3.1. Overview
Compared to the search for plumes at a ground station [
21], the processing of the CARIBIC data is complicated by the variability of the data over large distances in the upper troposphere and by the frequent changes of tropospheric and stratospheric air masses.
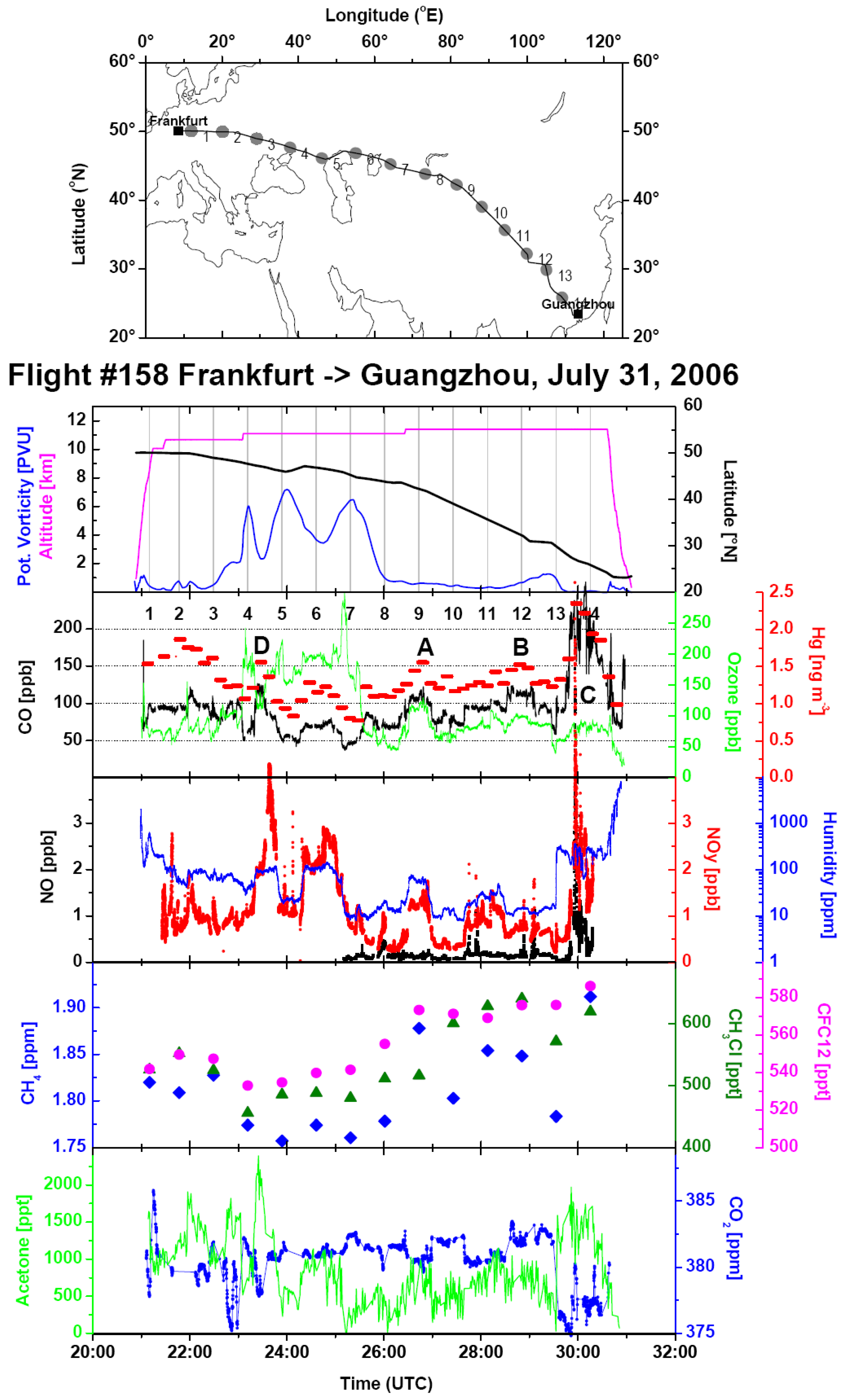
Figure 2 shows an overview of the data from Flight 158 from Frankfurt to Guangzhou on 31 July and 1 August 2006. The aircraft flew in the troposphere until ~23:00 UTC and then in the stratosphere until about 1:45 UTC on 1 August. The stratospheric section is evidenced by the high potential vorticity and O
3 mixing ratio, as well as the low CO mixing ratio shown in the upper two panels of the data time series. In the tropospheric section after about 1:45 UTC on 1 August 2006, mercury background concentrations vary between about 1.25 and 1.35 ng∙m
−3 (the second panel from the top of the data time series). Three events with elevated Hg concentrations, denoted as A, B and C, with peak Hg concentrations of 1.55, 1.5 and 2.3 ng∙m
−3 are observed at about 2:40, 4:50 and 6:00 UTC, respectively, on 1 August 2006. All events are accompanied by elevated CO (the second panel from the top), NOy, H
2O (middle panel) and acetone (bottom panel). We base our approach on the visual inspection of the data overview plots of each flight for the coincident occurrence of elevated Hg concentrations with elevated CO mixing ratios. Plumes identified in this way are cross-checked using variations of other tracers for anthropogenic emissions, such as NOy (middle panel), acetone (bottom panel), CH
4 (the second panel from the bottom), non-methane hydrocarbons (not available for this flight) and halocarbons (the second panel from the bottom). Humidity and cloud water content (determined as the difference between total water content and the water vapour mixing ratio) as tracers for convective processes are also sometimes useful.
An event within the stratospheric section of the flight at ~23:25 UTC (marked as D) has a similar characteristics as Events A, B and C and can easily be mistaken for a plume. The only pronounced difference is that the maximum of Hg concentration (and CO, acetone and H
2O mixing ratios) in Event D coincides with dips in potential vorticity and O
3 mixing ratios, both at higher levels characteristic for the lower stratosphere. Such dips in potential vorticity and O
3 indicate a crossing of a filament of tropospheric air in the stratosphere. The variation of all mentioned species during such crossing results from their strong gradients above the tropopause [
29]. Events of this type are thus not related to surface emissions and have to be eliminated from further consideration. Consequently, only events embedded in air with a potential vorticity of less than 1.5 PVU (1 PVU = 10
−6∙m
2∙K∙kg
−1∙s
−1) and/or less than 150 ppb O
3 were considered.
Because we rely mostly on Hg and CO as plume tracers, only those processes that emit Hg and CO, such as biomass burning, will be detected. This includes also collocated emissions of Hg and CO, CO
2 or CH
4, which applies for most of the urban and industrial emissions. However, emissions from mining and smelting, which emit hardly any CO, CO
2 or CH
4, will not be detected (unless collocated with other CO, CO
2 or CH
4 sources), because a suitable specific tracer for these processes, such as SO
2, is not on the otherwise comprehensive list of CARIBIC
in situ measurements. Lacking
in situ SO
2 measurements also prevents the direct detection and identification of volcanic Hg emissions. One such SO
2 plume was detected during the descent to Frankfurt airport on 15 August 2008, using remote SO
2 sensing by a nadir looking differential optical absorption system (DOAS) on-board CARIBIC and remote sensing satellite [
40]. Quantitative evaluation of this plume in terms of Hg emission, however, was not possible, because the DOAS measurement does not provide
in situ SO
2 concentrations and the elevated Hg concentrations were documented by only one Hg measurement point. Among the substances measured in the whole air samples are tracers for marine emissions, such as short-lived bromine and iodine containing halocarbons, but the low sampling frequency (28 samples taken over four intercontinental flights) makes them unsuitable as tracers for the detection of the Hg plumes of marine origin.
222Rn as a tracer for terrestrial Hg emissions [
22] is also not being measured on-board CARIBIC. Consequently, in this study, we can only distinguish between Hg plumes of biomass burning or urban/industry origin.
The short encounter with the Kasatochi volcano plume (~5 min) in 2008 during the descent to Frankfurt airport [
40] also illustrates some practical limits of our approach. The most severe limitation is given by the low temporal resolution of the Hg measurements of 5–15 min corresponding to a ~75–225 km flight distance. Statistically significant Hg
vs. CO, CO
2 and CH
4 correlations require at least three measurements. Thus, only plumes larger than ~300 km can be captured by our approach, which also means that many plumes encountered during the short aircraft ascents and descents cannot be resolved. The CARIBIC measurements start and stop at ~500 hPa to prevent the contamination of the CARIBIC system by polluted air in the boundary layer near the airports [
23]. Consequently, information for the boundary layer, the most polluted part of the troposphere, is missing in our data set. In addition, the Hg, CO, CO
2 and CH
4 data from ascents and descents through quasi-horizontal layers in the troposphere are likely to violate the assumption of a plume being embedded in a homogeneous air mass on which the Hg
vs. CO, CO
2 and CH
4 correlations are based.
Although almost all of the Hg plumes were accompanied by elevated CO mixing ratios, seven of them were observed during flight sections with nearly constant CO mixing ratios. All of these plumes were encountered over the equatorial Atlantic Ocean between 0° and 15° N during the flights to South American destinations. They were embedded in background mercury concentrations varying between ~0.95 and 1.3 ng∙m
−3, and the elevation above the background (ΔHg) varied between ~0.25 and 0.45 ng∙m
−3. In these events, elevated Hg concentrations were always accompanied by elevated humidity, frequently also with clouds and elevated NOy, as well as with low O
3 mixing ratios; frequently, only around 30 ppb. Such low O
3 mixing ratios are typical for the marine boundary layer over the equatorial Atlantic Ocean [
41]. Backward trajectories reveal contact with the equatorial Atlantic Ocean surface. Satellite images of cloud cover indicate that these events are due to the convection of the air masses from the marine boundary layer at the ITCZ. Elevated Hg concentration in these events encountered in the upper free troposphere can point to emissions of mercury by ocean [
42], but we lack highly resolved tracer data for air from the marine boundary layer to quantitatively describe them.
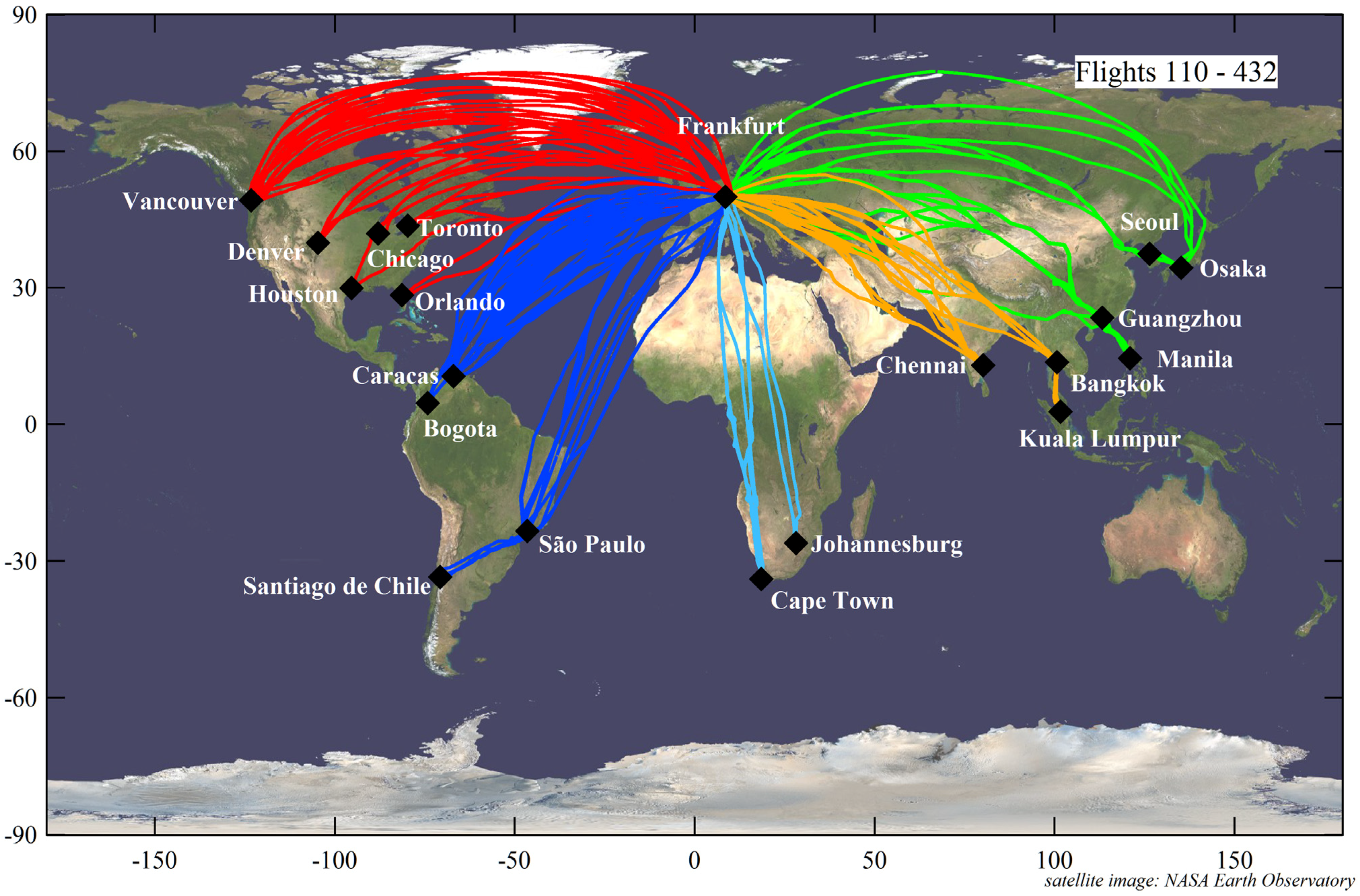
Despite these caveats and limitations, 98 encounters with plumes with elevated CO mixing ratios and simultaneously elevated Hg concentrations were counted during 309 CARIBIC flights with valid Hg measurements between May 2005 and June 2013. Taking into account the number of flights to the respective regions listed in
Table 1, the probability of plume encounters was highest during the flights to South Africa with 85% of the flights. The second highest probability of plume occurrence was over East Asia with 46%, followed by flights to South Asia with 26%, South America with 20% and North America with 17%. The low frequency of plume encounters over North America is partly due to the high northern latitude of the flight routes of these flights, which, at usual flight altitudes of 10–12 km, results in a high proportion of stratospheric sections with a potential vorticity >1.5 PVU. However, the high frequency of plume encounters during the flights to Osaka and Seoul with flight routes at similarly high northern latitudes shows that this bias alone cannot explain the low frequency of plume encounters over Europe and North America.
Figure 2.
Overview of the data from Flight 158 from Frankfurt to Guangzhou on 31 July 2006. (Top) Flight track and the locations of whole air samples. Time series plots are below: (uppermost panel): flight altitude (magenta) and latitude (black), potential vorticity (blue), sampling intervals (grey bars); (second panel from top): mixing ratios of CO (black), O3 (green) and Hg concentrations (red); (middle panel): mixing ratios of NO (black), NOy (red) and total water content (blue); (second panel from bottom): mixing ratios of CH4 (blue), CH3Cl (olive green) and CFC12 (CCl2F2, magenta)) in whole air samples; (bottom panel): mixing ratios of acetone (green) and CO2 (blue). The three identified plumes are marked with A, B and C in the second panel from top. Another event, due to a crossing of a filament of tropospheric air within the lower stratosphere, is marked with D. Although similar to Events A, B and C, this event has no relation to surface emissions (see the text).
Figure 2.
Overview of the data from Flight 158 from Frankfurt to Guangzhou on 31 July 2006. (Top) Flight track and the locations of whole air samples. Time series plots are below: (uppermost panel): flight altitude (magenta) and latitude (black), potential vorticity (blue), sampling intervals (grey bars); (second panel from top): mixing ratios of CO (black), O3 (green) and Hg concentrations (red); (middle panel): mixing ratios of NO (black), NOy (red) and total water content (blue); (second panel from bottom): mixing ratios of CH4 (blue), CH3Cl (olive green) and CFC12 (CCl2F2, magenta)) in whole air samples; (bottom panel): mixing ratios of acetone (green) and CO2 (blue). The three identified plumes are marked with A, B and C in the second panel from top. Another event, due to a crossing of a filament of tropospheric air within the lower stratosphere, is marked with D. Although similar to Events A, B and C, this event has no relation to surface emissions (see the text).
For 56 plume encounters (out of 98), the Hg
vs. CO correlations were statistically significant at a confidence level of at least 95%. For these correlations, data from the same plume encountered twice in the vicinity of an airport were combined, e.g., for a plume near Guangzhou encountered during the forward and return flight to Manila (Flights 203 and 204) or a plume encountered near São Paulo during Flights 123 (Frankfurt→São Paulo), 124 (São Paulo→Santiago de Chile) and 125 (Santiago de Chile→São Paulo). Seven extremely high Hg/CO slopes of 12.9–23.7 pg∙m
−3∙ppb
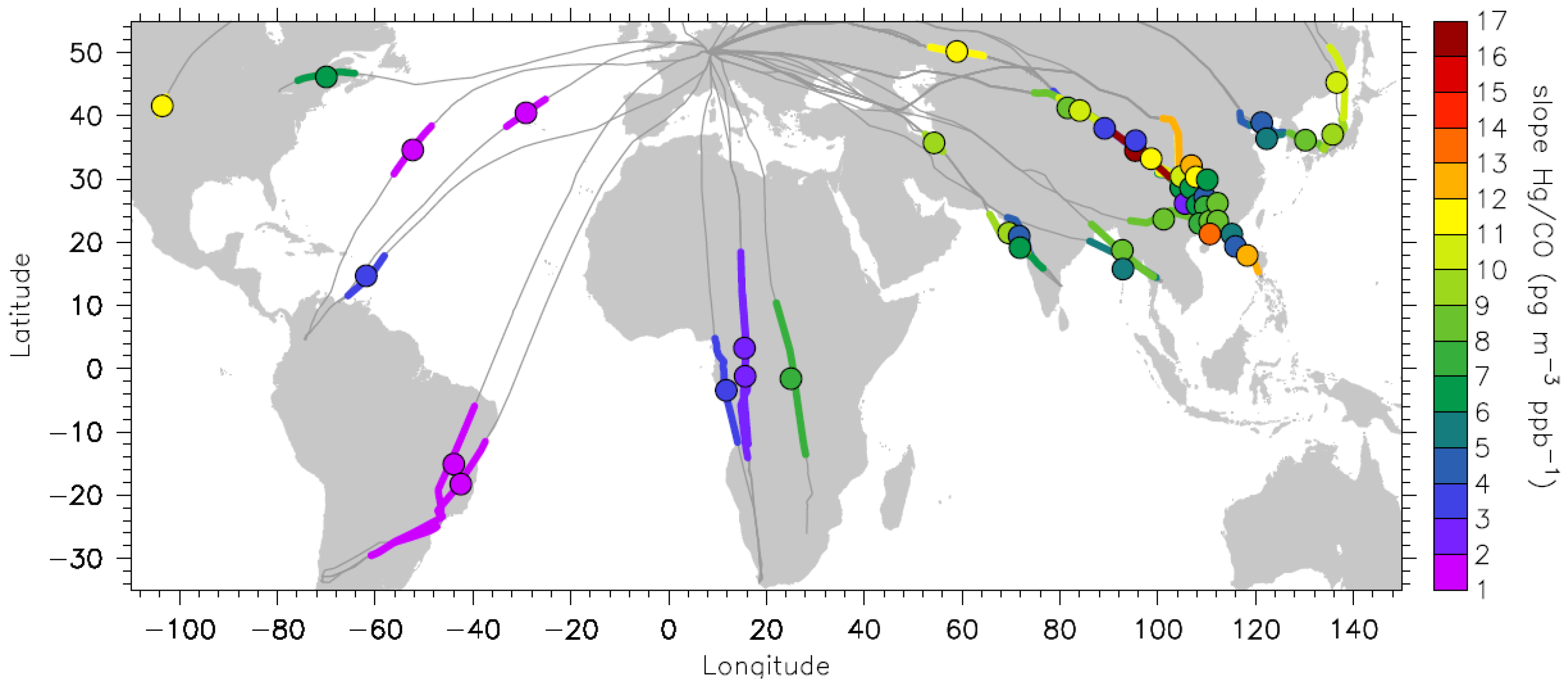
−1 were connected with physically unrealistic negative intercepts and are thus eliminated from the data set, leaving 49 plume encounters with valid Hg/CO slopes. The geographic distribution of these plumes is shown in
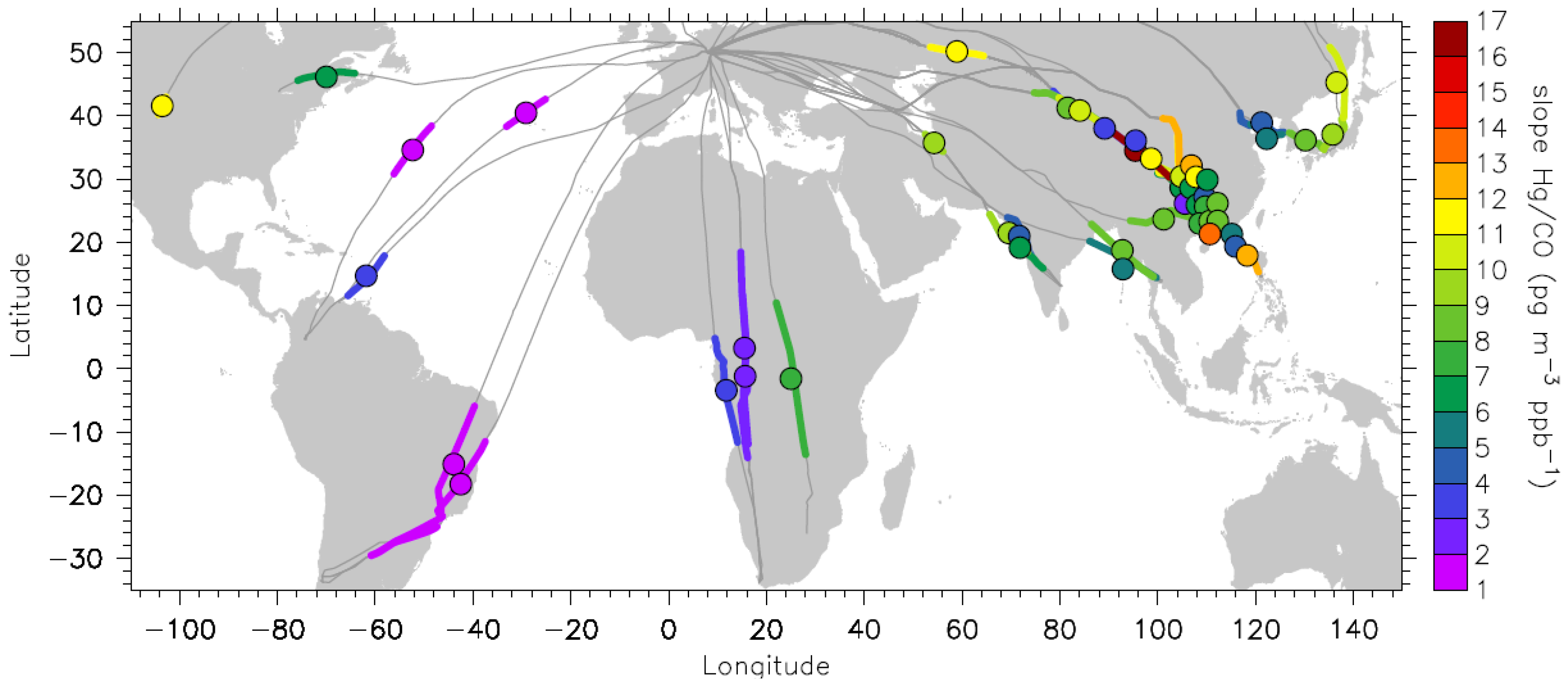
Figure 3.
Table 1.
Overview of encounters with plumes with elevated Hg concentrations and CO mixing ratios.
Table 1.
Overview of encounters with plumes with elevated Hg concentrations and CO mixing ratios.
| Destination Airport | Number of Flights | Number of Plume Encounters | Number of Plumes With Significant Hg vs. CO Correlations | Median and Range of Hg/CO Emission Ratios (pg·m−3·ppb−1) |
|---|
| South Africa | 13 | 11 | 4 | 2.9 (2.2–7.5) |
| East Asia | 101 | 46 | 31 | 8.2 (2.3–16.6) |
| South Asia | 57 | 15 | 6 | 7.4 (5.0–10.0) |
| South America | 90 | 18 | 5 | 1.3 (1.1–3.3) |
| North America | 48 | 8 | 2 | 9.2 (6.9 and 11.4) |
Figure 3.
Geographic distribution and the extension of the plumes with statistically significant Hg vs. CO correlations. The magnitude of Hg/CO emission ratios in pg∙m−3∙ppb−1 is colour coded.
Figure 3.
Geographic distribution and the extension of the plumes with statistically significant Hg vs. CO correlations. The magnitude of Hg/CO emission ratios in pg∙m−3∙ppb−1 is colour coded.
Most of the plumes with statistically significant Hg
vs. CO correlations (31 plumes) were encountered over the East Asian region, and these are listed in Table S1 (Supplementary Information). Table S2 lists 18 plumes with statistically significant Hg
vs. CO correlations for all other regions. Relative to the number of flights, the frequency of plumes with statistically significant Hg/CO correlations is the highest for the South African and East Asian flights, with each being 31%, followed by flights to South Asia with 11%, South America with 6% and North America with 4%. The high occurrence of plumes during the flights to South Africa in which Hg does not correlate with CO is caused by their large extension over a few thousands of km, changing the Hg and CO background from north to south hemispheric concentrations and the inhomogeneity of the plumes. This will be discussed later in
Section 3.3.
The colour code of
Figure 3 reveals a pronounced difference between the Hg/CO emission ratios in different regions. The Hg/CO emission ratios for plumes encountered over East Asia range from 2.3 to 16.6 pg∙m
−3∙ppb
−1 (median 8.2 pg∙m
−3∙ppb
−1) and are similar to those over South Asia, ranging from 5.0 to 10.0 pg∙m
−3∙ppb
−1 (median 7.4 pg∙m
−3∙ppb
−1). On the contrary, the Hg/CO emission ratios for plumes observed during the flights to South America and equatorial Africa range from 1.1 to 7.4 pg∙m
−3∙ppb
−1, with a median value of 1.8 pg∙m
−3∙ppb
−1 (a median of 1.3 pg∙m
−3∙ppb
−1 for South America and 2.9 pg∙m
−3∙ppb
−1 for equatorial Africa). These plumes include also two plumes encountered over the Atlantic Ocean, which, based on backward trajectories, can be attributed to forest fires in the USA, as will be discussed in
Section 3.4. The range and median of Hg/CO emission ratios observed over South America and equatorial Africa is similar to the one for plumes observed at Cape Point in South Africa [
21]. A compilation of previously published Hg/CO emission ratios reported for different processes and regions [
29] shows that biomass burning is characterized by ratios below 2 pg∙m
−3∙ppb
−1 [
43], whereas the ratios for urban/industrial emissions tend to be around 6 pg∙m
−3∙ppb
−1 and higher. Applying these criteria to the Hg/CO emission ratios shown in
Figure 3 thus leads to the conclusion that the plumes encountered during the flights to South America and equatorial Africa originate predominantly from biomass burning (see also [
44]), whereas the plumes observed over East Asia, South Asia and North America are of industrial/urban or mixed origin. This preliminary classification is supported by the detailed discussion in
Section 3.2,
Section 3.3 and
Section 3.4.
CO
2 emissions are better known than those of CO, and thus, the Hg/CO
2 emission ratios have a potential to provide a more accurate estimate of Hg emissions [
21]. Unfortunately, CO
2 data were available for only 69 (out of 98) of the encountered plumes, of which nine were too narrow for Hg
vs. CO
2 correlations. Statistically significant correlations of Hg
vs. CO
2 were only found for the 17 plumes listed in Table S3. These correlations will be discussed in
Section 3.5.
Mercury was also found to correlate frequently with methane at Cape Point, which proved to be useful for constraining the mercury emissions in South Africa [
21]. Methane data were available only for flights since October 2010, and altogether, 26 correlations of Hg
vs. CH
4 could be calculated, of which, only the six listed in Table S4 were statistically significant. These will be discussed in
Section 3.6.
3.2. Plumes Encountered during the East Asian Flights
The East Asian plumes were encountered during the flights from Frankfurt to Manila with a stopover at Guangzhou and during the flights to Osaka and Seoul. Apart from a few plumes over Central Asia, most of them were encountered within a ~2000-km distance from the airports at Guangzhou, Osaka and Seoul. Data from one of the flights have already been shown in
Figure 2. The Hg/CO emission ratios were 8.8 ± 1.5, 11.3 ± 2.0 and 7.49 ± 1.0 pg∙m
−3∙ppb
−1 for Events A, B and C, respectively. Air sample analyses (the second panel from the bottom) show high CH
4 in Sample 9 (Event A, sample numbers are marked in the uppermost panel), 11 and 12 (Event B), and the highest level in Sample 14 (Event C). Acetonitrile as a tracer for biomass burning is not available for this flight section, but elevated CH
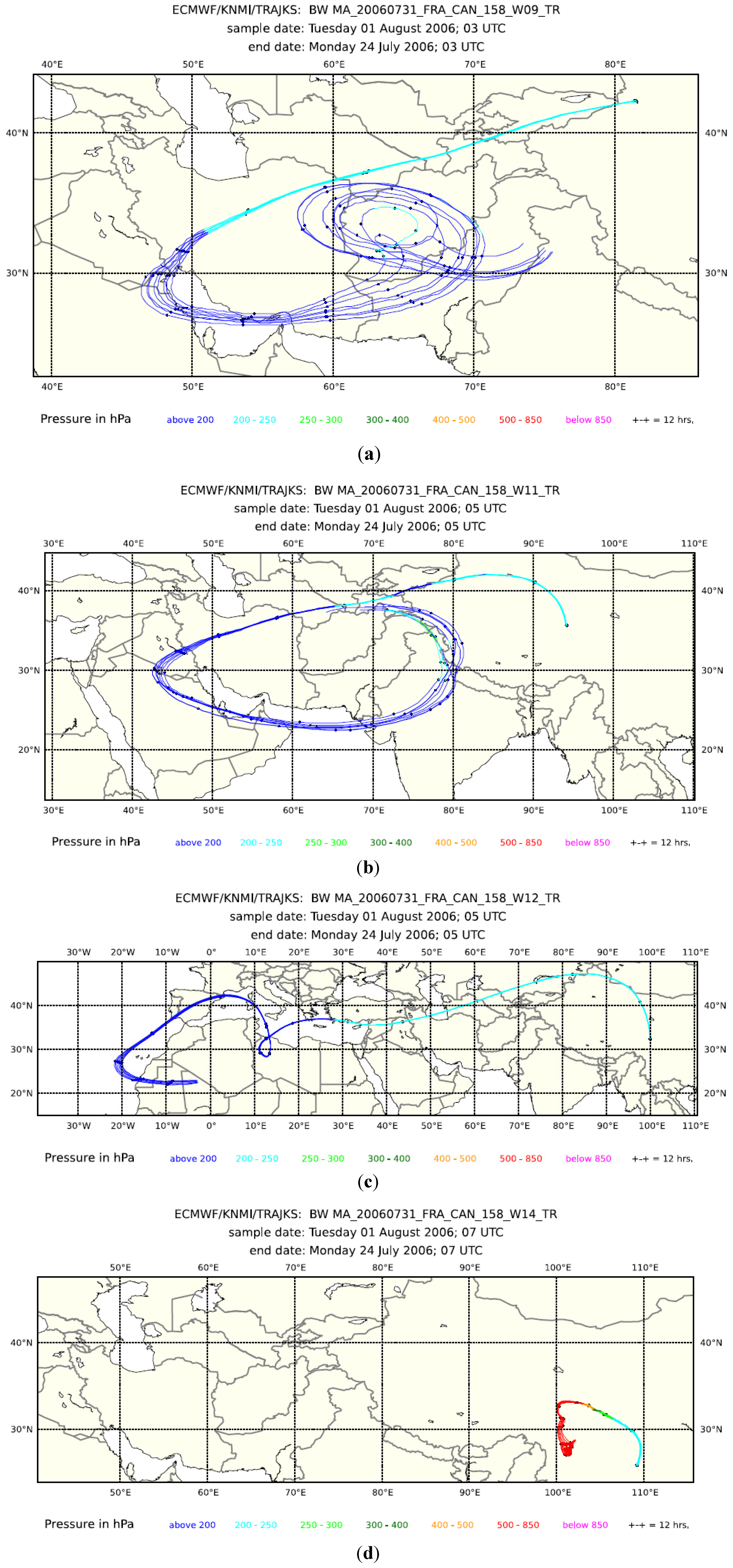
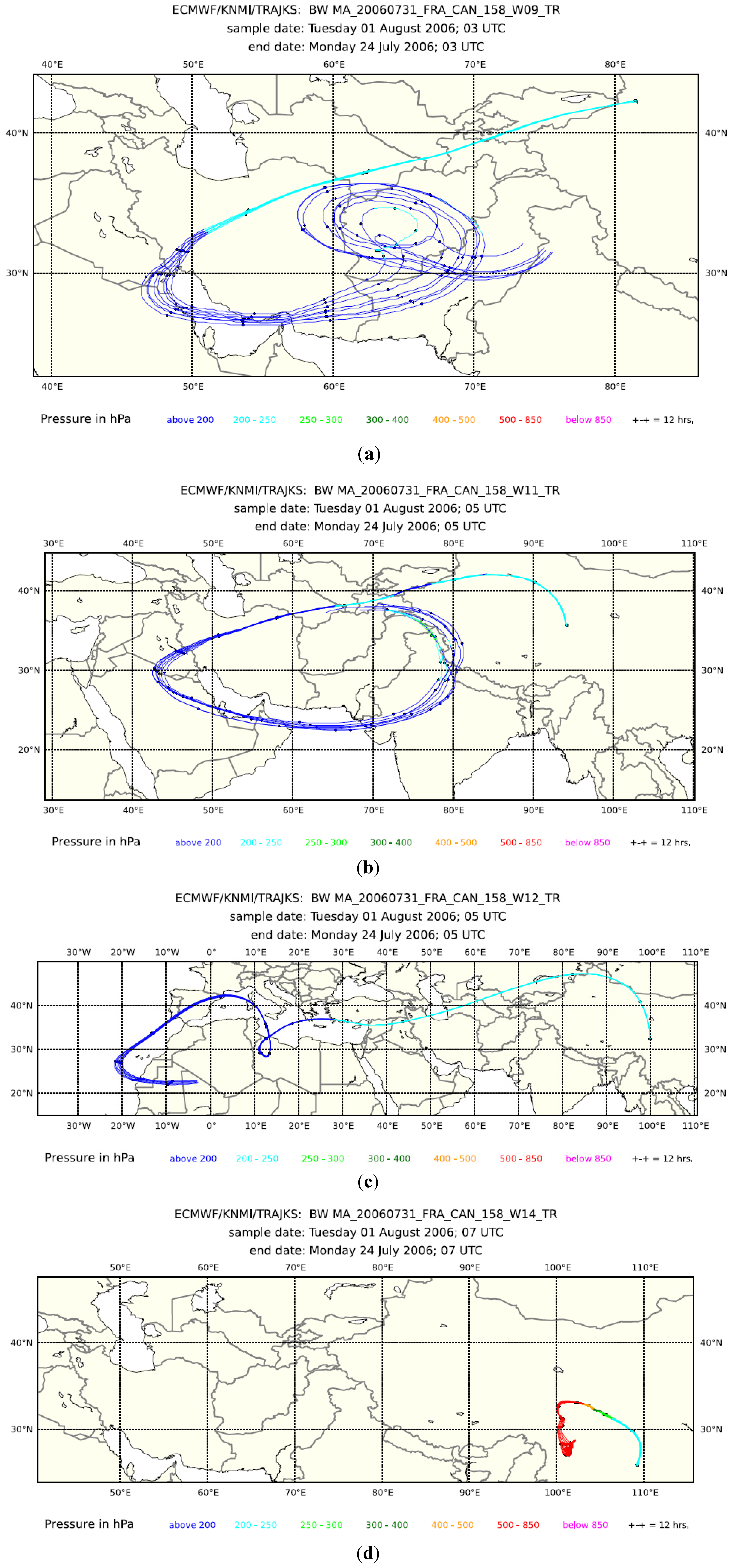
3Cl mixing ratios (dark green triangles in the second panel from the bottom) indicate some influence of biomass/biofuel burning in Samples 10, 11, 12 and 14, but not in Sample 9. Backward trajectories for the preceding eight days for Samples 9, 11 and 12 in
Figure 4 all show that transport took place at a higher altitude (<250 hPa). Notwithstanding, elevated H
2O mixing ratios and satellite cloud images (not shown) indicate convection in the area above the Persian Gulf and the Gulf of Oman for Samples 9 and 11 and above the Iberian Peninsula for Sample 12. Backward trajectories for Sample 14 had a surface contact over Sichuan, China. Fire maps (not shown) show fire counts in Oman and the Indus River Valley, which are reached by the trajectories of Sample 11, but not of 9. The biomass burning contribution for Sample 12 probably originates from fire activity in north-western Spain. No fire counts were reported for Sichuan during this period, but the observed influence of biomass burning could originate from biofuel use.
The composition and origin of the plumes over southern China and the Philippines (around the airport of Guangzhou, east of 103° E and south of 32° N) were analysed in detail by Lai
et al. [
45]. According to their chemical signatures, the 51 identified high CO events were attributed to biomass/biofuel burning, anthropogenic emissions or a mixture of both. The backward trajectories pointed to three source areas, namely: southern China, the Indochinese Peninsula and the Philippines/Indonesia. The emissions from southern China were found to be dominated by urban/industrial emissions, while emissions from biomass/biofuel burning contributed substantially to plumes from the Indochinese Peninsula. Mixed emissions were attributed to plumes originating from the Philippines/Indonesia.
Figure 4.
Eight-day backward trajectories for whole air Samples 9 (a), 11 (b), 12 (c) and 14 (d) taken during Flight 158.
Figure 4.
Eight-day backward trajectories for whole air Samples 9 (a), 11 (b), 12 (c) and 14 (d) taken during Flight 158.
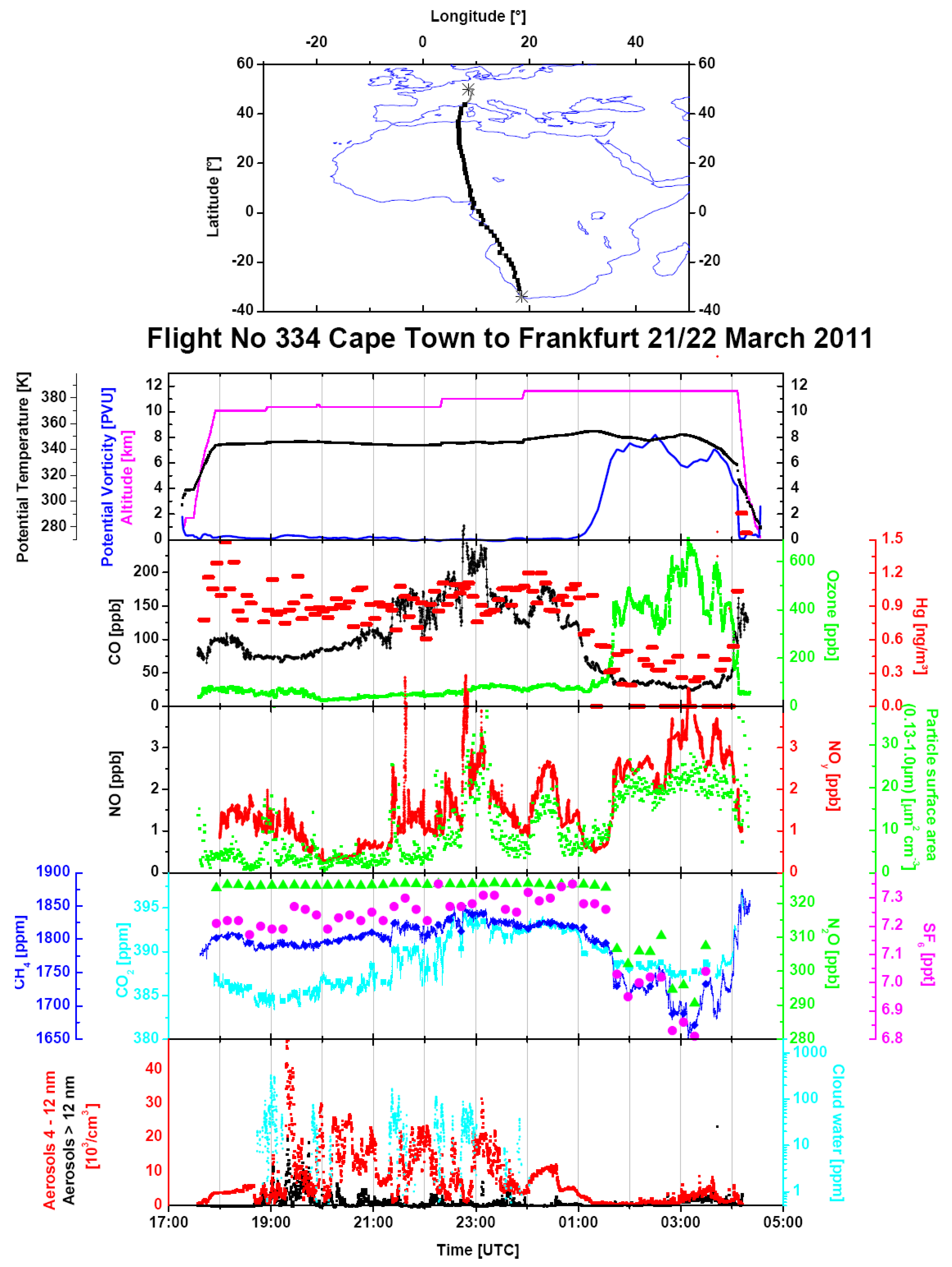
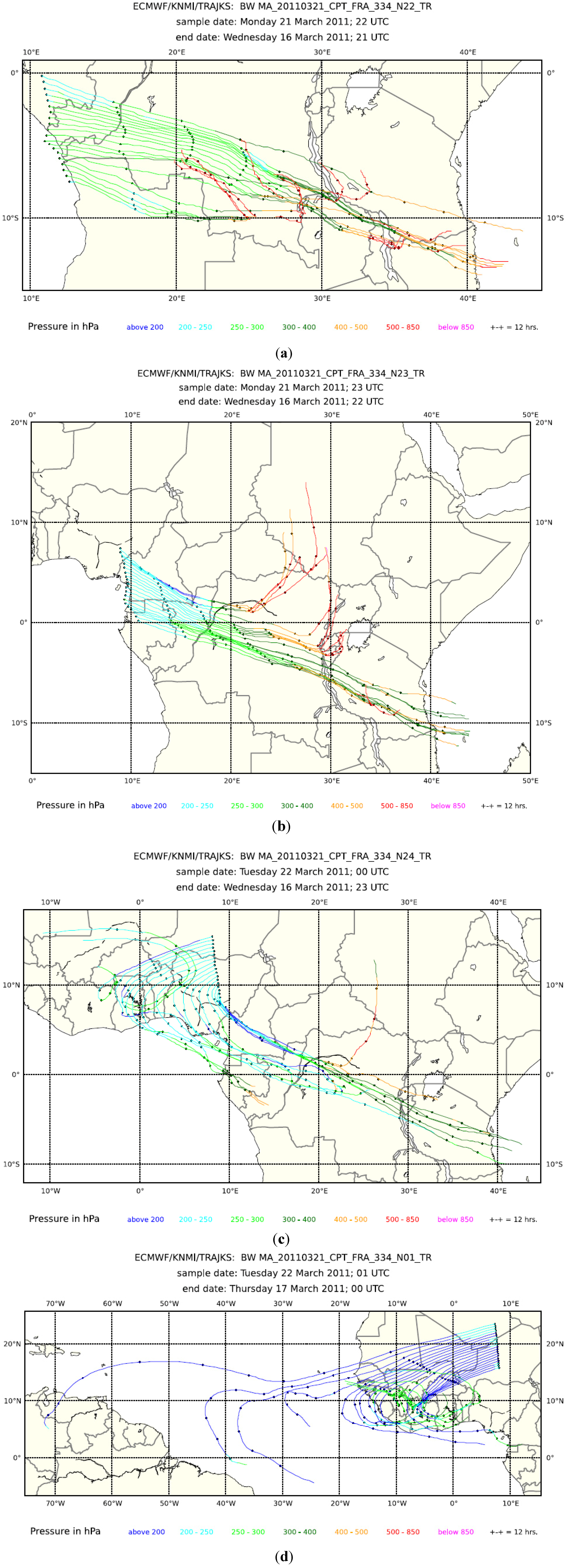
Figure 5.
The overview of the data from Flight 334 from Cape Town to Frankfurt on 21/22 March 2011. The parameters displayed here are similar to those in
Figure 2. (
Middle) The time series plots additionally show the particle surface area concentrations (green); (
second panel from the bottom) mixing ratios of SF
6 (magenta) and N
2O (green) in whole air samples, as well as continuously measured mixing ratios of CH
4 (dark blue) and CO
2 (light blue); (
bottom) cloud water content (light blue) and concentrations of particles within the 4–12 nm size range (red) and larger than 12 nm (black).
Figure 5.
The overview of the data from Flight 334 from Cape Town to Frankfurt on 21/22 March 2011. The parameters displayed here are similar to those in
Figure 2. (
Middle) The time series plots additionally show the particle surface area concentrations (green); (
second panel from the bottom) mixing ratios of SF
6 (magenta) and N
2O (green) in whole air samples, as well as continuously measured mixing ratios of CH
4 (dark blue) and CO
2 (light blue); (
bottom) cloud water content (light blue) and concentrations of particles within the 4–12 nm size range (red) and larger than 12 nm (black).
A dense plume observed during Flight 300 from Osaka to Frankfurt on 24 June 2010, covering a distance of about 1000 km over the Korean Peninsula and Yellow Sea, was characterized by CO mixing ratios of ~240 ppb, Hg concentrations of 2.25 ng∙m
−3 and an Hg/CO emission ratio of 4.4 pg∙m
−3∙ppb
−1. High mixing ratios of biomass burning tracers, such as CH
3Cl, and of tracers of anthropogenic origin, such as SF
6, together with elevated levels of pollutants, which may originate from both biomass burning and anthropogenic processes, such as ethyne and propane, point to the mixed origin of this plume, both from anthropogenic processes and from biomass burning. Backward trajectories and a map of fire counts for 20–24 June 2010 (not shown, FIRMS (Fire Information for Resource Management System) web fire mapper
http://firefly.geog.umd.edu, accessed on 28 October 2010), indicate that the biomass burning component originated most likely from a region with a high burning density in Shandong, Henan, Shanxi and Hebei provinces of China and possibly from some fires in southern Siberia. The anthropogenic component most likely originated from the Chinese provinces mentioned above.
The plumes observed near Osaka during two flights, 331 and 332, on 26/27 February 2011, are characterised by similar CO mixing ratios as in June 2010, but higher Hg concentrations of ~2.7 ng∙m−3, resulting in the higher Hg/CO emission ratios of 8.4 and 10.0 pg∙m−3∙ppb−1, which point to urban/industrial origin. Air samples taken within the plume had elevated mixing ratios of SF6 (~7.6 ppt), ethyne (~500 ppt), CH4 (~1870 ppb), CO2 (~397 ppm) and several other hydrocarbons, documenting the urban/industrial component of the plumes. Biomass burning also contributed to this plume, as evidenced by high CH3Cl mixing ratios of ~700 ppt. However, the high Hg/CO slope of ~9 pg∙m−3∙ppb−1 and the high SF6 mixing ratios imply urban/industrial origin to be dominating. Backward trajectories for the plume observed during Flight 331 show a fast transport from the west with surface contact over northern India, southern Pakistan and southern Iran (within ~3 days) and a transport at high altitudes afterwards. Backward trajectories for the plume observed during Flight 332 are similar, but because of their lower altitude contributions from sources in China, cannot be ruled out. However, based on the trajectories from Flight 331 and the low probability of convection in February, we deem northern India and southern Pakistan to be the major source of the observed plumes.
A narrow plume observed during the descent to Seoul during Flight 383 on 29 March 2012, is characterised by very high CO mixing ratios of up to 357 ppb, an Hg concentration of up to 2.49 ng∙m−3 and an Hg/CO emission ratio of 5.6 pg∙m−3∙ppb−1. Apart from high NOy and CO2, there are no other measurements available to characterise this plume. The medium Hg/CO slope indicates a mixture of emissions from biomass burning and industrial/urban emissions. The backward trajectories are changing during the descent and point to northern China or/and southern Siberia as possible source areas.
Several plumes were also encountered over central Asia on the way to Guangzhou and back, especially during Flights 158, 161 (both flights were on 1 August 2006), 166 and 169 (both flights were on October 20, 2006). The events during the outward bound flight on October 20 at 3:53–4:53 UTC and during the return flight on the same day at 17:49–20:29 UTC have very similar Hg/CO emission ratios of 3.5 ± 0.7 and 3.2 ± 1.1 pg∙m
−3∙ppb
−1, respectively, and originate, with little doubt, from one and the same plume. This plume is analysed in detail by Baker
et al. [
46]. The low Hg/CO emission ratio and elevated acetonitrile, CH
3Cl and CH
3Br mixing ratios suggest that the plume originates partly from biomass/biofuel burning, whereas elevated mixing ratios of C
2Cl
4 and toluene, which are used as solvents, indicate anthropogenic contributions. Backward trajectories for the samples taken in this plume pass over Afghanistan, Pakistan and a region of Northern India, where extensive fire activity was recorded during 17–29 October 2006 [
46]. Another plume with an Hg/CO emission ratio of 11.2 ± 3.4 pg∙m
−3∙ppb
−1 and very high acetone mixing ratios was encountered during Flight 161 on 1 August 2006, at 22:07–23:07 UTC. No air samples were taken during this event. Backward trajectories pass at high altitude (<300 hPa) over the Black Sea partly to northern Europe and partly to the European Mediterranean coast. Elevated H
2O mixing ratios and satellite images indicate some convective activity west of the Black Sea. Extensive fire activity was recorded during this time for a broad area along the northern coast of the Black Sea and for the Mediterranean coast.
3.3. Plumes over Africa
Data are available for a total of 13 flights en route between Frankfurt and Cape Town or Johannesburg. As the South African airports were served by Lufthansa Airbus A340-600 aircraft only during the winter flight schedule, the data cover the months from November to March (March 2009–March 2011) encompassing the austral summer. Elevated CO mixing ratios over equatorial Africa were encountered during all flights with peak values varying between ~120 and ~250 ppb, but for only four flights, the Hg vs. CO correlation was statistically significant. The reasons for such a low yield are discussed below.
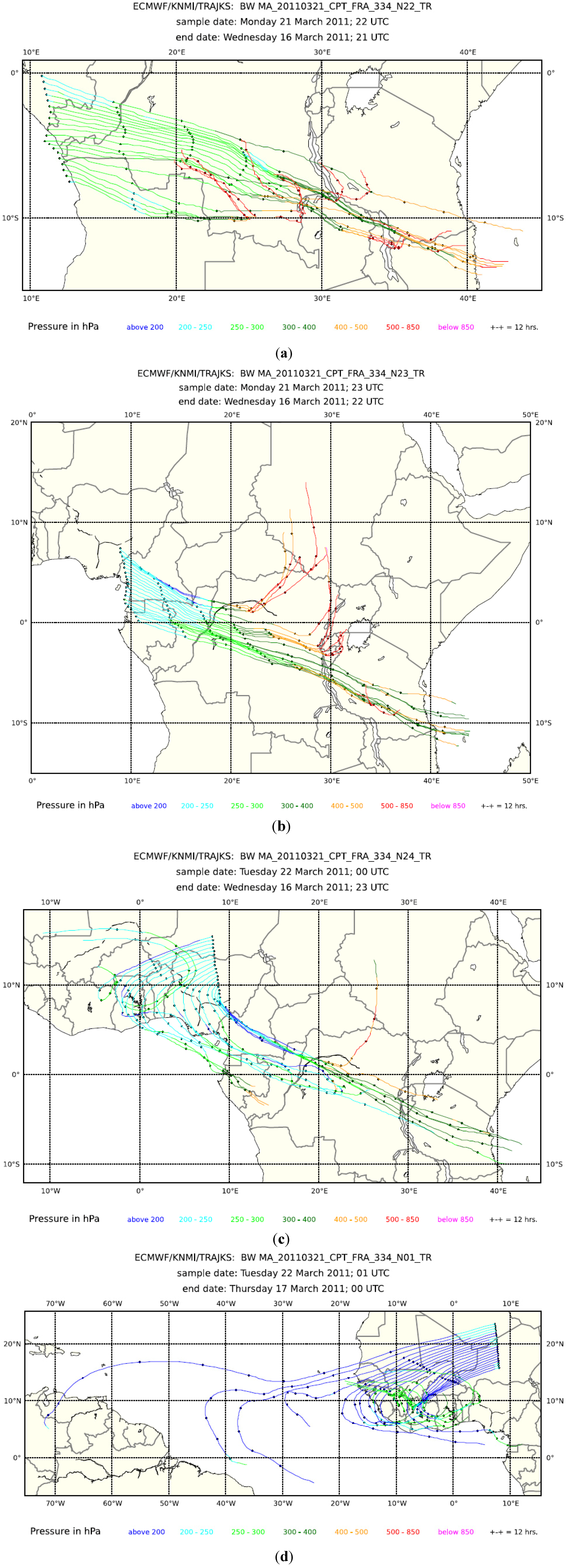
Figure 6.
Five-day backward trajectories (every 3 min) for Flight 334 from Cape Town to Frankfurt on 21 and 22 March 2011: (a) 21–22 UTC; (b) 22–23 UTC; (c) 23–24 UTC; and (d) 0–1 UTC.
Figure 6.
Five-day backward trajectories (every 3 min) for Flight 334 from Cape Town to Frankfurt on 21 and 22 March 2011: (a) 21–22 UTC; (b) 22–23 UTC; (c) 23–24 UTC; and (d) 0–1 UTC.
Figure 5 and Figure S1 show a typical example of the data obtained during Flight 334 from Cape Town to Frankfurt on 20 and 21 March 2011. The second panel from the top of the data time series in
Figure 5 shows somewhat elevated CO mixing ratios of ~100 ppb after ascent, decreasing to a southern hemispheric background of ~ 75 ppb after 18:30 UTC. CO then increases in the course of the flight up to a maximum of ~240 ppb between 22:45 and 23:10 UTC to subsequently decrease, with another smaller peak, with a maximum of ~175 ppb around 0:20 UTC, to ~125 ppb, before the aircraft crosses the tropopause into the lower stratosphere at ~1:00 UTC. The NOy mixing ratio and particle surface area concentration (the third panel from the top) display a similar pattern, even in the finer structure, whereas CO
2 and CH
4 show only a broad maximum with a somewhat different shape, peaking shortly before 23:00 UTC. The SF
6 mixing ratio increases gradually from ~7.2 ppt after ascent to ~7.3 ppt before entering the stratosphere. Although small, this increase documents that all plume observations are embedded in a broad gradient between southern and northern hemispheric air masses [
47]. Mixing ratios of ethane, propane and ethyne, shown in Figure S1, broadly follow the CO pattern, but high mixing ratios of short-lived n-butane and i-butane around 21:15 UTC and at 23:00 UTC indicate the admixture of freshly polluted air. The CH
3Cl mixing ratio (Figure S1, lowermost panel) of ~ 650 ppt in the tropospheric section of the flight is substantially higher than the background mixing ratio of ~550 ppt, and this implies a large-scale influence of biomass burning. The highest CH
3Cl mixing ratios of almost 700 ppt are found in two samples taken after midnight. They coincide with elevated mixing ratios of CO, ethane, propane, ethyne and NOy (
Figure 5, middle panel), but the short-lived butanes have almost disappeared. Such a coincidence is characteristic for aged air from another regional biomass burning plume. Consequently, the CO bulge over equatorial Africa has to be viewed as a composite of several regional plumes. Backward trajectories, shown in
Figure 6 for 21–22 UTC (a), 22–23 UTC (b), 23–24 UTC (c) and 0–1 UTC (d), support this view by pointing to different source regions in different sections of the flight. Several of the trajectories exhibit rather fast upward transport from the boundary layer and lower troposphere (purple and red colours). Cloud water content (
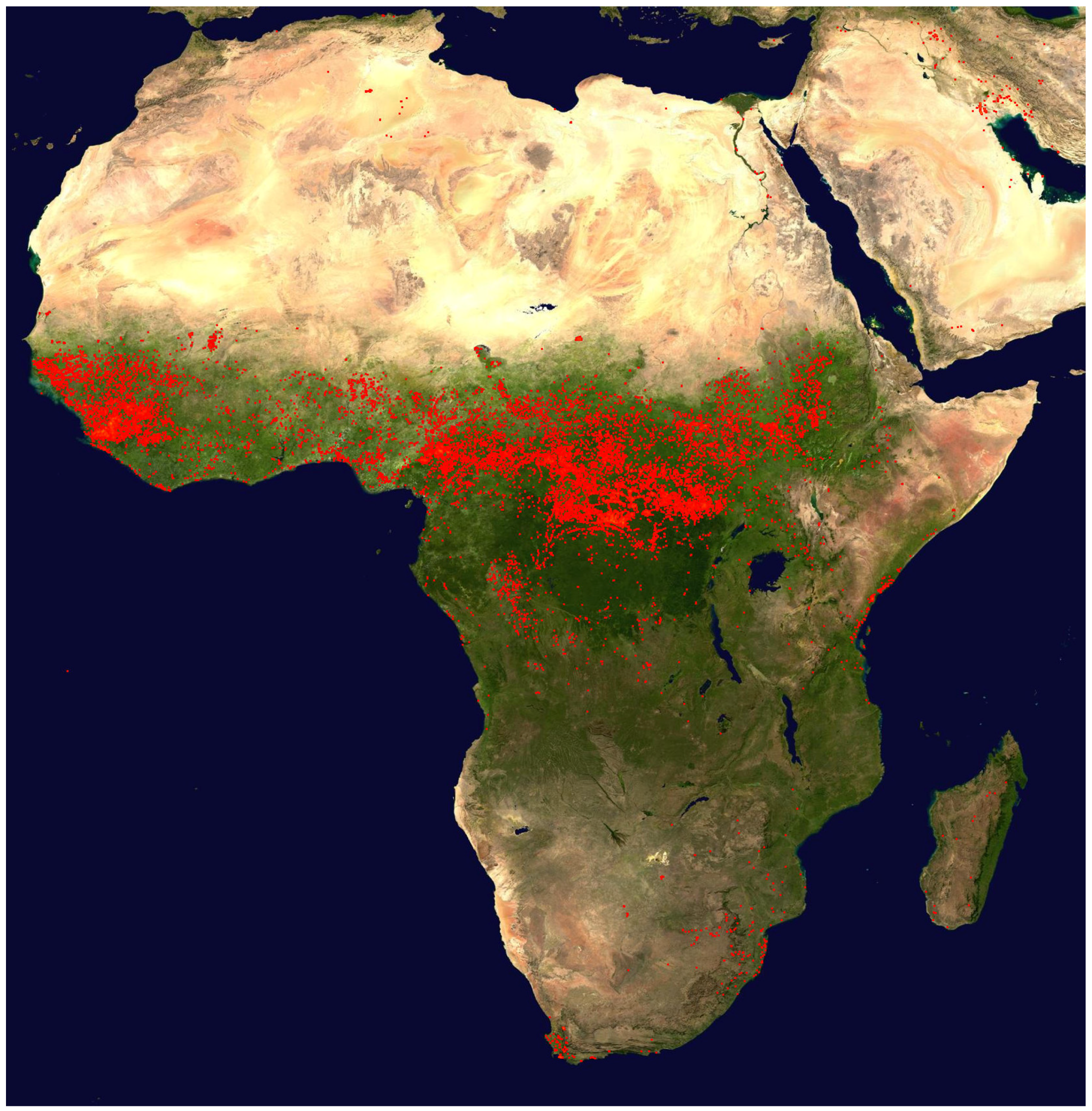
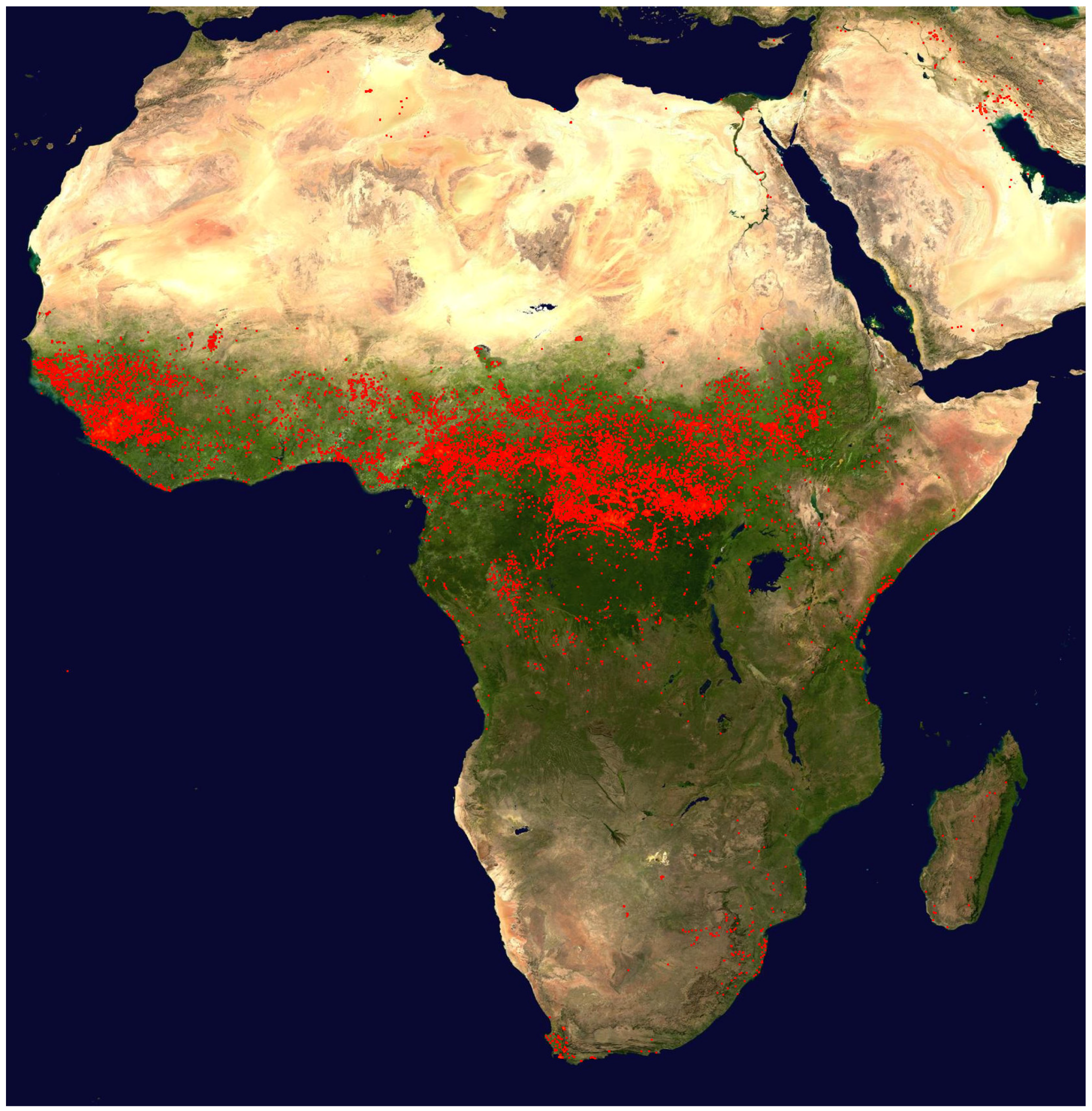
Figure 5, bottom panel) in several sections of the flight is a sign of convective activity in these areas. The fire map displayed in
Figure 7 and the trajectories show that emissions from biomass burning have, to a varying degree, influenced all observations between ~21:00 UTC on 20 March to ~1:00 UTC on 21 March.
The correlation of Hg vs. CO was statistically non-significant for the whole plume starting at 21:16:30 and ending at 01:01:30 UTC, as well as for sections of it, such as between 23:56:30 and 00:56:30 UTC or between 21:21:30 and 23:56:30 UTC. The correlation of Hg vs. CO2 was statistically significant for the whole plume (239.4 ± 94.4 pg∙m−3∙ppm−1 at > 95% level), but statistically non-significant for the sections mentioned above.
In summary, the scarcity of statistically significant Hg vs. CO and Hg vs. CO2 correlations in the plumes observed over equatorial Africa is a result of several factors. The plumes are embedded in broad north-south gradients, which violates the assumption of a constant background. Due to their large extent, they consist of a multitude of overlapping smaller plumes from different regions and sources and are thus not homogeneous. In addition, the CO enhancements (ΔCO) against the background are rather small, varying between ~45 ppb during Flights 290 and 291 to Cape Town on 27 and 28 October 2009, to ~165 ppb during Flights 333 and 334 to Cape Town on 20 and 21 March 2011. Assuming that the plumes originate predominantly from biomass burning with a typical Hg/CO emission ratio of 1 pg∙m−3∙ppb−1, the CO enhancements of this magnitude would produce Hg enhancements of only 0.045 to 0.165 ng∙m−3. Such enhancements are difficult to detect with a precision of 0.05 ng∙m−3 of the mercury measurements, even if the background were constant and the plumes homogeneous. For all of these reasons, the Hg/CO emission ratios derived from these flights will be substantially more uncertain than their statistical uncertainty stated in Table S2.
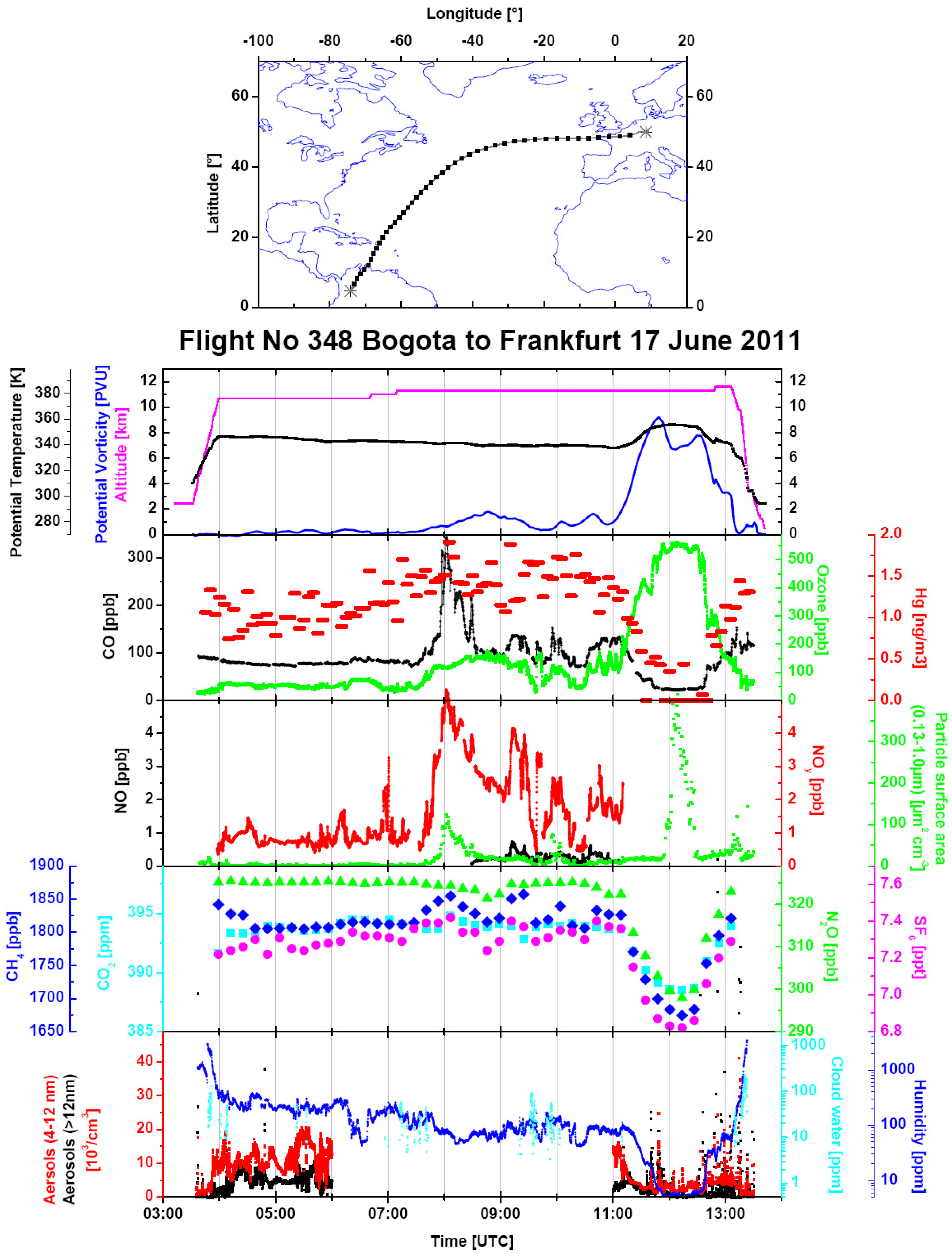
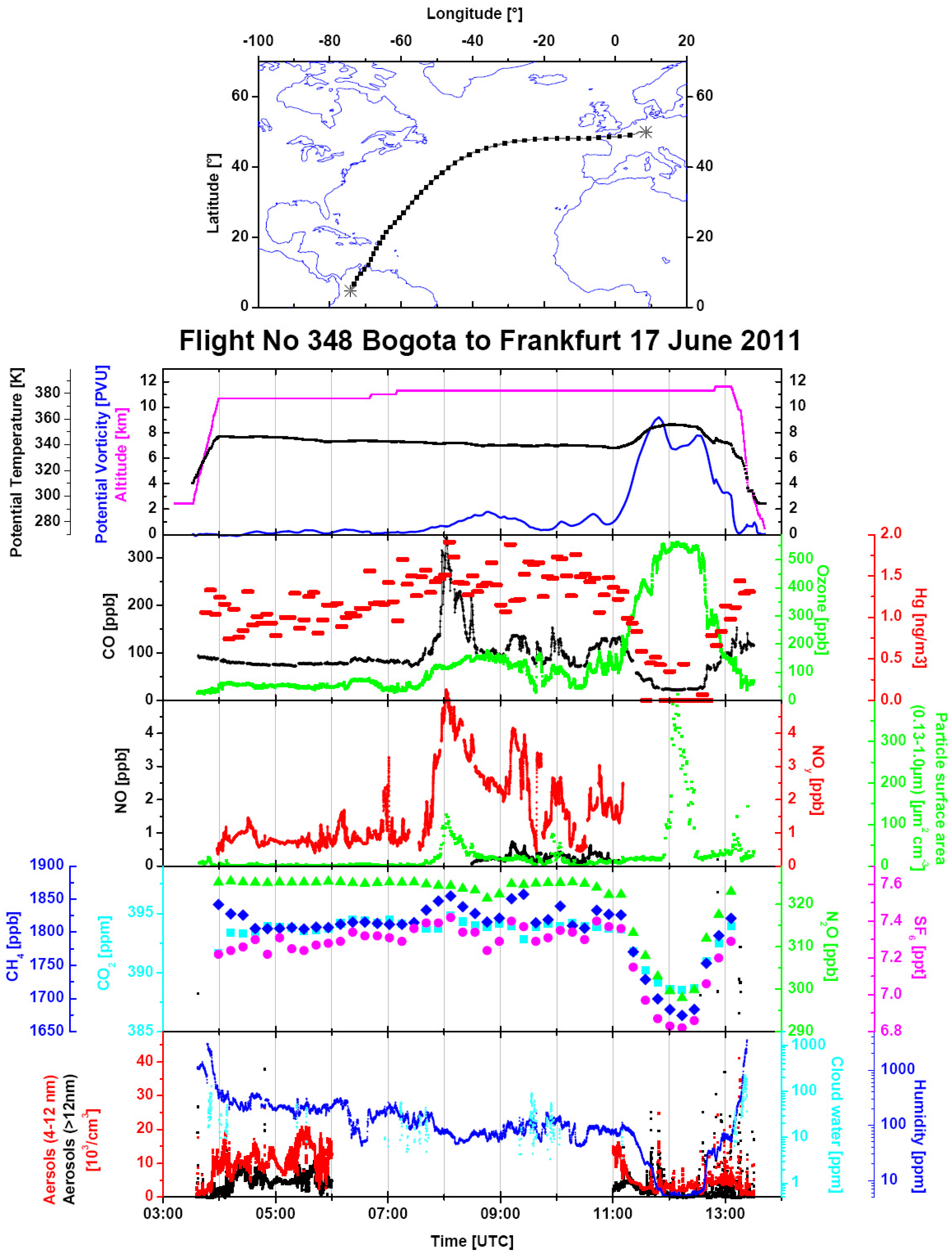
Figure 8.
An overview of the data from Flight 348 from Bogotá to Frankfurt on 17 June 2011. The same parameters are displayed as in
Figure 5. Additionally, total water content (dark blue) is shown in the bottom panel.
Figure 8.
An overview of the data from Flight 348 from Bogotá to Frankfurt on 17 June 2011. The same parameters are displayed as in
Figure 5. Additionally, total water content (dark blue) is shown in the bottom panel.
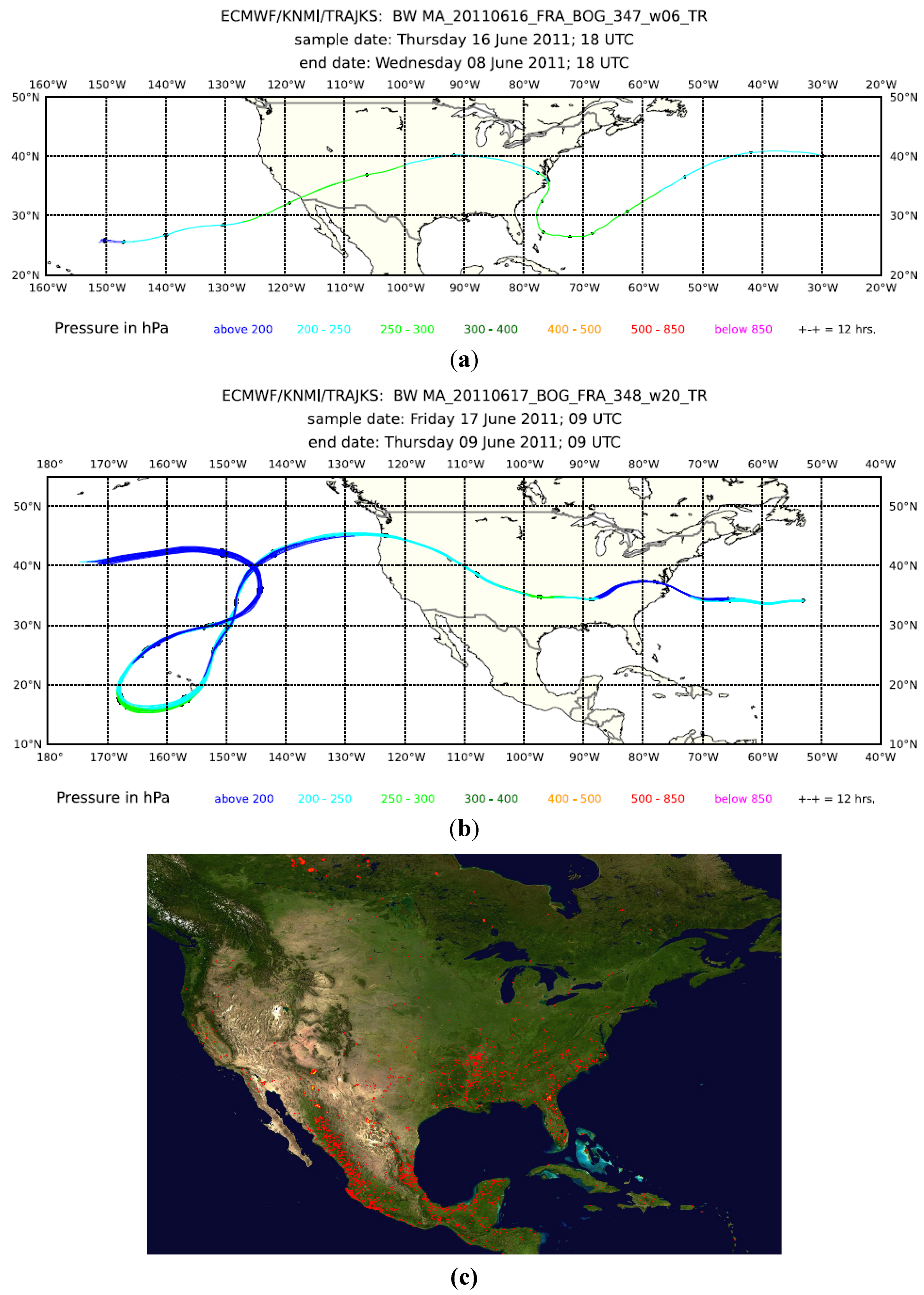
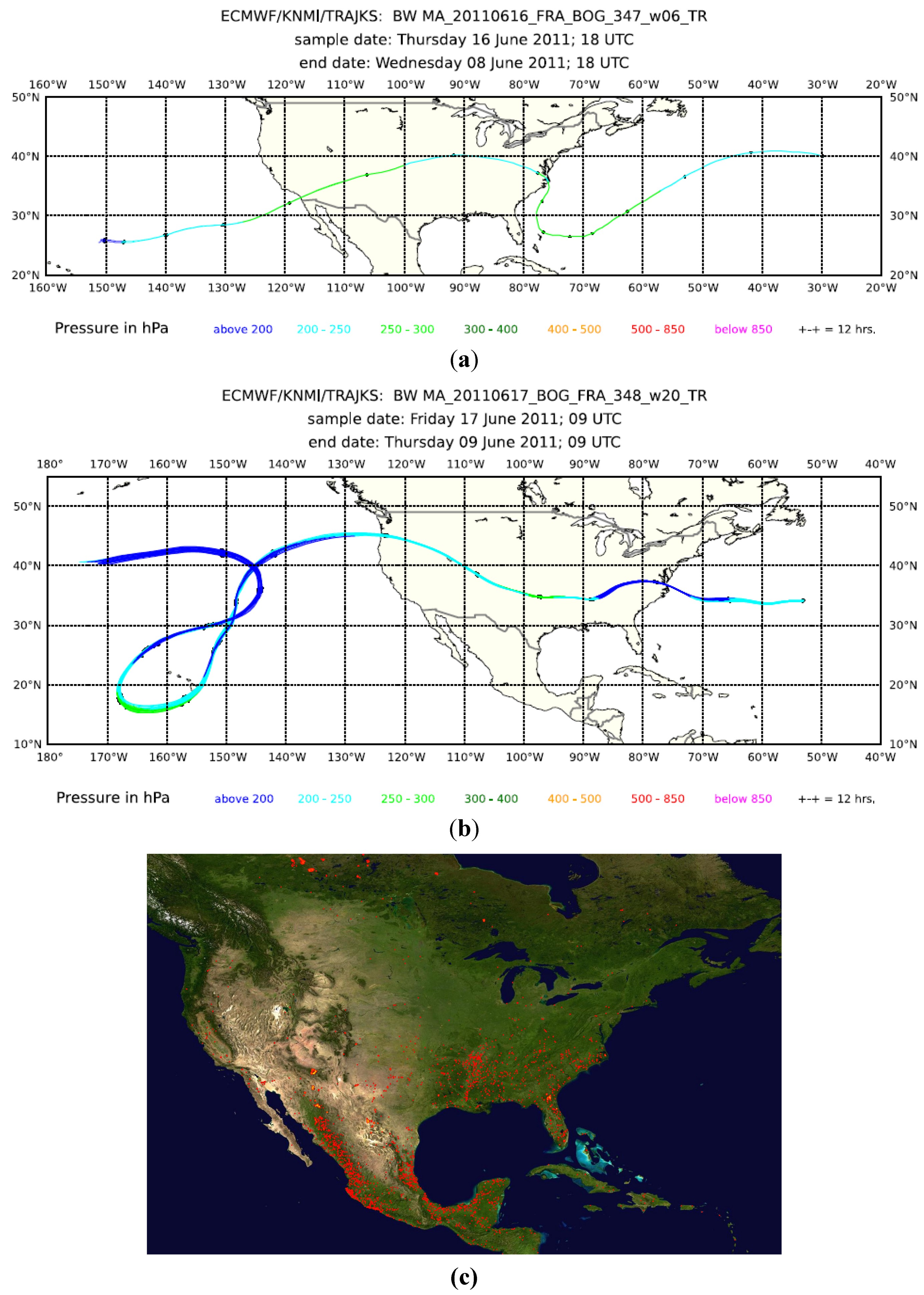
Figure 9.
(
a) eight-day backward trajectories for whole air Sample 6 from the CO peak encountered around 17:20 UTC during Flight 347 from Frankfurt to Bogota on 16 June 2011; and (
b) for whole air Sample 20 from the CO peak encountered around 8:05 UTC during Flight 348 from Bogota to Frankfurt on 17 June 2011. (
c) The map of the fire counts for 10–19 June 2011 (
http://rapidfire.sci.gsfc.nasa.gov/firemaps/, accessed on 10 October 2013).
Figure 9.
(
a) eight-day backward trajectories for whole air Sample 6 from the CO peak encountered around 17:20 UTC during Flight 347 from Frankfurt to Bogota on 16 June 2011; and (
b) for whole air Sample 20 from the CO peak encountered around 8:05 UTC during Flight 348 from Bogota to Frankfurt on 17 June 2011. (
c) The map of the fire counts for 10–19 June 2011 (
http://rapidfire.sci.gsfc.nasa.gov/firemaps/, accessed on 10 October 2013).
Three of the four statistically significant Hg vs. CO correlations for plumes observed over equatorial Africa yield Hg/CO emission ratios of 2.18–3.36 pg∙m−3∙ppb−1, which, again, points to the predominant contribution of emissions from biomass burning.
3.5. Hg/CO2 Emission Ratios
Hg/CO
2 emission ratios are potentially more useful for constraining the mercury emissions, because CO
2 inventories tend to be more accurate than those of CO [
21]. Unfortunately, only a few Hg/CO
2 emission ratios have been reported, so far. Table S3 displays the events with significant Hg
vs. CO
2 correlations. CO
2 data were available only for 46 plume encounters. Among these, for only 17 encounters, the Hg
vs. CO
2 correlations were statistically significant (significance level ≥95%). The yield of statistically significant Hg
vs. CO
2 correlations is thus somewhat smaller than for Hg
vs. CO, and the significance of these correlations with mostly only 95% tends also to somewhat smaller values. Eleven of the statistically significant correlations were found for plumes with significant Hg
vs. CO correlations.
The Hg/CO2 emission ratios vary over a broad range, from 14.4 to 964 pg∙m−3∙ppm−1, and those observed during the flights to East Asian destinations vary between 107 and 964 pg∙m−3∙ppm−1. The Hg/CO2 emission ratio from the plume observed during Flight 334 to Frankfurt immediately after ascent from Cape Town on 21 March 2011, and during Flight 373 to Chennai after ascent from Frankfurt on 16 January 2012, also fit the range of East Asian plumes. The lowest Hg/CO2 emission ratios of 14.4 and 21.9 pg∙m−3∙ppm−1 were both derived from plumes encountered during Flights 329 and 334 over equatorial Africa on 24 February 2011 and 21 March 2011, respectively.
The low Hg/CO
2 emission ratios in the plumes of equatorial Africa are comparable to the median of 34.1 pg∙m
−3∙ppm
−1 (average: 62.7 ± 80.2 pg∙m
−3∙ppm
−1) of emission ratios observed in the plumes encountered at Cape Point, which, according to their Hg/CO emission ratios, seem to originate predominantly from biomass burning [
21]. The Hg/CO
2 emission ratio from the only plume clearly attributed to biomass burning near Cape Point was somewhat higher with 109 ± 27 pg∙m
−3∙ppm
−1 [
49], but this is comparable to 131 ± 53 pg∙m
−3∙ppm
−1, observed in the plume from biomass burning in the south-eastern U.S. in June 2011 (Flights 347 and 348). Based on the coal mercury content of 0.15–0.45 µg∙Hg∙g
−1 and a flue cleaning efficiency for mercury of 50%–90%, Brunke
et al. [
21] predicted an Hg/CO
2 emission ratio to be within the range of 2–30 pg∙m
−3∙ppm
−1. The Hg content in coal consumed in China varies from 0.027 to 0.369 µg∙g
−1 [
50] and is not much different from that in South Africa. The flue cleaning efficiency for mercury in China is with up to 57% somewhat lower [
50], but this difference cannot explain the Hg/CO
2 emission ratios larger than 100 pg∙m
−3∙ppm
−1 observed over East Asia, Europe and at Cape Point in South Africa [
21]. If confirmed by further measurements, high Hg/CO
2 emission ratios would imply a substantial contribution of emissions from other sources than coal burning.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}