The Cloud Nucleating Properties and Mixing State of Marine Aerosols Sampled along the Southern California Coast

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Overview of Measurements Conducted on the Scripps Pier
2.2. Chemical Analysis of Marine Aerosols Using ATOFMS
2.3. Hygroscopicity Measurements of Marine Aerosols
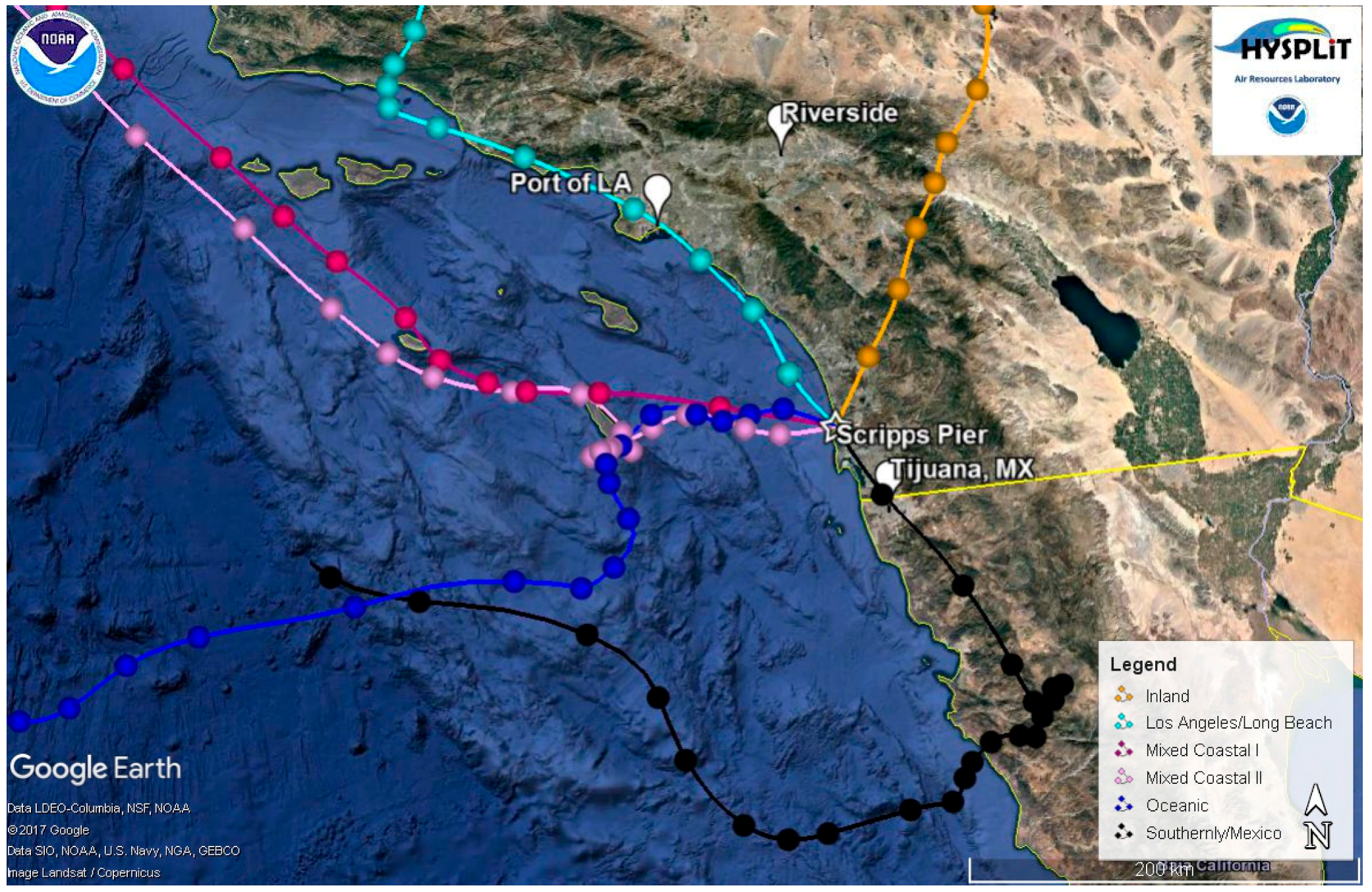
2.4. Classification of Air Masses
3. Results and Discussion
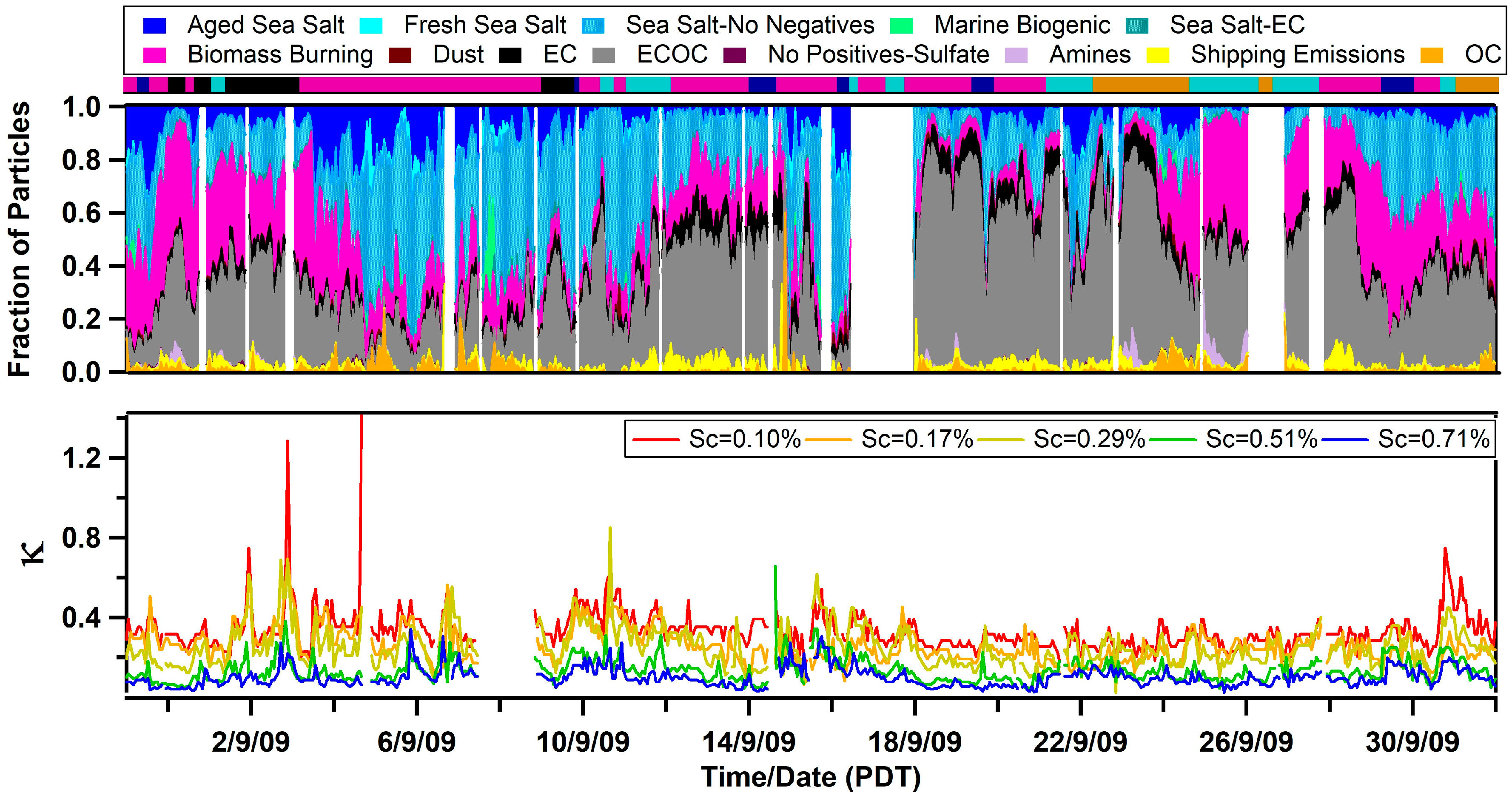
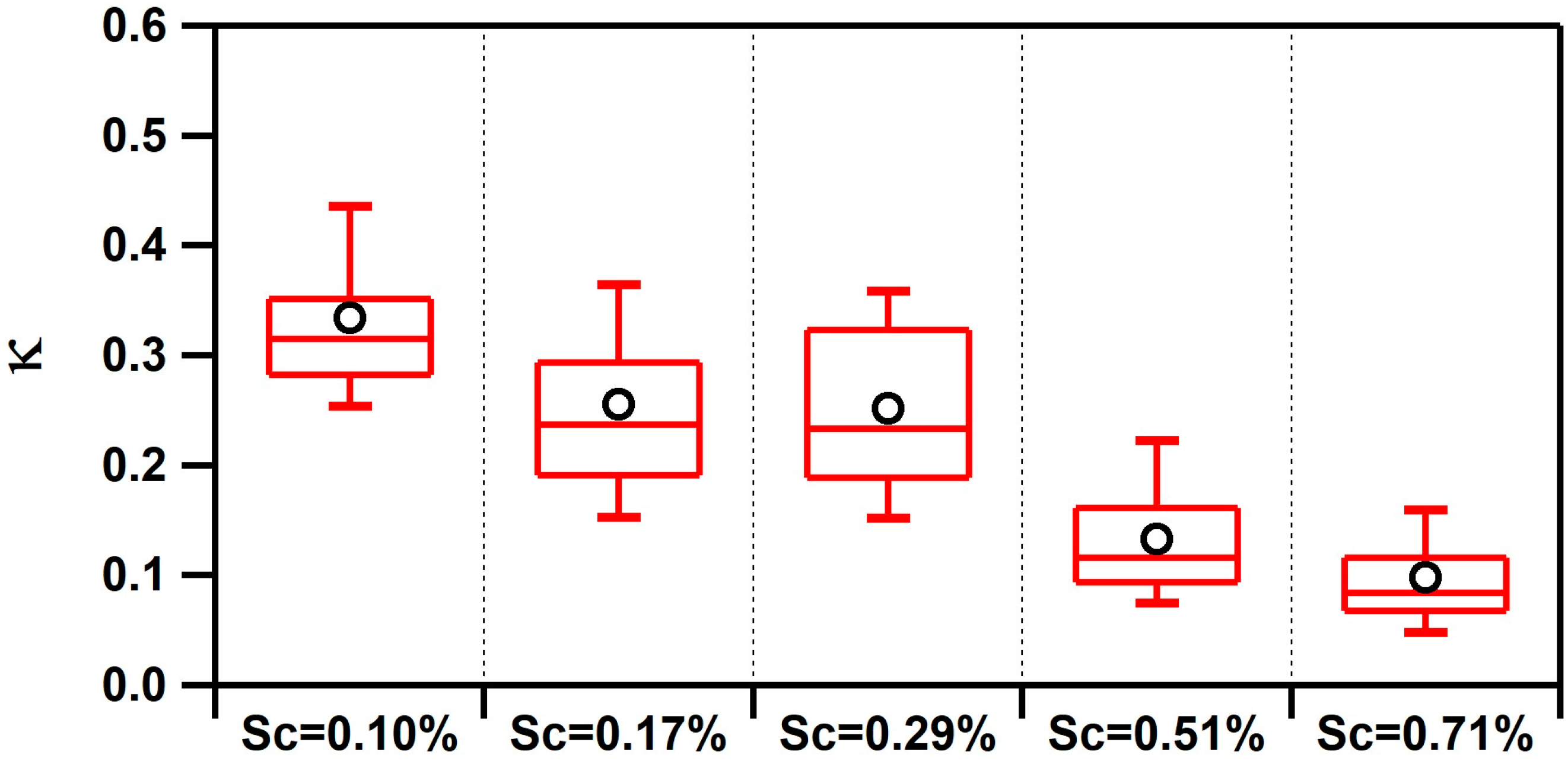
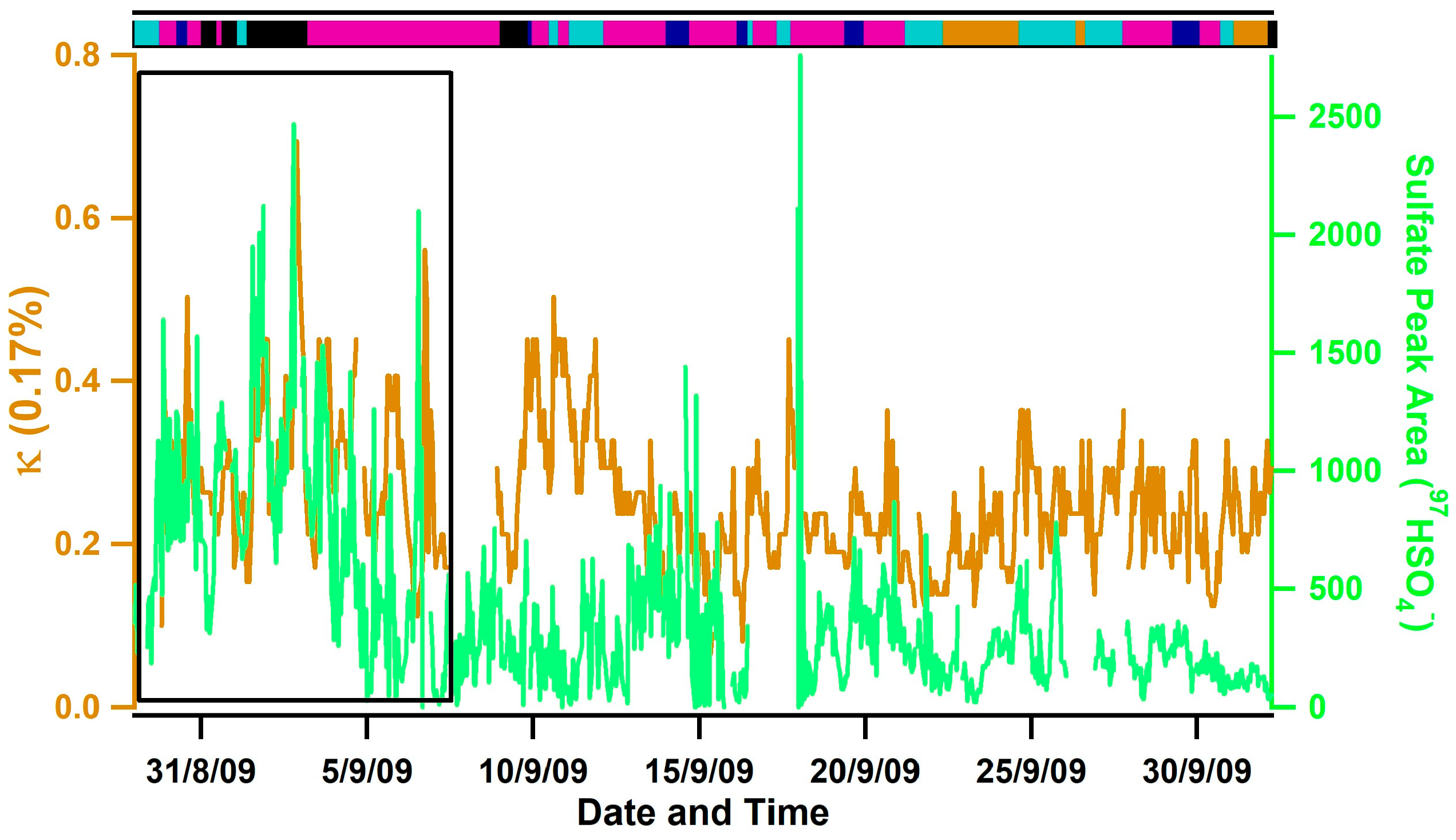
3.1. Observed Hygroscopicity of Marine Aerosols
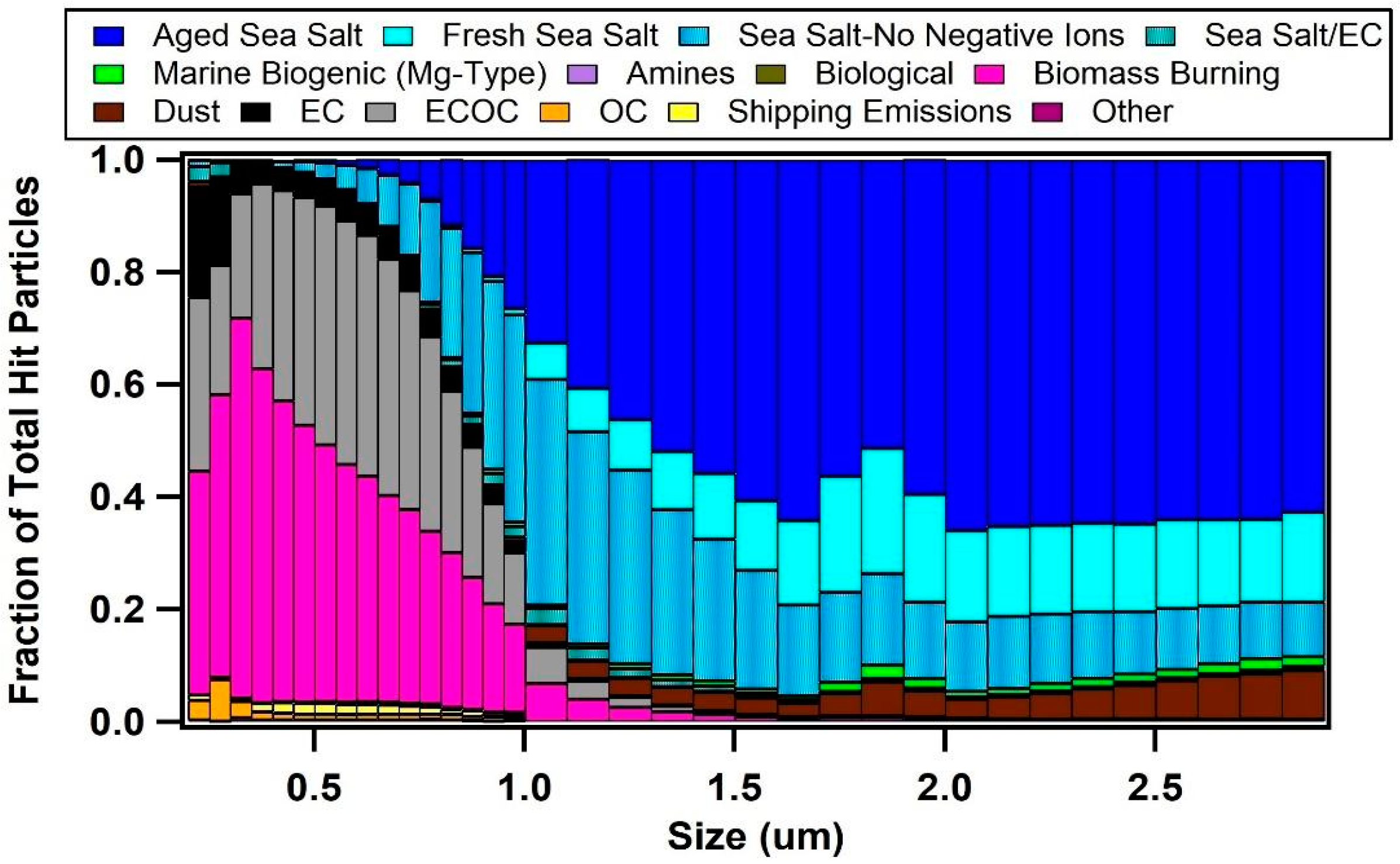
3.2. Single Particle Composition of Marine Aerosols
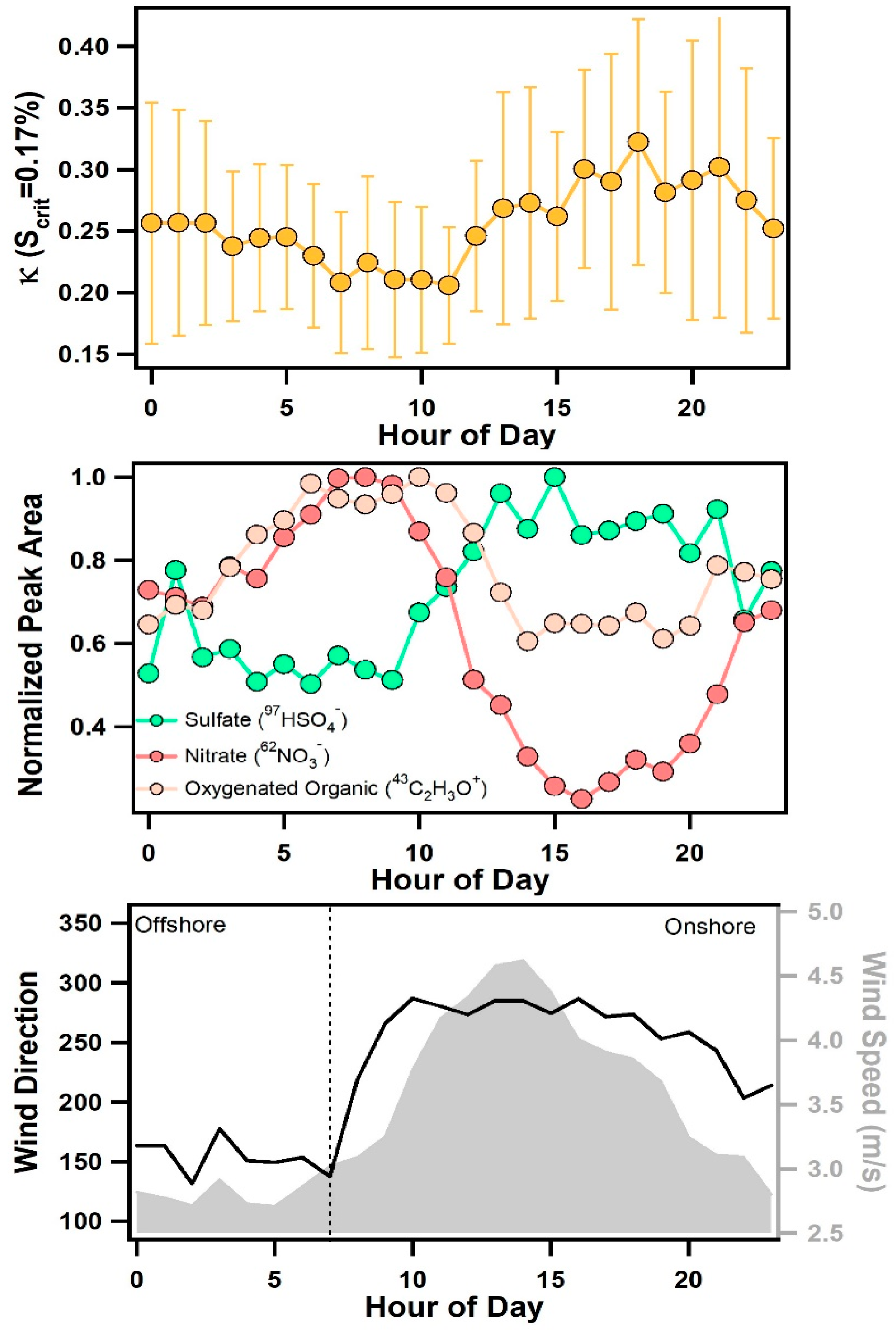
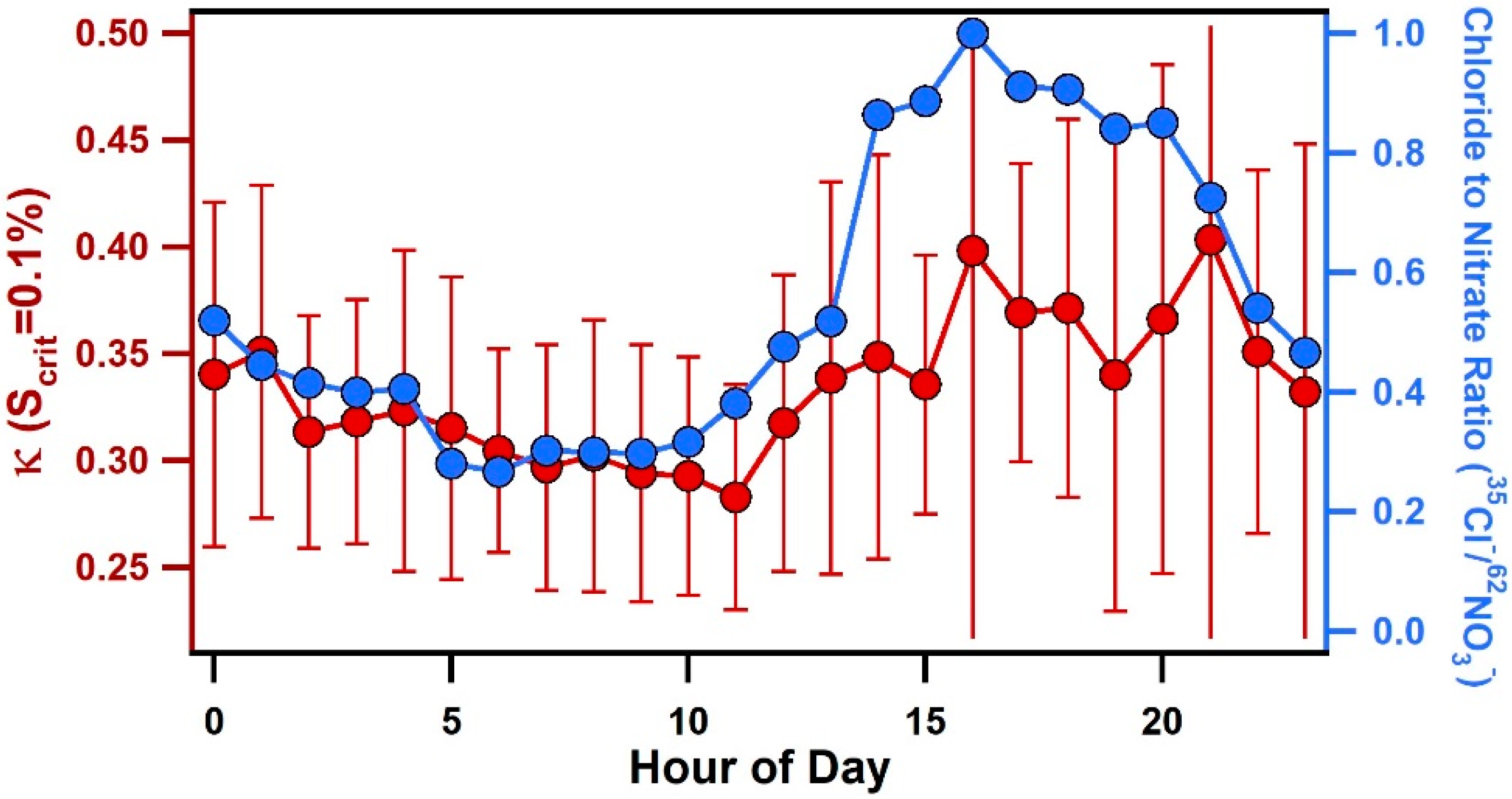
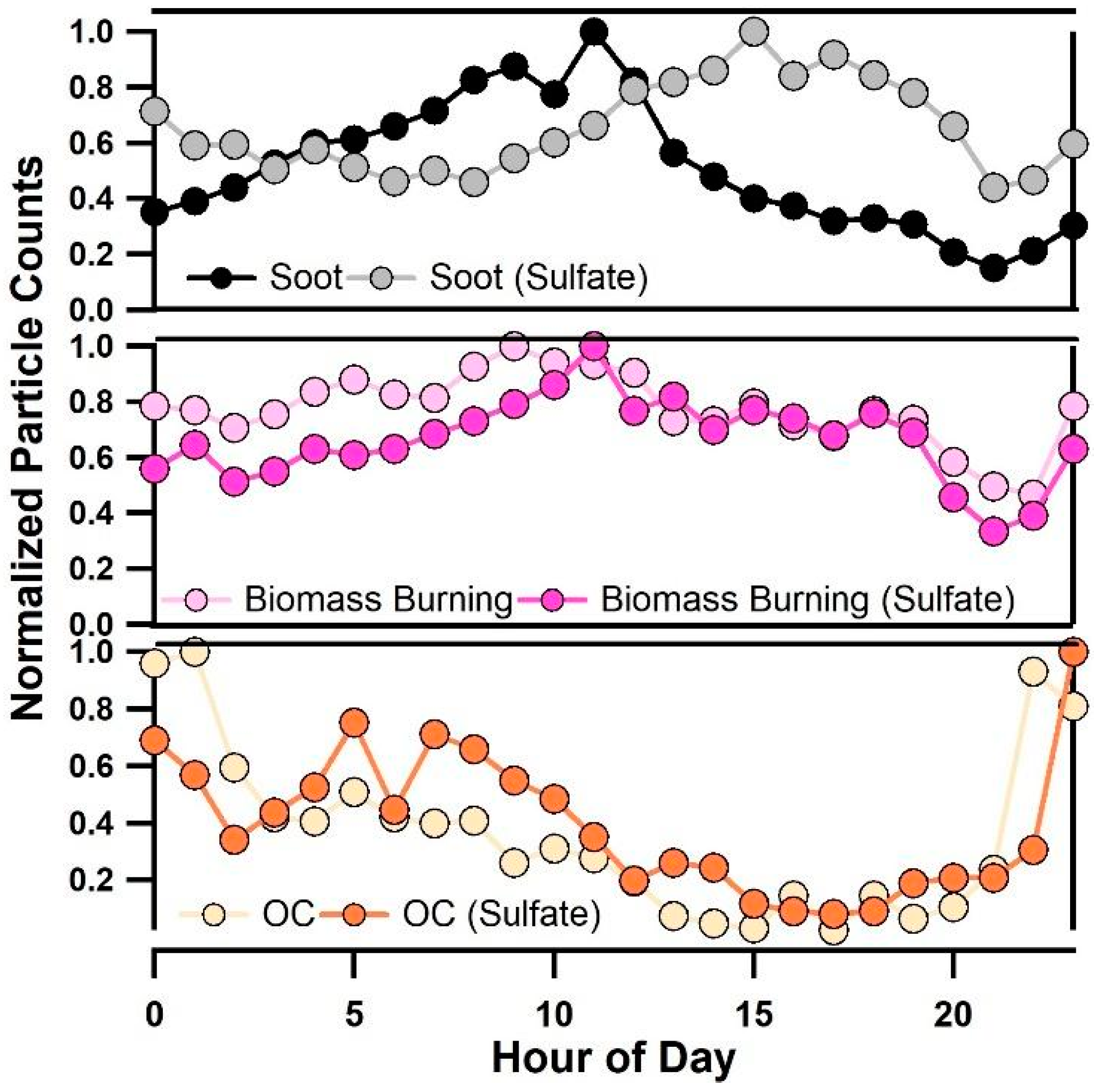
3.3. Diurnal Variability of Κ and the Role of Chemical Aging
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Myhre, G.; Shindell, D.; Bréon, F.-M.; Collins, W.; Fuglestvedt, J.; Huang, J.; Koch, D.; Lamarque, J.-F.; Lee, D.; Mendoza, B.; et al. Anthropogenic and Natural Radiative Forcing; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013. [Google Scholar]
- McFiggans, G.; Artaxo, P.; Baltensperger, U.; Coe, H.; Facchini, M.C.; Feingold, G.; Fuzzi, S.; Gysel, M.; Laaksonen, A.; Lohmann, U.; et al. The effect of physical and chemical aerosol properties on warm cloud droplet activation. Atmos. Chem. Phys. 2006, 6, 2593–2649. [Google Scholar] [CrossRef]
- Pruppacher, H.R.; Klett, J.D. Microphysics of Clouds and Precipitation, 2nd ed.; Springer: Berlin, Germany, 1996. [Google Scholar]
- Quinn, P.K.; Bates, T.S.; Coffman, D.J.; Covert, D.S. Influence of particle size and chemistry on the cloud nucleating properties of aerosols. Atmos. Chem. Phys. 2008, 8, 1029–1042. [Google Scholar] [CrossRef]
- Petters, M.D.; Kreidenweis, S.M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007, 7, 1961–1971. [Google Scholar] [CrossRef]
- Andreae, M.O.; Rosenfeld, D. Aerosol-cloud-precipitation interactions. Part 1. The nature and sources of cloud-active aerosols. Earth-Sci. Rev. 2008, 89, 13–41. [Google Scholar] [CrossRef]
- Pringle, K.J.; Tost, H.; Pozzer, A.; Poschl, U.; Lelieveld, J. Global distribution of the effective aerosol hygroscopicity parameter for CCN activation. Atmos. Chem. Phys. 2010, 10, 5241–5255. [Google Scholar] [CrossRef]
- Collins, D.B.; Ault, A.P.; Moffet, R.C.; Ruppel, M.J.; Cuadra-Rodriguez, L.A.; Guasco, T.L.; Corrigan, C.E.; Pedler, B.E.; Azam, F.; Aluwihare, L.I.; et al. Impact of marine biogeochemistry on the chemical mixing state and cloud forming ability of nascent sea spray aerosol. J. Geophys. Res. Atmos. 2013, 118, 8553–8565. [Google Scholar] [CrossRef]
- Good, N.; Topping, D.O.; Allan, J.D.; Flynn, M.; Fuentes, E.; Irwin, M.; Williams, P.I.; Coe, H.; McFiggans, G. Consistency between parameterisations of aerosol hygroscopicity and CCN activity during the RHaMBLe discovery cruise. Atmos. Chem. Phys. 2010, 10, 3189–3203. [Google Scholar] [CrossRef]
- Hudson, J.G. Variability of the relationship between particle size and cloud-nucleating ability. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef]
- Martin, M.; Chang, R.Y.W.; Sierau, B.; Sjogren, S.; Swietlicki, E.; Abbatt, J.P.D.; Leck, C.; Lohmann, U. Cloud condensation nuclei closure study on summer arctic aerosol. Atmos. Chem. Phys. 2011, 11, 11335–11350. [Google Scholar] [CrossRef]
- Bougiatioti, A.; Nenes, A.; Fountoukis, C.; Kalivitis, N.; Pandis, S.N.; Mihalopoulos, N. Size-resolved CCN distributions and activation kinetics of aged continental and marine aerosol. Atmos. Chem. Phys. 2011, 11, 8791–8808. [Google Scholar] [CrossRef]
- Roberts, G.C.; Day, D.A.; Russell, L.M.; Dunlea, E.J.; Jimenez, J.L.; Tomlinson, J.M.; Collins, D.R.; Shinozuka, Y.; Clarke, A.D. Characterization of particle cloud droplet activity and composition in the free troposphere and the boundary layer during INTEX-B. Atmos. Chem. Phys. 2010, 10, 6627–6644. [Google Scholar] [CrossRef]
- Moore, R.H.; Cerully, K.; Bahreini, R.; Brock, C.A.; Middlebrook, A.M.; Nenes, A. Hygroscopicity and composition of California CCN during summer 2010. J. Geophys. Res. Atmos. 2012, 117. [Google Scholar] [CrossRef]
- Furutani, H.; Dall’osto, M.; Roberts, G.C.; Prather, K.A. Assessment of the relative importance of atmospheric aging on CCN activity derived from field observations. Atmos. Environ. 2008, 42, 3130–3142. [Google Scholar] [CrossRef]
- Blanchard, D.C.; Woodcock, A.H. Bubble formation and modification in the sea and its meteorological significance. Tellus 1957, 9, 145–158. [Google Scholar] [CrossRef]
- O’Dowd, C.D.; De Leeuw, G. Marine aerosol production: A review of the current knowledge. Philos. Trans. A 2007, 365, 1753–1774. [Google Scholar] [CrossRef] [PubMed]
- Prather, K.A.; Bertram, T.H.; Grassian, V.H.; Deane, G.B.; Stokes, M.D.; DeMott, P.J.; Aluwihare, L.I.; Palenik, B.P.; Azam, F.; Seinfeld, J.H.; et al. Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol. Proc. Natl. Acad. Sci. USA 2013, 110, 7550–7555. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Butcher, A.C.; Rosenoern, T.; Coz, E.; Lieke, K.I.; de Leeuw, G.; Nilsson, E.D.; Bilde, M. Investigating Primary Marine Aerosol Properties: CCN Activity of Sea Salt and Mixed Inorganic-Organic Particles. Environ. Sci. Technol. 2012, 46, 10405–10412. [Google Scholar] [CrossRef] [PubMed]
- Coggon, M.M.; Sorooshian, A.; Wang, Z.; Craven, J.S.; Metcalf, A.R.; Lin, J.J.; Nenes, A.; Jonsson, H.H.; Flagan, R.C.; Seinfeld, J.H. Observations of continental biogenic impacts on marine aerosol and clouds off the coast of California. J. Geophys. Res. 2014, 119, 6724–6748. [Google Scholar] [CrossRef]
- Hu, Q.-H.; Xie, Z.-Q.; Wang, X.-M.; Kang, H.; Zhang, P. Levoglucosan indicates high levels of biomass burning aerosols over oceans from the Arctic to Antarctic. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Brioude, J.; Cooper, O.R.; Feinfeld, G.; Trainer, M.; Freitas, S.R.; Kowal, D.; Ayers, J.R.; Prins, E.; Minnis, P.; McKeen, S.A.; et al. Effect of biomass burning on marine stratocumulus clouds off the California coast. Atmos. Chem. Phys. 2009, 9, 8841–8856. [Google Scholar] [CrossRef]
- Petters, M.D.; Carrico, C.M.; Kreidenweis, S.M.; Prenni, A.J.; DeMott, P.J.; Collett, J.L.; Moosmuller, H. Cloud condensation nucleation activity of biomass burning aerosol. J. Geophys. Res. Atmos. 2009, 114. [Google Scholar] [CrossRef]
- Engelhart, G.J.; Hennigan, C.J.; Miracolo, M.A.; Robinson, A.L.; Pandis, S.N. Cloud condensation nuclei activity of fresh primary and aged biomass burning aerosol. Atmos. Chem. Phys. 2012, 12, 7285–7293. [Google Scholar] [CrossRef]
- Lathem, T.; Beyersdorf, A.J.; Thornhill, K.L.; Winstead, E.L.; Cubison, M.J.; Hecobian, A.; Jimenez, J.L.; Weber, R.J.; Anderson, B.E.; Nenes, A. Analysis of CCN activity of Arctic aerosol and Canadian biomass burning during summer 2008. Atmos. Chem. Phys. 2013, 13, 2735–2756. [Google Scholar] [CrossRef]
- Dua, S.K.; Hopke, P.K.; Raunemaa, T. Hygroscopicity of diesel aerosols. Water Air Soil Pollut. 1999, 112, 247–257. [Google Scholar] [CrossRef]
- Coggon, M.M.; Sorooshian, A.; Wang, Z.; Metcalf, A.R.; Frossard, A.A.; Lin, J.J.; Craven, J.S.; Nenes, A.; Jonsson, H.H.; Russell, L.M.; et al. Ship impacts on the marine atmosphere: Insights into the contribution of shipping emissions to the properties of marine aerosol and clouds. Atmos. Chem. Phys. 2012, 12, 8439–8458. [Google Scholar] [CrossRef]
- Hegg, D.A.; Covert, D.S.; Jonsson, H.H.; Woods, R.K. The contribution of anthropogenic aerosols to aerosol light-scattering and CCN activity in the California coastal zone. Atmos. Chem. Phys. 2010, 10, 7341–7351. [Google Scholar] [CrossRef]
- Langley, L.; Leaitch, W.R.; Lohmann, U.; Shantz, N.C.; Worsnop, D. Contributions from DMS and ship emissions to CCN observed over the summertime North Pacific. Atmos. Chem. Phys. 2010, 10, 1287–1314. [Google Scholar] [CrossRef]
- Gaston, C.J.; Quinn, P.K.; Bates, T.S.; Gilman, J.B.; Bon, D.M.; Kuster, W.C.; Prather, K.A. The impact of shipping, agricultural, and urban emissions on single particle chemistry observed aboard the R/V Atlantis during CalNex. J. Geophys. Res. Atmos. 2013, 118. [Google Scholar] [CrossRef]
- Gard, E.E.; Kleeman, M.J.; Gross, D.S.; Hughes, L.S.; Allen, J.O.; Morrical, B.D.; Fergenson, D.P.; Dienes, T.; Galli, M.E.; Johnson, R.J.; et al. Direct observation of heterogeneous chemistry in the atmosphere. Science 1998, 279, 1184–1187. [Google Scholar] [CrossRef] [PubMed]
- Saul, T.D.; Tolocka, M.P.; Johnston, M.V. Reactive uptake of nitric acid onto sodium chloride aerosols across a wide range of relative humidities. J. Phys. Chem. A 2006, 110, 7614–7620. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.A.; Abbatt, J.P.D. N2O5 reaction on submicron sea salt aerosol: Kinetics, products, and the effect of surface active organics. J. Phys. Chem. A 2005, 109, 10004–10012. [Google Scholar] [CrossRef] [PubMed]
- Farmer, D.K.; Cappa, C.D.; Kreidenweis, S.M. Atmospheric processes and their controlling influence on cloud condensation nuclei activity. Chem. Rev. 2015, 115, 4199–4217. [Google Scholar] [CrossRef] [PubMed]
- Ault, A.P.; Gaston, C.J.; Wang, Y.; Dominguez, G.; Thiemens, M.H.; Prather, K.A. Characterization of the single particle mixing state of individual ship plume events measured at the Port of Los Angeles. Environ. Sci. Technol. 2010, 44, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Gaston, C.J.; Furutani, H.; Guazzotti, S.A.; Coffee, K.R.; Bates, T.S.; Quinn, P.K.; Aluwihare, L.I.; Mitchell, B.G.; Prather, K.A. Unique ocean-derived particles serve as a proxy for changes in ocean chemistry. J. Geophys. Res. Atmos. 2011, 116, D18310. [Google Scholar] [CrossRef]
- Guazzotti, S.A.; Coffee, K.R.; Prather, K.A. Continuous measurements of size-resolved particle chemistry during INDOEX-Intensive Field Phase 99. J. Geophys. Res. Atmos. 2001, 106, 28607–28627. [Google Scholar] [CrossRef]
- Sullivan, R.C.; Guazzotti, S.A.; Sodeman, D.A.; Tang, Y.; Carmichael, G.R.; Prather, K.A. Mineral dust is a sink for chlorine in the marine boundary layer. Atmos. Environ. 2007, 41, 7166–7179. [Google Scholar] [CrossRef]
- The Scripps Pier Page. Available online: http://www.sccoos.org/data/piers/ (accessed on 30 November 2017).
- Gard, E.; Mayer, J.E.; Morrical, B.D.; Dienes, T.; Fergenson, D.P.; Prather, K.A. Real-time analysis of individual atmospheric aerosol particles: Design and performance of a portable ATOFMS. Anal. Chem. 1997, 69, 4083–4091. [Google Scholar] [CrossRef]
- Dall’Osto, M.; Harrison, R.M.; Beddows, D.C.S.; Freney, E.J.; Heal, M.R.; Donovan, R.J. Single-particle detection efficiencies of aerosol time-of-flight mass spectrometry during the North Atlantic marine boundary layer experiment. Environ. Sci. Technol. 2006, 40, 5029–5035. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Bhave, P.V.; Prather, K.A. Comparison of two methods for obtaining quantitative mass concentrations from aerosol time-of-flight mass spectrometry measurements. Anal. Chem. 2006, 78, 6169–6178. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, R.J.; Liu, D.Y.; Edgerton, E.S.; Prather, K.A. Aerosol time-of-flight mass spectrometry during the Atlanta Supersite Experiment: 2. Scaling procedures. J. Geophys. Res. Atmos. 2003, 108, 8427. [Google Scholar] [CrossRef]
- Noble, C.A.; Prather, K.A. Real-time measurement of correlated size and composition profiles of individual atmospheric aerosol particles. Environ. Sci. Technol. 1996, 30, 2667–2680. [Google Scholar] [CrossRef]
- Neubauer, K.R.; Johnston, M.V.; Wexler, A.S. Humidity effects on the mass spectra of single aerosol particles. Atmos. Environ. 1998, 32, 2521–2529. [Google Scholar] [CrossRef]
- Neubauer, K.R.; Johnston, M.V.; Wexler, A.S. On-line analysis of aqueous aerosols by laser desorption ionization. Int. J. Mass Spectrom. Ion Process. 1997, 163, 29–37. [Google Scholar] [CrossRef]
- Allen, J.O. YAADA Software Toolkit to Analyze Single-Particle Mass Spectral Data: Reference Manual Version 1.1. Arizona State University, 2002. Available online: http://www.yaada.org (accessed on 30 November 2017).
- Song, X.H.; Hopke, P.K.; Fergenson, D.P.; Prather, K.A. Classification of single particles analyzed by ATOFMS using an artificial neural network, ART-2a. Anal. Chem. 1999, 71, 860–865. [Google Scholar] [CrossRef]
- Roberts, G.C.; Nenes, A. A continuous-flow streamwise thermal-gradient CCN chamber for atmospheric measurements. Aerosol Sci. Technol. 2005, 39, 206–221. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. Noaa’s Hysplit Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-time Environmental Applications and Display sYstem: READY. Environ. Model. Softw. 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Liu, S.; Day, D.A.; Shields, J.E.; Russell, L.M. Ozone-driven daytime formation of secondary organic aerosol containing carboxylic acid groups and alkane groups. Atmos. Chem. Phys. 2011, 11, 8321–8341. [Google Scholar] [CrossRef]
- Qin, X.; Pratt, K.A.; Shields, L.G.; Toner, S.M.; Prather, K.A. Seasonal comparisons of single-particle chemical mixing state in Riverside, CA. Atmos. Environ. 2012, 59, 587–596. [Google Scholar] [CrossRef]
- Pastor, S.H.; Allen, J.O.; Hughes, L.S.; Bhave, P.; Cass, G.R.; Prather, K.A. Ambient single particle analysis in Riverside, California by aerosol time-of-flight mass spectrometry during the SCOS97-NARSTO. Atmos. Environ. 2003, 37, 239–258. [Google Scholar] [CrossRef]
- Denkenberger, K.A.; Moffet, R.C.; Holecek, J.C.; Rebotier, T.P.; Prather, K.A. Real-time, single-particle measurements of oligomers in aged ambient aerosol particles. Environ. Sci. Technol. 2007, 41, 5439–5446. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Hayes, P.L.; Ortega, A.; Taylor, J.W.; Allan, J.D.; Gilman, J.; Kuster, W.; de Gouw, J.; Jimenez, J.L.; Wang, J. Droplet activation properties of organic aerosols observed at an urban site during CalNex-LA. J. Geophys. Res. Atmos. 2013, 118, 2903–2917. [Google Scholar] [CrossRef]
- Mei, F.; Setyan, A.; Zhang, Q.; Wang, J. CCN activity of organic aerosols observed downwind of urban emissions during CARES. Atmos. Chem. Phys. 2013, 13, 12155–12169. [Google Scholar] [CrossRef]
- Cerully, K.M.; Raatikainen, T.; Lance, S.; Tkacik, D.; Tiitta, P.; Petaja, T.; Ehn, M.; Kulmala, M.; Worsnop, D.R.; Laaksonen, A.; et al. Aerosol hygroscopicity and CCN activation kinetics in a boreal forest environment during the 2007 EUCAARI campaign. Atmos. Chem. Phys. 2011, 11, 12369–12386. [Google Scholar] [CrossRef]
- Gaston, C.J.; Pratt, K.A.; Qin, X.Y.; Prather, K.A. Real-time detection and mixing state of methanesulfonate in single particles at an inland urban location during a phytoplankton bloom. Environ. Sci. Technol. 2010, 44, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Ault, A.P.; Moore, M.J.; Furutani, H.; Prather, K.A. Impact of emissions from the Los Angeles port region on San Diego air quality during regional transport events. Environ. Sci. Technol. 2009, 43, 3500–3506. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.J.; Prather, K.A. Interpretation of mass spectra from organic compounds in aerosol time-of-flight mass spectrometry. Anal. Chem. 2000, 72, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Shields, L.G.; Suess, D.T.; Prather, K.A. Determination of single particle mass spectral signatures from heavy-duty diesel vehicle emissions for PM2.5 source apportionment. Atmos. Environ. 2007, 41, 3841–3852. [Google Scholar] [CrossRef]
- Toner, S.M.; Shields, L.G.; Sodeman, D.A.; Prather, K.A. Using mass spectral source signatures to apportion exhaust particles from gasoline and diesel powered vehicles in a freeway study using UF-ATOFMS. Atmos. Environ. 2008, 42, 568–581. [Google Scholar] [CrossRef]
- Bhave, P.V.; Allen, J.O.; Morrical, B.D.; Fergenson, D.P.; Cass, G.R.; Prather, K.A. A field-based approach for determining ATOFMS instrument sensitivities to ammonium and nitrate. Environ. Sci. Technol. 2002, 36, 4868–4879. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gross, D.S.; Galli, M.E.; Silva, P.J.; Prather, K.A. Relative sensitivity factors for alkali metal and ammonium cations in single particle aerosol time-of-flight mass spectra. Anal. Chem. 2000, 72, 416–422. [Google Scholar] [CrossRef] [PubMed]
- CARB. Final Regulation Order. Fuel Sulfur and Other Operational Requirements for Ocean-Going Vessels within California Waters and 24 Nautical Miles of the California Baseline; California Air Resources Board: Sacramento, CA, USA, 2009.
- Roberts, G.C.; Artaxo, P.; Zhou, J.C.; Swietlicki, E.; Andreae, M.O. Sensitivity of CCN spectra on chemical and physical properties of aerosol: A case study from the Amazon Basin. J. Geophys. Res. Atmos. 2002, 107, 8070. [Google Scholar] [CrossRef]
- Wang, J.; Lee, Y.N.; Daum, P.H.; Jayne, J.; Alexander, M.L. Effects of aerosol organics on cloud condensation nucleus (CCN) concentration and first indirect aerosol effect. Atmos. Chem. Phys. 2008, 8, 6325–6339. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Air Mass | BC (ng/m3) | κ | fCCN | Dcrit (nm) | Submicron Particle Types |
|---|---|---|---|---|---|
| Oceanic | 128.8 (216.3) | 0.23 (0.07) | 0.46 (0.15) | 130 (15) | Sea salts, EC |
| Mixed Coastal I (Cleaner) | 229.5 (375.4) | 0.28 (0.09) | 0.36 (0.14) | 122 (14) | Sea salts, OC, ECOC |
| Mixed Coastal II (Dirtier) | 313.9 (301.7) | 0.22 (0.06) | 0.36 (0.15) | 132 (12) | Aged sea salts, shipping emissions, ECOC |
| Los Angeles/Long Beach | 362.6 (411.8) | 0.26 (0.08) | 0.33 (0.11) | 125 (13) | Shipping emissions |
| Southerly/Mexico | 331.5 (236.5) | 0.31 (0.11) | 0.38 (0.11) | 118 (13) | Biomass burning, No positives-sulfate |
| Inland | 687.6 (492.9) | 0.21 (0.05) | 0.19 (0.10) | 132 (10) | ECOC, EC, OC, Dust |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaston, C.J.; Cahill, J.F.; Collins, D.B.; Suski, K.J.; Ge, J.Y.; Barkley, A.E.; Prather, K.A. The Cloud Nucleating Properties and Mixing State of Marine Aerosols Sampled along the Southern California Coast. Atmosphere 2018, 9, 52. https://doi.org/10.3390/atmos9020052
Gaston CJ, Cahill JF, Collins DB, Suski KJ, Ge JY, Barkley AE, Prather KA. The Cloud Nucleating Properties and Mixing State of Marine Aerosols Sampled along the Southern California Coast. Atmosphere. 2018; 9(2):52. https://doi.org/10.3390/atmos9020052
Chicago/Turabian StyleGaston, Cassandra J., John F. Cahill, Douglas B. Collins, Kaitlyn J. Suski, Jimmy Y. Ge, Anne E. Barkley, and Kimberly A. Prather. 2018. "The Cloud Nucleating Properties and Mixing State of Marine Aerosols Sampled along the Southern California Coast" Atmosphere 9, no. 2: 52. https://doi.org/10.3390/atmos9020052
APA StyleGaston, C. J., Cahill, J. F., Collins, D. B., Suski, K. J., Ge, J. Y., Barkley, A. E., & Prather, K. A. (2018). The Cloud Nucleating Properties and Mixing State of Marine Aerosols Sampled along the Southern California Coast. Atmosphere, 9(2), 52. https://doi.org/10.3390/atmos9020052