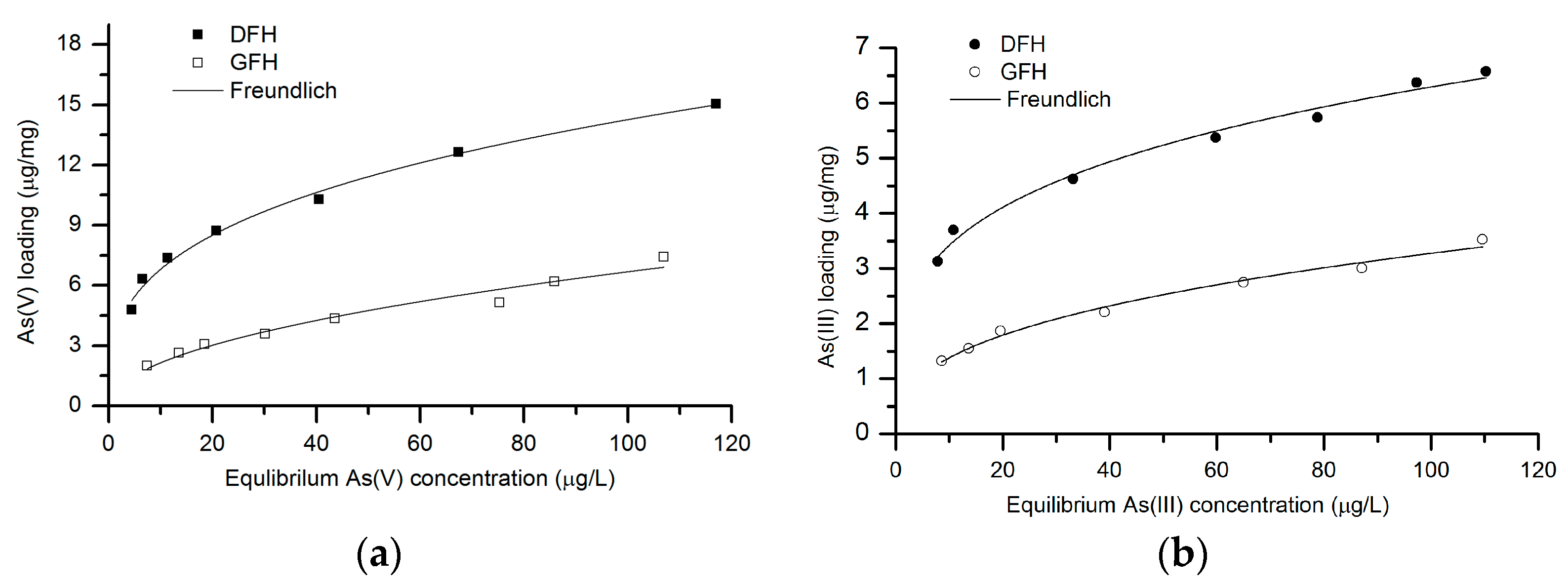
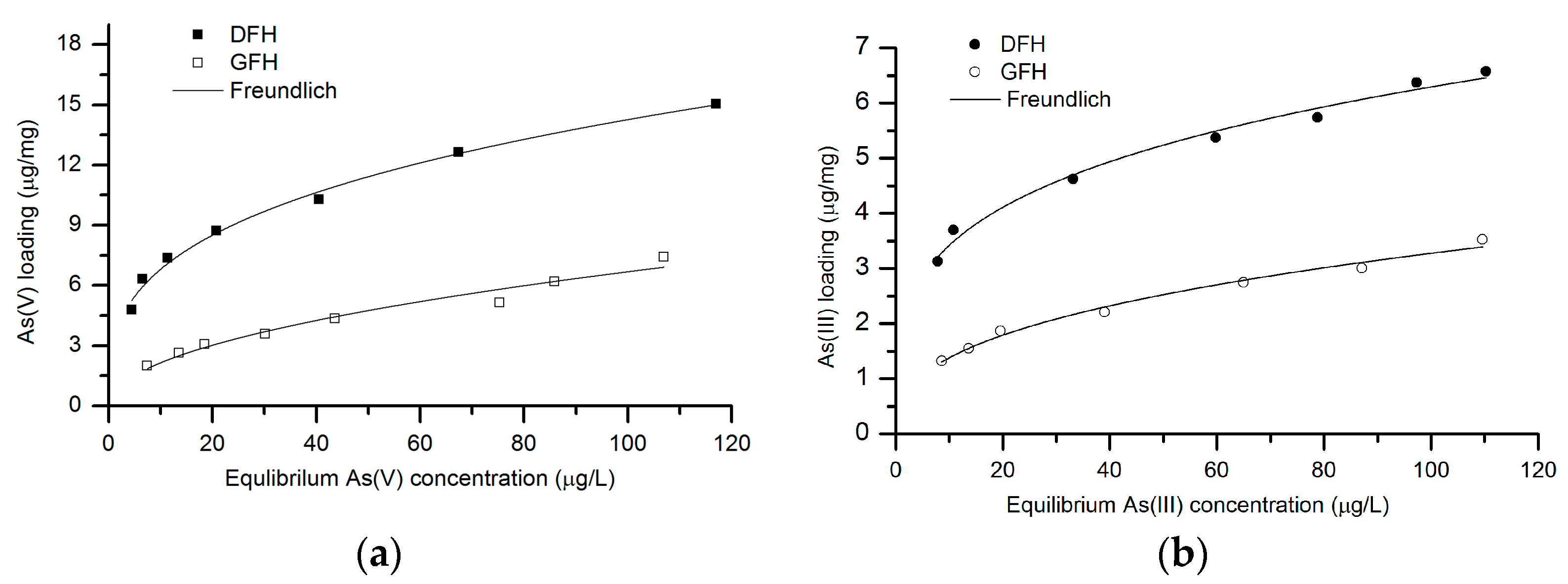
3.2. Batch Isotherm Studies
3.2.1. As(V) Adsorption
3.2.2. As(III) Adsorption
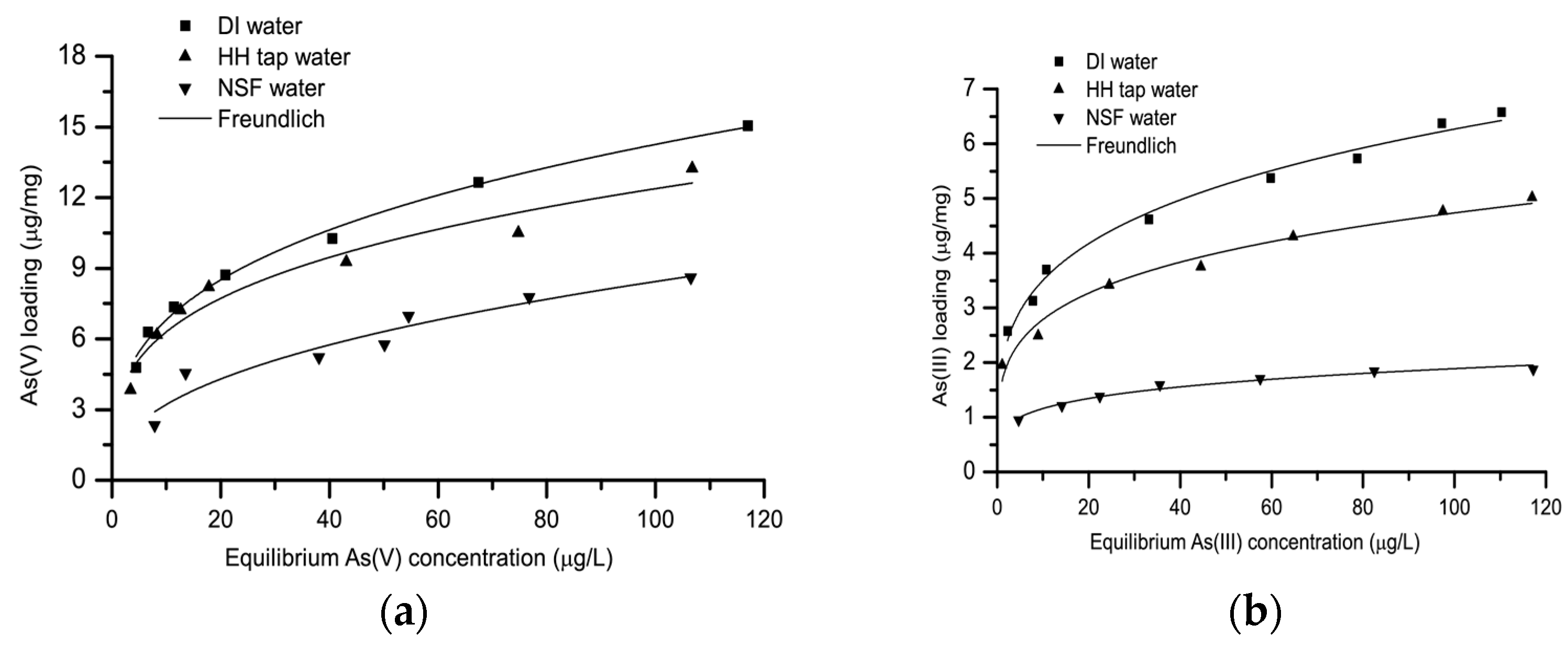
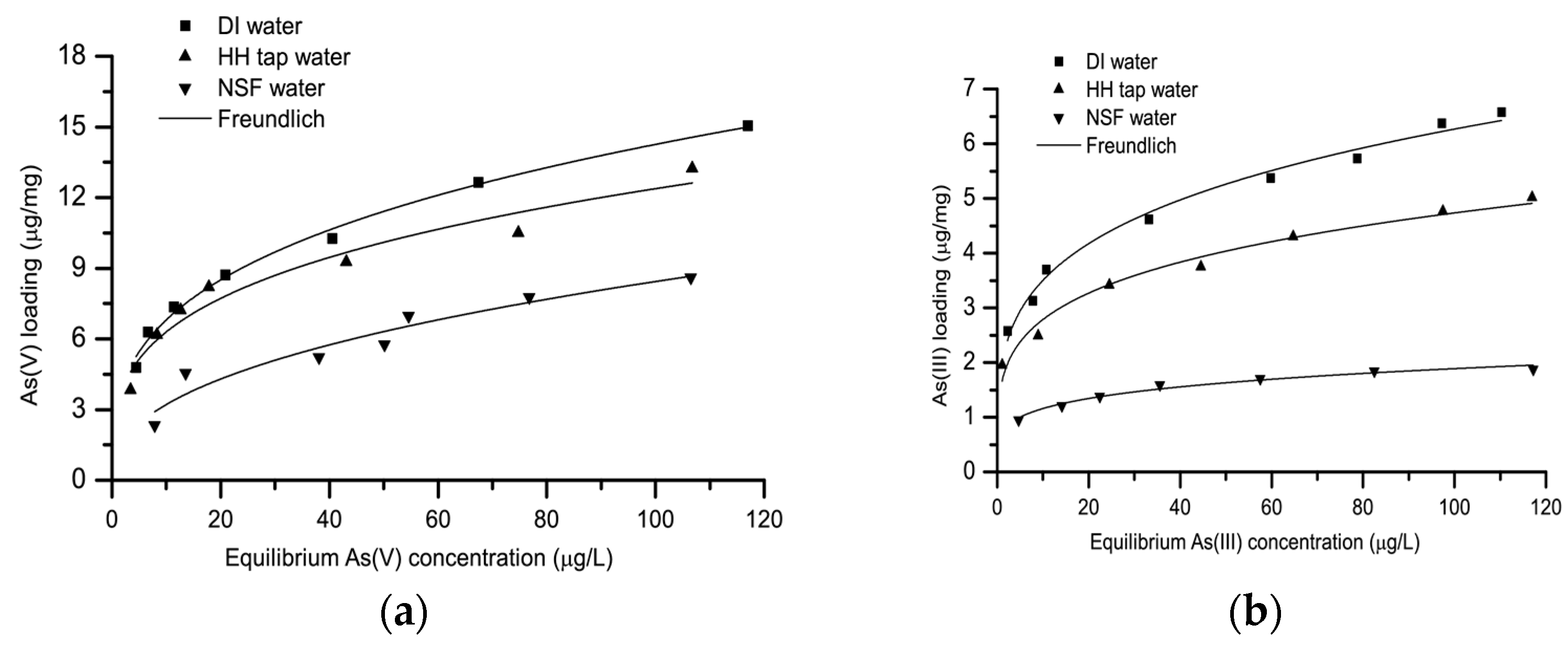
3.3. Effect of Water Matrix on Arsenic Adsorption
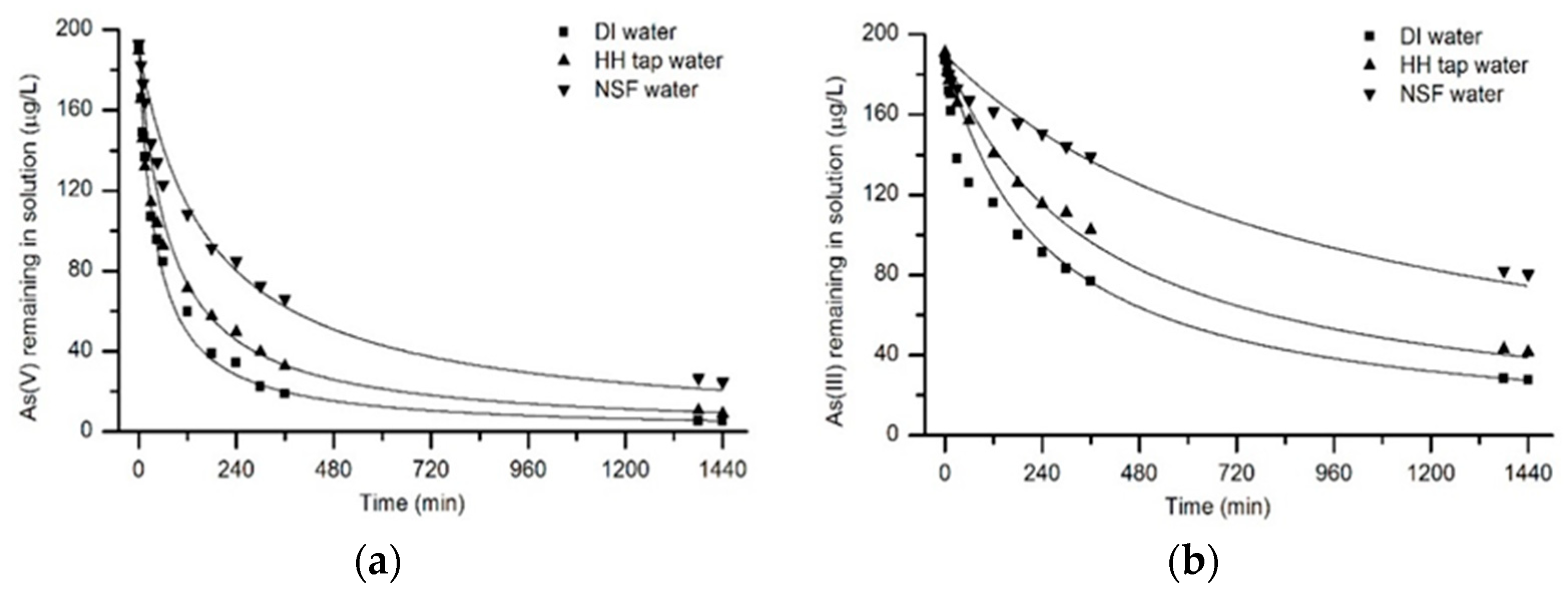
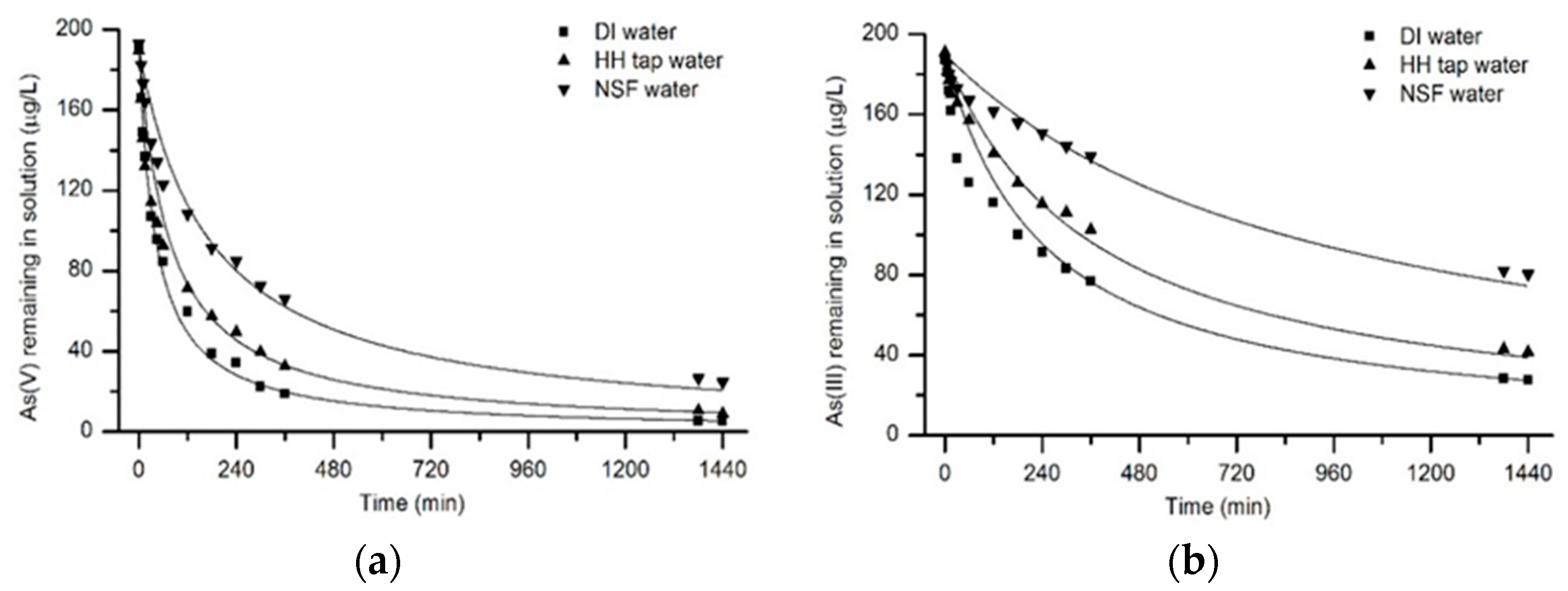
3.4. Arsenic Removal Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Smith, A.H.; Hopenhayn-Rich, C.; Bates, M.N.; Goeden, H.M.; Hertz-Picciotto, I.; Duggan, H.M.; Wood, R.; Kosnett, M.J.; Smith, M.T. Cancer risks from arsenic in drinking water. Environ. Health Perspect. 1992, 97, 259. [Google Scholar] [CrossRef] [PubMed]
- Violante, A.; Ricciardella, M.; Del Gaudio, S.; Pigna, M. Coprecipitation of arsenate with metal oxides: Nature, mineralogy, and reactivity of aluminum precipitates. Environ. Sci. Technol. 2006, 40, 4961–4967. [Google Scholar] [CrossRef] [PubMed]
- Tantry, B.A.; Shrivastava, D.; Taher, I.; Nabi Tantry, M. Arsenic exposure: Mechanisms of action and related health effects. J. Environ. Anal. Toxicol. 2015, 5, 1. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sengupta, M.K.; Hossain, M.A.; Ahamed, S.; Das, B.; Nayak, B.; Lodh, D.; Rahman, M.M.; Chakraborti, D. Arsenic contamination in groundwater: A global perspective with emphasis on the Asian scenario. J. Health Popul. Nutr. 2006, 142–163. [Google Scholar]
- Garelick, H.; Jones, H.; Dybowska, A.; Valsami-Jones, E. Arsenic pollution sources. In Reviews of Environmental Contamination; Springer: New York, NY, USA, 2009; Volume 197, pp. 17–60. [Google Scholar]
- Ware, G.W.; Albert, L.A.; Crosby, D.G.; Voogt, d.P.; Hutzinger, O.; Knaak, J.B.; Mayer, F.L.; Morgan, D.P.; Park, D.L.; Tjeerdema, R.S.; et al. Reviews of Environmental Contamination and Toxicology; Springer: New York, NY, USA, 2005. [Google Scholar]
- Mandal, S.; Sahu, M.K.; Patel, R.K. Adsorption studies of arsenic(III) removal from water by zirconium polyacrylamide hybrid material (ZrPACM-43). Water Resour. Ind. 2013, 4, 51–67. [Google Scholar] [CrossRef]
- Abejón, R.; Garea, A. A bibliometric analysis of research on arsenic in drinking water during the 1992–2012 period: An outlook to treatment alternatives for arsenic removal. J. Water Process Eng. 2015, 6, 105–119. [Google Scholar] [CrossRef]
- Organization, W.H. Guidelines for Drinking-Water Quality; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Sanjrani, M.A.; Mek, T.; Sanjrani, N.D.; Leghari, S.J.; Moryani, H.T.; Shabnam, A.B. Current situation of aqueous arsenic contamination in Pakistan, focused on Sindh and Punjab Province, Pakistan: A review. J. Pollut. Eff. Cont. 2017, 5, 207. [Google Scholar]
- Podgorski, J.E.; Eqani, S.A.M.A.S.; Khanam, T.; Ullah, R.; Shen, H.; Berg, M. Extensive arsenic contamination in high-pH unconfined aquifers in the Indus Valley. Sci. Adv. 2017, 3, e1700935. [Google Scholar] [CrossRef] [PubMed]
- Hering, J.G.; Katsoyiannis, I.A.; Theoduloz, G.A.; Berg, M.; Hug, S.J. Arsenic removal from drinking water: Experiences with technologies and constraints in practice. J. Environ. Eng. 2017, 143, 3117002. [Google Scholar] [CrossRef]
- Wang, L.; Chen, A.S.C.; Sorg, T.J.; Fields, K.A. Field evaluation of as removal by IX and AA. J. Am. Water Work. Assoc. 2002, 94, 161–173. [Google Scholar] [CrossRef]
- Tresintsi, S.; Simeonidis, K.; Estradé, S.; Martinez-Boubeta, C.; Vourlias, G.; Pinakidou, F.; Katsikini, M.; Paloura, E.C.; Stavropoulos, G.; Mitrakas, M. Tetravalent manganese feroxyhyte: A novel nanoadsorbent equally selective for As(III) and As(V) removal from drinking water. Environ. Sci. Technol. 2013, 47, 9699–9705. [Google Scholar] [CrossRef] [PubMed]
- Tresintsi, S.; Simeonidis, K.; Zouboulis, A.; Mitrakas, M. Comparative study of As(V) removal by ferric coagulation and oxy-hydroxides adsorption: Laboratory and full-scale case studies. Desalt. Water Treat. 2013, 51, 2872–2880. [Google Scholar] [CrossRef]
- Amy, G.L.; Chen, H.-W.; Dinzo, A.; Brandhuber, P. Adsorbent Treatment Technologies for Arsenic Removal; American Water Works Association: Denver, CO, USA, 2005. [Google Scholar]
- Ćurko, J.; Matošić, M.; Crnek, V.; Stulić, V.; Mijatović, I. Adsorption characteristics of different adsorbents and iron(III) salt for removing As(V) from water. Food Technol. Biotechnol. 2016, 54, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, O.S.; Viraraghavan, T.; Subramanian, K.S. Arsenic removal from drinking water using granular ferric hydroxide. Water Sa 2003, 29, 161–170. [Google Scholar] [CrossRef]
- Katsoyiannis, I.A.; Mitrakas, M.; Zouboulis, A.I. Arsenic occurrence in Europe: Emphasis in Greece and description of the applied full-scale treatment plants. Desalt. Water Treat. 2015, 54, 2100–2107. [Google Scholar] [CrossRef]
- An, B.; Steinwinder, T.R.; Zhao, D. Selective removal of arsenate from drinking water using a polymeric ligand exchanger. Water Res. 2005, 39, 4993–5004. [Google Scholar] [CrossRef] [PubMed]
- Abejón, A.; Garea, A.; Irabien, A. Arsenic removal from drinking water by reverse osmosis: Minimization of costs and energy consumption. Sep. Purif. Technol. 2015, 144, 46–53. [Google Scholar] [CrossRef]
- Víctor-Ortega, M.D.; Ratnaweera, H.C. Double filtration as an effective system for removal of arsenate and arsenite from drinking water through reverse osmosis. Process Saf. Environ. Prot. 2017, 111, 399–408. [Google Scholar] [CrossRef]
- Nidheesh, P.V.; Singh, T.S.A. Arsenic removal by electrocoagulation process: Recent trends and removal mechanism. Chemosphere 2017, 181, 418–432. [Google Scholar] [CrossRef] [PubMed]
- Lata, S.; Samadder, S.R. Removal of arsenic from water using nano adsorbents and challenges: A review. J. Environ. Manag. 2016, 166, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Driehaus, W.; Jekel, M.; Hildebrandt, U. Granular ferric hydroxide—A new adsorbent for the removal of arsenic from natural water. J. Water Supply Res. Technol. AQUA 1998, 47, 30–35. [Google Scholar] [CrossRef]
- Pal, B.N. Granular ferric hydroxide for elimination of arsenic from drinking water. Technol. Arsen. Remov. Drink. Water 2001, 59–68. [Google Scholar]
- Katsoyiannis, I.A.; Zouboulis, A.I.; Mitrakas, M.; Althoff, H.W.; Bartel, H. A hybrid system incorporating a pipe reactor and microfiltration for biological iron, manganese and arsenic removal from anaerobic groundwater. Fresenius Environ. Bull. 2013, 22, 3848–3853. [Google Scholar]
- Kalaruban, M.; Loganathan, P.; Shim, W.; Kandasamy, J.; Vigneswaran, S. Mathematical modelling of nitrate removal from water using a submerged membrane adsorption hybrid system with four adsorbents. Appl. Sci. 2018, 8, 194. [Google Scholar] [CrossRef]
- Badruzzaman, M.; Westerhoff, P.; Knappe, D.R.U. Intraparticle diffusion and adsorption of arsenate onto granular ferric hydroxide (GFH). Water Res. 2004, 38, 4002–4012. [Google Scholar] [CrossRef] [PubMed]
- Kersten, M.; Karabacheva, S.; Vlasova, N.; Branscheid, R.; Schurk, K.; Stanjek, H. Surface complexation modeling of arsenate adsorption by akagenéite (β-FeOOH)-dominant granular ferric hydroxide. Colloids Surf. A Physicochem. Eng. Asp. 2014, 448, 73–80. [Google Scholar] [CrossRef]
- Kosmulski, M. Surface Charging and Points of Zero Charge; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Simeonidis, K.; Papadopoulou, V.; Tresintsi, S.; Kokkinos, E.; Katsoyiannis, I.; Zouboulis, A.; Mitrakas, M. Efficiency of iron-based oxy-hydroxides in removing antimony from groundwater to levels below the drinking water regulation limits. Sustainability 2017, 9, 238. [Google Scholar] [CrossRef]
- Simeonidis, K.; Mourdikoudis, S.; Kaprara, E.; Mitrakas, M.; Polavarapu, L. Inorganic engineered nanoparticles in drinking water treatment: A critical review. Environ. Sci. Water Res. Technol. 2016, 2, 43–70. [Google Scholar] [CrossRef]
- Skoog, D.A.; Leary, J.J. Principles of instrumental analysis. Clin. Chem.-Ref. Ed. 1994, 40, 1612. [Google Scholar]
- Banerjee, K.; Nour, S.; Selbie, M.; Prevost, M.; Blumenschein, C.D.; Chen, H.W.; Amy, G.L. Optimization of process parameters for arsenic treatment with granular ferric hydroxide. In Proceedings of the AWWA Annual Conference, Anaheim, CA, USA, 15–19 June 2003. [Google Scholar]
- Tresintsi, S.; Mitrakas, M.; Simeonidis, K.; Kostoglou, M. Kinetic modeling of AS(III) and AS(V) adsorption by a novel tetravalent manganese feroxyhyte. J. Colloid Interface Sci. 2015, 460, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.N.; You, S.-J.; Hosseini-Bandegharaei, A.; Chao, H.-P. Mistakes and inconsistencies regarding adsorption of contaminants from aqueous solutions: A critical review. Water Res. 2017, 120, 88–116. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Amy, G.L.; Prevost, M.; Nour, S.; Jekel, M.; Gallagher, P.M.; Blumenschein, C.D. Kinetic and thermodynamic aspects of adsorption of arsenic onto granular ferric hydroxide (GFH). Water Res. 2008, 42, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.A.; Fendorf, S.E.; Goldberg, S. Surface structures and stability of arsenic(III) on goethite: Spectroscopic evidence for inner-sphere complexes. Environ. Sci. Technol. 1998, 32, 2383–2388. [Google Scholar] [CrossRef]
- Bolisetty, S.; Reinhold, N.; Zeder, C.; Orozco, M.N.; Mezzenga, R. Efficient purification of arsenic-contaminated water using amyloid-carbon hybrid membranes. Chem. Commun. 2017, 53, 5714–5717. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Fan, M.; Banerjee, K. Modeling of arsenic (V) adsorption onto granular ferric hydroxide. J. Am. Water Works Assoc. 2007, 99, 92–102. [Google Scholar] [CrossRef]
- Nguyen, V.L.; Chen, W.-H.; Young, T.; Darby, J. Effect of interferences on the breakthrough of arsenic: Rapid small scale column tests. Water Res. 2011, 45, 4069–4080. [Google Scholar] [CrossRef] [PubMed]
- Genz, A.; Kornmüller, A.; Jekel, M. Advanced phosphorus removal from membrane filtrates by adsorption on activated aluminium oxide and granulated ferric hydroxide. Water Res. 2004, 38, 3523–3530. [Google Scholar] [CrossRef] [PubMed]
- Tresintsi, S.; Simeonidis, K.; Vourlias, G.; Stavropoulos, G.; Mitrakas, M. Kilogram-scale synthesis of iron oxy-hydroxides with improved arsenic removal capacity: Study of Fe(II) oxidation—Precipitation parameters. Water Res. 2012, 46, 5255–5267. [Google Scholar] [CrossRef] [PubMed]
- Eljamal, O.; Sasaki, K.; Tsuruyama, S.; Hirajima, T. Kinetic model of arsenic sorption onto zero-valent iron (ZVI). Water Qual. Expo. Health 2011, 2, 125–132. [Google Scholar] [CrossRef]
- Saldaña-Robles, A.; Saldaña-Robles, N.; Saldaña-Robles, A.L.; Damian-Ascencio, C.; Rangel-Hernández, V.H.; Guerra-Sanchez, R. Arsenic removal from aqueous solutions and the impact of humic and fulvic acids. J. Clean. Prod. 2017, 159, 425–431. [Google Scholar] [CrossRef]
- Smith, E.H. Surface complexation modeling of metal removal by recycled iron sorbent. J. Environ. Eng. 1998, 124, 913–920. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Value | Literature Value |
|---|---|---|
| Chemical composition | β-FeOOH and Fe(OH) | |
| Dry solids content (%) | ~50 a | ~50 b |
| Moisture content (%) | ~50 a | ~50 b |
| Particle size (µm), dp | 7.4–250 a | 1.8–250 b |
| Mean particle size (µm) | 78.40 | − |
| Point of zero charge (PZC) | 5.3 ± 0.2 | ~5.5 c |
| Isoelectric point (IEP) | 7.8 ± 0.2 | ~7.8 c |
| Surface charge density | 0.9 mmol [OH−]/g | − |
| Parameter. (mg/L) | Water Matrixs. | |
|---|---|---|
| HH Tap Water * | NSF Challenge Water | |
| Na+ | 14 | 73.7 |
| Ca2+ | 42 | 40.1 |
| Mg2+ | 4 | 12.6 |
| HCO3− | 150–300 | 183.0 |
| Cl− | 19 | 71.0 |
| SO42− | 23 | 50.0 |
| NO3− | 0.62 | 2.0 |
| F− | 0.13 | 1.0 |
| PO43− | 0.05–0.15 | 0.123 |
| SiO2 | 16.6–18.5 | 20 |
| DOC | 0.8 ± 0.2 | − |
| Water Matrix | Adsorbent | n (−) | KF * | Q10 (μg/mg) | R2 | |
|---|---|---|---|---|---|---|
| DI water | GFH | 2.03 | 0.68 | 2.1 | 0.960 | 0.136 |
| DI water | DFH | 3.10 | 3.25 | 6.9 | 0.991 | 0.117 |
| HH tap water | DFH | 3.41 | 3.20 | 6.3 | 0.941 | 0.551 |
| NSF water | DFH | 2.39 | 1.22 | 3.2 | 0.918 | 0.367 |
| Water Matrix | Adsorbent | n (−) | KF * | Q10 (μg/mg) | R2 | |
|---|---|---|---|---|---|---|
| DI water | GFH | 2.66 | 0.58 | 1.4 | 0.985 | 0.01 |
| DI water | DFH | 3.96 | 1.96 | 3.5 | 0.987 | 0.023 |
| HH tap water | DFH | 4.34 | 1.64 | 2.8 | 0.972 | 0.036 |
| NSF water | DFH | 4.39 | 0.64 | 1.1 | 0.970 | 0.005 |
| Water Matrix | As(V) | As(III) | ||||||
|---|---|---|---|---|---|---|---|---|
| First Order | Second Order | First Order | Second Order | |||||
| R2 | SE | R2 | SE | R2 | SE | R2 | SE | |
| DI water | 0.529 | 59.99 | 0.998 | 4.93 | 0.802 | 35.31 | 0.996 | 14.52 |
| HH tap water | 0.579 | 57.62 | 0.994 | 14.31 | 0.769 | 21.59 | 0.969 | 6.72 |
| NSF water | 0.714 | 23.76 | 0.992 | 10.68 | 0.905 | 14.45 | 0.980 | 8.12 |
| Water Matrix | As(V) | As(III) |
|---|---|---|
| k2 (h−1) | k2 (h−1) | |
| DI water | 7.51 × 10−3 | 1.34 × 10−3 |
| HH tap water | 4.23 × 10−3 | 0.85 × 10−3 |
| NSF water | 1.82 × 10−3 | 0.34 × 10−3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usman, M.; Katsoyiannis, I.; Mitrakas, M.; Zouboulis, A.; Ernst, M. Performance Evaluation of Small Sized Powdered Ferric Hydroxide as Arsenic Adsorbent. Water 2018, 10, 957. https://doi.org/10.3390/w10070957
Usman M, Katsoyiannis I, Mitrakas M, Zouboulis A, Ernst M. Performance Evaluation of Small Sized Powdered Ferric Hydroxide as Arsenic Adsorbent. Water. 2018; 10(7):957. https://doi.org/10.3390/w10070957
Chicago/Turabian StyleUsman, Muhammad, Ioannis Katsoyiannis, Manassis Mitrakas, Anastasios Zouboulis, and Mathias Ernst. 2018. "Performance Evaluation of Small Sized Powdered Ferric Hydroxide as Arsenic Adsorbent" Water 10, no. 7: 957. https://doi.org/10.3390/w10070957
APA StyleUsman, M., Katsoyiannis, I., Mitrakas, M., Zouboulis, A., & Ernst, M. (2018). Performance Evaluation of Small Sized Powdered Ferric Hydroxide as Arsenic Adsorbent. Water, 10(7), 957. https://doi.org/10.3390/w10070957