
From Target Prediction to Mechanistic Insights: Revealing Air Pollution-Driven Mechanisms in Endometrial Cancer via Interpretable Machine Learning and Molecular Docking


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source and Processing
2.1.1. Collection of Air Pollutant-Related Target Genes
2.1.2. Collection of EC-Related Genes
2.1.3. TCGA Dataset Acquisition and Preprocessing
2.2. Toxicity Evaluation of Air Pollutants
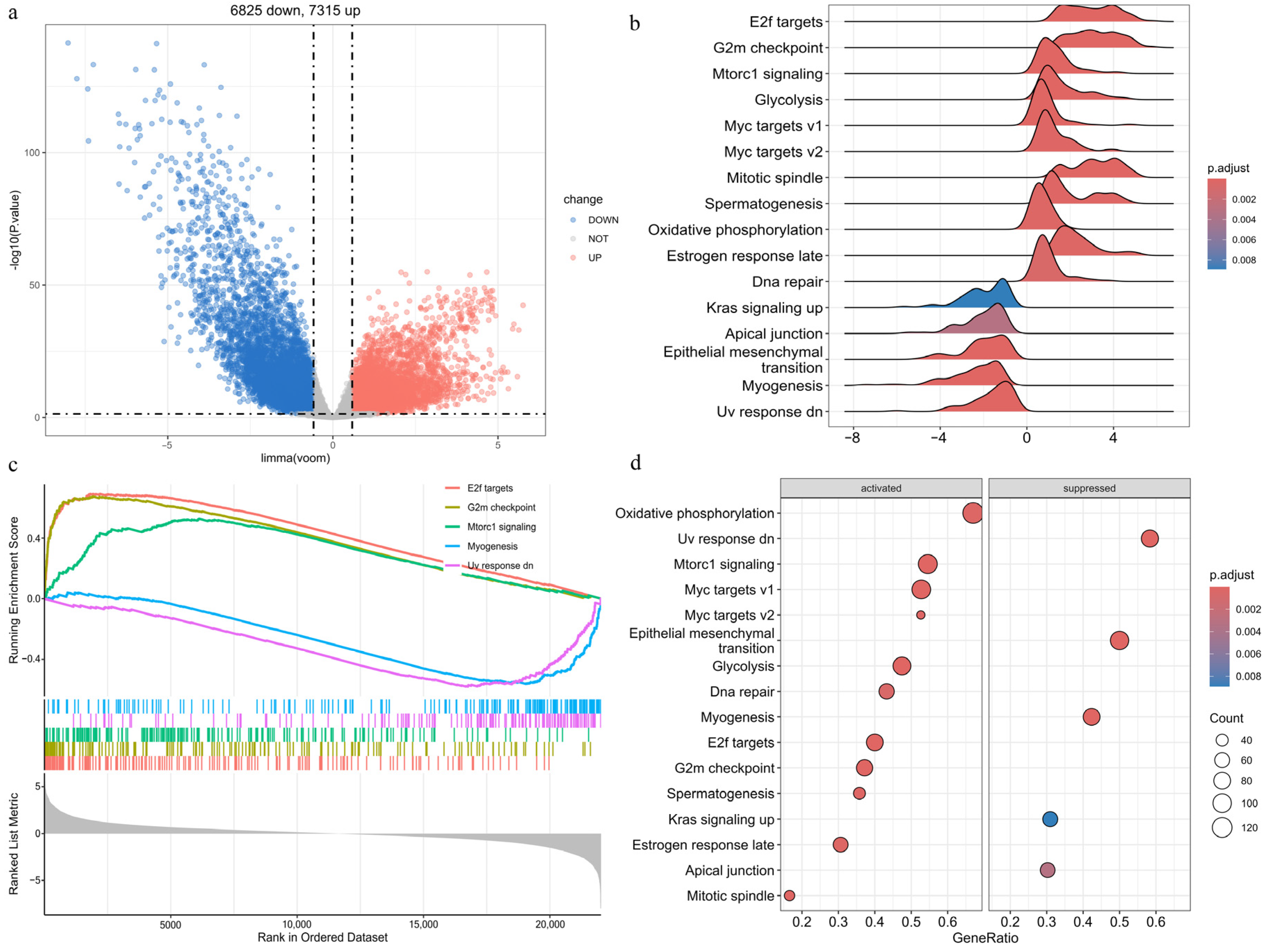
2.3. Identification of DEGs in EC
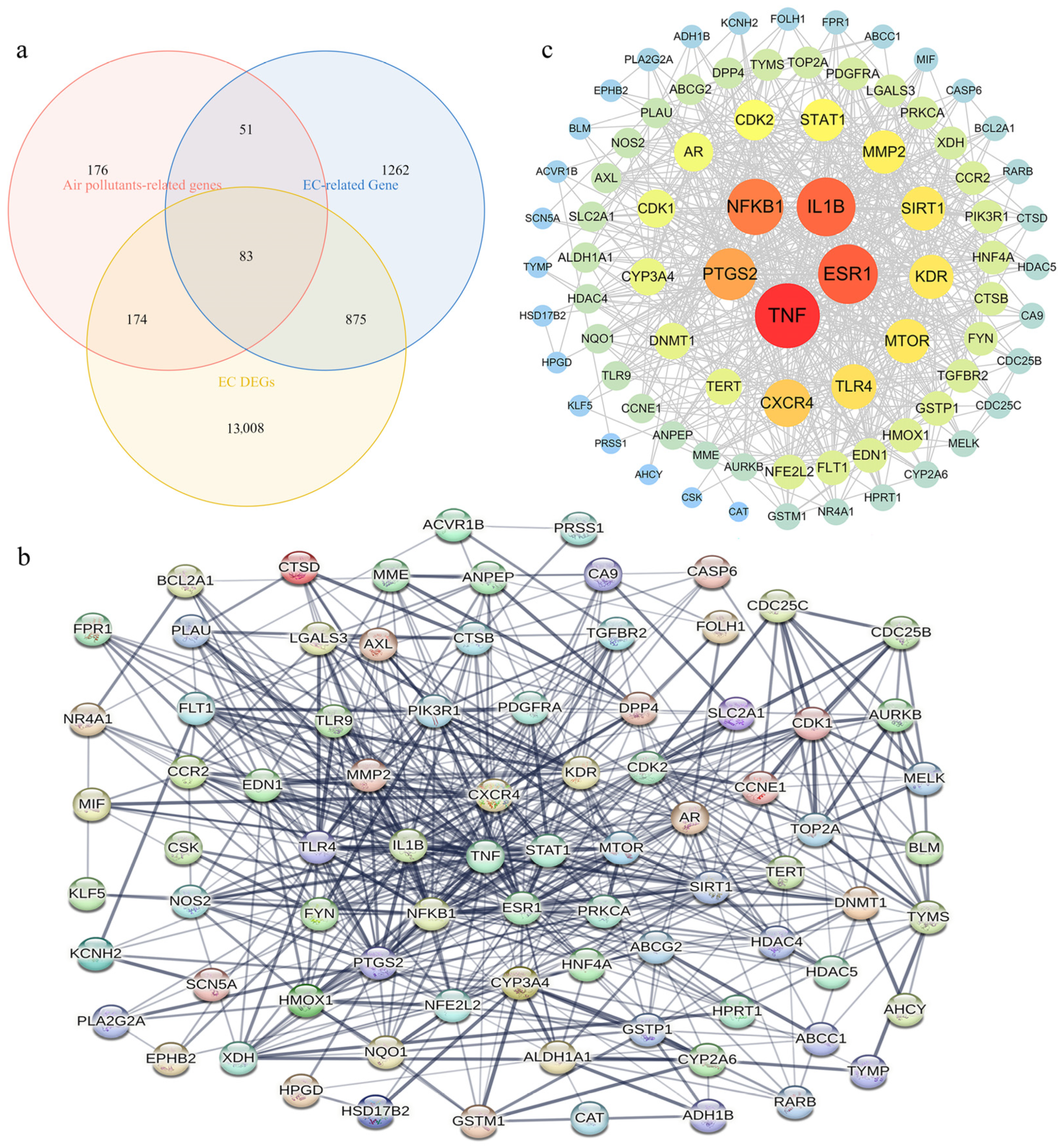
2.4. Screening of APECGs and Construction of PPI Network
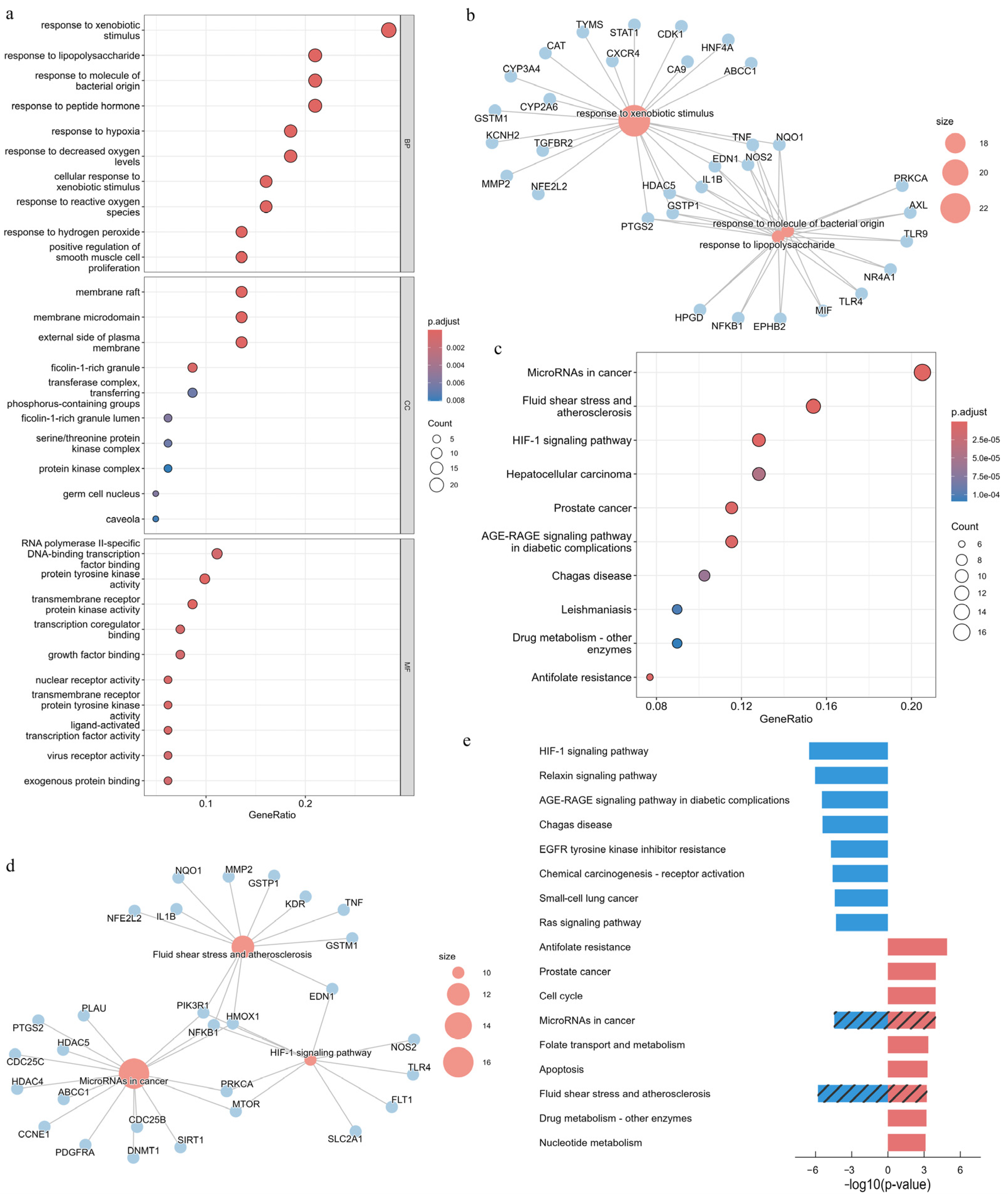
2.5. Functional Enrichment Analysis
2.5.1. Gene Set Enrichment Analysis (GSEA)
2.5.2. Overrepresentation Analysis (ORA)
2.6. Development and Evaluation of the Optimal Prognostic Model Using 117 Machine Learning Combinations
2.7. Construction and Validation of the Risk Score Model
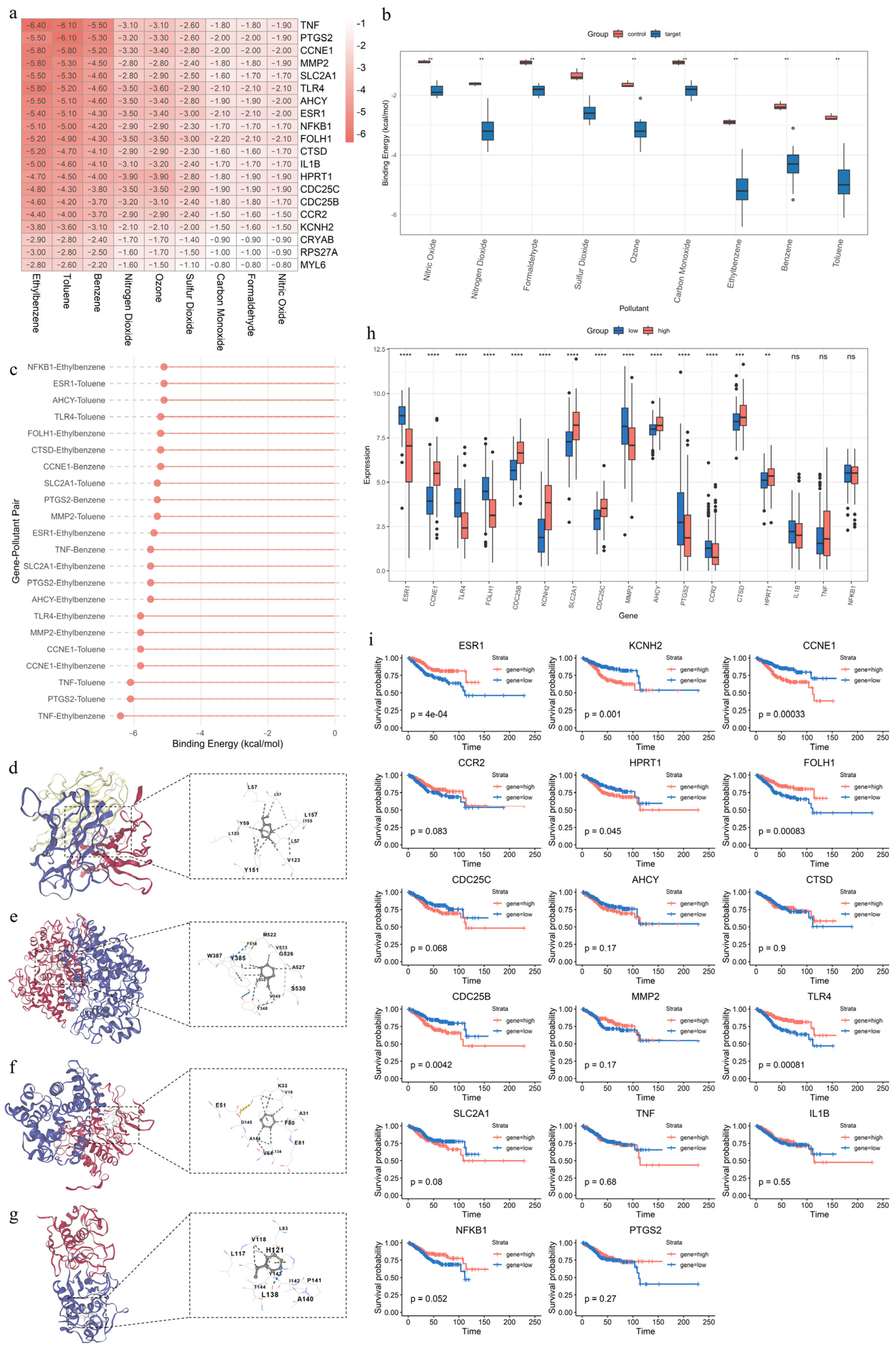
2.8. Molecular Docking
2.9. Pathway Enrichment Analysis Between Risk Groups
2.10. Construction of Random Forest Models for Risk-Associated Pathways
2.11. SHAP-Based Interpretation of Pathway Contributions
2.12. Statistical Analyses
3. Results
3.1. Toxicity Assessment of Air Pollutants
3.2. Overview of Air Pollutant-Related Target Genes Identified
3.3. Overview of EC-Related Genes Identified
3.4. Differential Expression and Enrichment Analyses in EC
3.5. Identification of APECGs and Hub Genes from the PPI Network
3.6. GO and KEGG Enrichment Analyses of APECGs
3.7. Development and Evaluation of an Optimal Prognostic Model Using 117 Machine Learning Combinations
3.8. Risk Stratification Using a 13-Gene APECG Model
3.9. Molecular Docking, Expression Patterns, and Prognostic Significance of Key APECGs
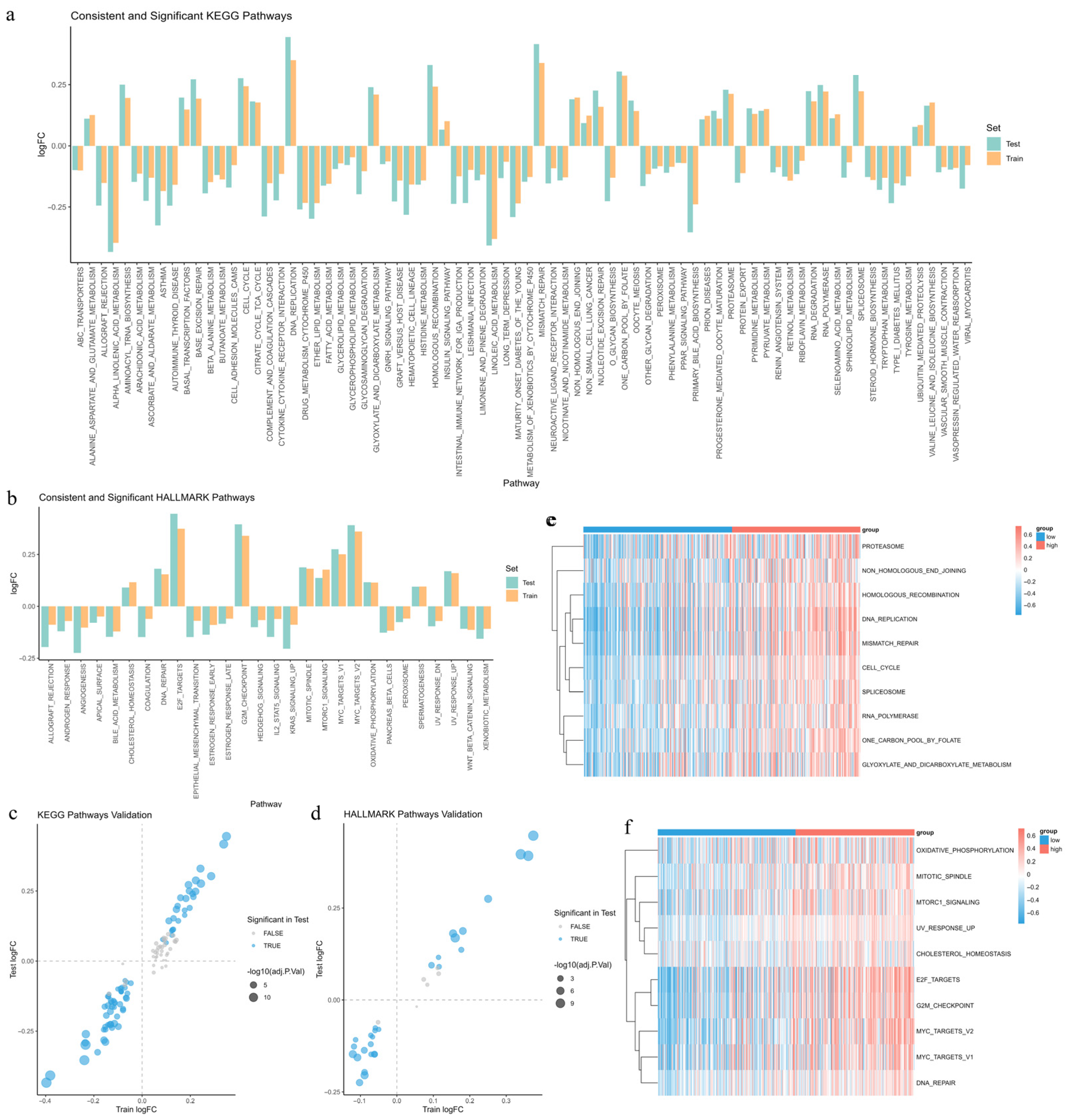
3.10. Identification of Differentially Enriched Pathways Between Risk Groups
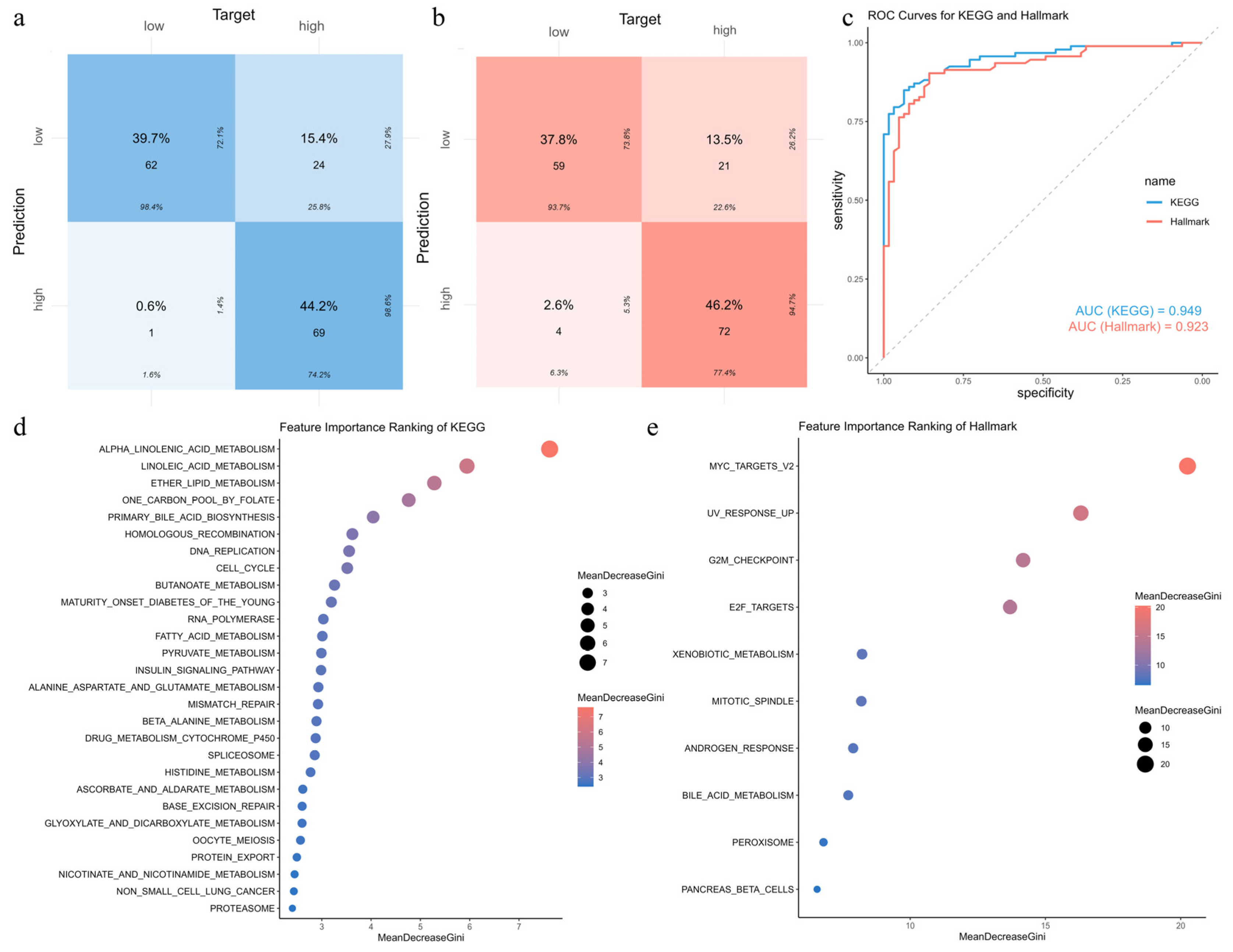
3.11. Construction and Validation of Random Forest Models for Risk-Associated Pathways
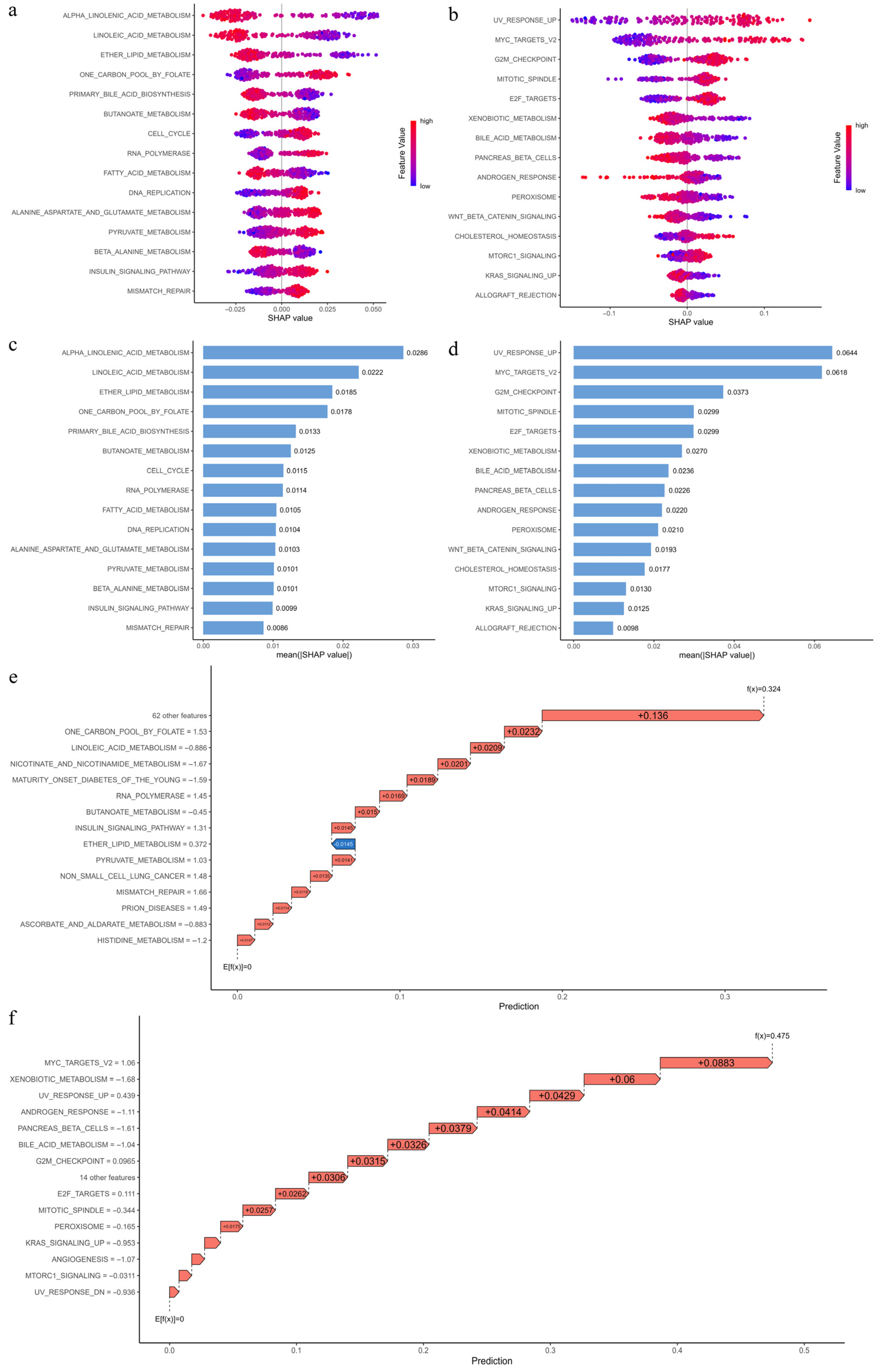
3.12. Interpretation of Pathway Importance in Risk Prediction Using SHAP Analysis
4. Discussion
- (1)
- Initial Exposure and Local Disturbance: Gaseous pollutants—particularly benzene, toluene, and ethylbenzene—enter systemic circulation and accumulate in metabolically active tissues such as the endometrium. These compounds generate reactive intermediates and electrophilic metabolites, inducing oxidative stress, forming DNA and protein adducts, and disrupting local redox balance. Several pollutants also exhibit weak estrogenic activity, potentially altering estrogen receptor signaling and hormonal regulation in the endometrium.
- (2)
- DNA Damage and Genomic Instability: These genotoxic insults activate damage response pathways while overwhelming the DNA repair machinery. In parallel, upregulation of cell cycle drivers such as CCNE1 and CDC25B/C enables continued proliferation despite DNA damage, compromising checkpoint fidelity and accelerating mutation accumulation—key events that drive malignant transformation.
- (3)
- Metabolic Reprogramming and Epigenetic Alterations: To adapt to persistent stress and elevated biosynthetic demands, endometrial cells undergo a metabolic shift toward aerobic glycolysis, characterized by increased glucose uptake and enhanced glycolytic flux. This shift is accompanied by suppression of lipid-based energy metabolism, including α-linolenic acid metabolism, linoleic acid metabolism, and fatty acid oxidation, as well as downregulation of detoxification-related pathways such as peroxisome function and bile acid metabolism. Together, these changes reduce metabolic flexibility, impair redox buffering, and favor a glycolysis-dominant, pro-tumor phenotype. At the same time, dysregulated one-carbon metabolism—driven by increased AHCY activity—alters DNA and histone methylation patterns, enabling epigenetic reprogramming that supports oncogene activation and silencing of tumor suppressor genes.
- (4)
- Immune Dysregulation and Chronic Inflammation: Air pollutants suppress key components of the immune system by downregulating genes involved in leukocyte recruitment and antigen presentation, such as TLR4, CCR2, and FOLH1. This leads to reduced cytotoxic T-cell infiltration and impaired immune surveillance. Additionally, reduced synthesis of polyunsaturated fatty acids (PUFAs) limits the production of anti-inflammatory lipid mediators—such as resolvins and lipoxins—compromising inflammation resolution. Persistent activation of inflammatory signaling pathways, including those regulated by TNF, IL1B, and NFKB1, perpetuates low-grade chronic inflammation, reinforcing a tumor-promoting immune microenvironment.
- (5)
- Systemic Imbalance and Malignant Progression: These molecular disruptions interact synergistically to establish a self-reinforcing loop of DNA damage, metabolic reprogramming, epigenetic dysregulation, immune escape, and unresolved inflammation. Impaired detoxification capacity further limits the clearance of lipophilic toxins, while endocrine and immune imbalances exacerbate local stress. Collectively, these changes reshape the endometrial microenvironment, enabling sustained tumor growth, invasion, and potential metastasis.
- (1)
- Experimental validation of key mechanisms
- (2)
- Modeling real-world exposure complexity
- (3)
- Integration of epidemiological exposure data
- (4)
- Refinement and external validation of the prognostic model
- (5)
- Multi-omics and single-cell dissection of pollutant effects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schraufnagel, D.E.; Balmes, J.R.; Cowl, C.T.; De Matteis, S.; Jung, S.H.; Mortimer, K.; Perez-Padilla, R.; Rice, M.B.; Riojas-Rodriguez, H.; Sood, A.; et al. Air Pollution and Noncommunicable Diseases: A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 2: Air Pollution and Organ Systems. Chest 2019, 155, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Schraufnagel, D.E.; Balmes, J.R.; Cowl, C.T.; De Matteis, S.; Jung, S.H.; Mortimer, K.; Perez-Padilla, R.; Rice, M.B.; Riojas-Rodriguez, H.; Sood, A.; et al. Air Pollution and Noncommunicable Diseases: A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 1: The Damaging Effects of Air Pollution. Chest 2019, 155, 409–416. [Google Scholar] [CrossRef]
- Barbier, E.; Carpentier, J.; Simonin, O.; Gosset, P.; Platel, A.; Happillon, M.; Alleman, L.Y.; Perdrix, E.; Riffault, V.; Chassat, T.; et al. Oxidative stress and inflammation induced by air pollution-derived PM(2.5) persist in the lungs of mice after cessation of their sub-chronic exposure. Environ. Int. 2023, 181, 108248. [Google Scholar] [CrossRef]
- Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K.; et al. The carcinogenicity of outdoor air pollution. Lancet Oncol. 2013, 14, 1262–1263. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.C.; Andersen, Z.J.; Baccarelli, A.; Diver, W.R.; Gapstur, S.M.; Pope, C.A., 3rd; Prada, D.; Samet, J.; Thurston, G.; Cohen, A. Outdoor air pollution and cancer: An overview of the current evidence and public health recommendations. CA Cancer J. Clin. 2020, 70, 460–479. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Q.; Su, Z.; Liu, Y.; Yan, T.; Zhang, Y.; Wang, T.; Wei, X.; Chen, Z.; Hu, G.; et al. Genetic damage and potential mechanism exploration under different air pollution patterns by multi-omics. Environ. Int. 2022, 170, 107636. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, L.; Zhang, Y.; Zhao, Y.; Liu, Y. Air pollution: A culprit of lung cancer. J. Hazard. Mater. 2022, 434, 128937. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Crosbie, E.J.; Kitson, S.J.; McAlpine, J.N.; Mukhopadhyay, A.; Powell, M.E.; Singh, N. Endometrial cancer. Lancet 2022, 399, 1412–1428. [Google Scholar] [CrossRef]
- Lortet-Tieulent, J.; Ferlay, J.; Bray, F.; Jemal, A. International Patterns and Trends in Endometrial Cancer Incidence, 1978–2013. J. Natl. Cancer Inst. 2018, 110, 354–361. [Google Scholar] [CrossRef]
- Onstad, M.A.; Schmandt, R.E.; Lu, K.H. Addressing the Role of Obesity in Endometrial Cancer Risk, Prevention, and Treatment. J. Clin. Oncol. 2016, 34, 4225–4230. [Google Scholar] [CrossRef] [PubMed]
- Papatla, K.; Huang, M.; Slomovitz, B. The obese endometrial cancer patient: How do we effectively improve morbidity and mortality in this patient population? Ann. Oncol. 2016, 27, 1988–1994. [Google Scholar] [CrossRef]
- Simpson, A.N.; Lennox, G. Highlighting obesity as a risk factor for endometrial cancer. CMAJ 2021, 193, E58. [Google Scholar] [CrossRef] [PubMed]
- Brinton, L.A.; Trabert, B.; Anderson, G.L.; Falk, R.T.; Felix, A.S.; Fuhrman, B.J.; Gass, M.L.; Kuller, L.H.; Pfeiffer, R.M.; Rohan, T.E.; et al. Serum Estrogens and Estrogen Metabolites and Endometrial Cancer Risk among Postmenopausal Women. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1081–1089. [Google Scholar] [CrossRef]
- Kim, J.J.; Kurita, T.; Bulun, S.E. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr. Rev. 2013, 34, 130–162. [Google Scholar] [CrossRef] [PubMed]
- Peltomaki, P.; Nystrom, M.; Mecklin, J.P.; Seppala, T.T. Lynch Syndrome Genetics and Clinical Implications. Gastroenterology 2023, 164, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chen, L.; Zang, Y.; Liu, W.; Liu, S.; Teng, F.; Xue, F.; Wang, Y. Endometrial cancer in Lynch syndrome. Int. J. Cancer 2022, 150, 7–17. [Google Scholar] [CrossRef]
- Brown, J.A.; Ish, J.L.; Chang, C.J.; Bookwalter, D.B.; O’Brien, K.M.; Jones, R.R.; Kaufman, J.D.; Sandler, D.P.; White, A.J. Outdoor air pollution exposure and uterine cancer incidence in the Sister Study. J. Natl. Cancer Inst. 2024, 116, 948–956. [Google Scholar] [CrossRef]
- Craver, A.; Luo, J.; Kibriya, M.G.; Randorf, N.; Bahl, K.; Connellan, E.; Powell, J.; Zakin, P.; Jones, R.R.; Argos, M.; et al. Air quality and cancer risk in the All of Us Research Program. Cancer Causes Control. 2024, 35, 749–760. [Google Scholar] [CrossRef]
- Li, W.; Wang, W. Causal effects of exposure to ambient air pollution on cancer risk: Insights from genetic evidence. Sci. Total Environ. 2024, 912, 168843. [Google Scholar] [CrossRef]
- Iwai, K.; Mizuno, S.; Miyasaka, Y.; Mori, T. Correlation between suspended particles in the environmental air and causes of disease among inhabitants: Cross-sectional studies using the vital statistics and air pollution data in Japan. Environ. Res. 2005, 99, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Pan, Y. Unraveling the mechanisms underlying diabetic cataracts: Insights from Mendelian randomization analysis. Redox Rep. 2024, 29, 2420563. [Google Scholar] [CrossRef]
- Shao, X.; Wang, Y.; Geng, Z.; Liang, G.; Zhu, X.; Liu, L.; Meng, M.; Duan, L.; Zhu, G. Novel therapeutic targets for major depressive disorder related to oxidative stress identified by integrative multi-omics and multi-trait study. Transl. Psychiatry 2024, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, W.; Zhang, Y.; Adegboro, A.A.; Fasoranti, D.O.; Dai, L.; Pan, Z.; Liu, H.; Xiong, Y.; Li, W.; et al. Mime: A flexible machine-learning framework to construct and visualize models for clinical characteristics prediction and feature selection. Comput. Struct. Biotechnol. J. 2024, 23, 2798–2810. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Ray, I.; Meira, L.B.; Michael, A.; Ellis, P.E. Adipocytokines and disease progression in endometrial cancer: A systematic review. Cancer Metastasis Rev. 2022, 41, 211–242. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Wang, X.; Zhang, Y.; Zhao, J.; Han, R.; Dong, Y. DNMT3A/3B overexpression might be correlated with poor patient survival, hypermethylation and low expression of ESR1/PGR in endometrioid carcinoma: An analysis of The Cancer Genome Atlas. Chin. Med. J. 2019, 132, 161–170. [Google Scholar] [CrossRef]
- Tian, W.; Teng, F.; Gao, J.; Gao, C.; Liu, G.; Zhang, Y.; Yu, S.; Zhang, W.; Wang, Y.; Xue, F. Estrogen and insulin synergistically promote endometrial cancer progression via crosstalk between their receptor signaling pathways. Cancer Biol. Med. 2019, 16, 55–70. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, D.; Yang, X.; Song, Y. Estrogen receptor alpha activates MAPK signaling pathway to promote the development of endometrial cancer. J. Cell Biochem. 2019, 120, 17593–17601. [Google Scholar] [CrossRef]
- Backes, F.J.; Walker, C.J.; Goodfellow, P.J.; Hade, E.M.; Agarwal, G.; Mutch, D.; Cohn, D.E.; Suarez, A.A. Estrogen receptor-alpha as a predictive biomarker in endometrioid endometrial cancer. Gynecol. Oncol. 2016, 141, 312–317. [Google Scholar] [CrossRef]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen Signaling in Endometrial Cancer: A Key Oncogenic Pathway with Several Open Questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, Z.; Vahrenkamp, J.M.; Berrett, K.C.; Arnesen, S.; Gertz, J. Estrogen-independent molecular actions of mutant estrogen receptor 1 in endometrial cancer. Genome Res. 2019, 29, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kaneuchi, M.; Fujimoto, S.; Tanaka, Y.; Dahiya, R. Hypermethylation can selectively silence multiple promoters of steroid receptors in cancers. Mol. Cell Endocrinol. 2003, 202, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Wik, E.; Raeder, M.B.; Krakstad, C.; Trovik, J.; Birkeland, E.; Hoivik, E.A.; Mjos, S.; Werner, H.M.; Mannelqvist, M.; Stefansson, I.M.; et al. Lack of estrogen receptor-alpha is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin. Cancer Res. 2013, 19, 1094–1105. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Rebe, C.; Ghiringhelli, F. Interleukin-1beta and Cancer. Cancers 2020, 12, 1791. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Garlanda, C.; Mantovani, A. Interleukin-1 in tumor progression, therapy, and prevention. Cancer Cell 2021, 39, 1023–1027. [Google Scholar] [CrossRef]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Concetti, J.; Wilson, C.L. NFKB1 and Cancer: Friend or Foe? Cells 2018, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- Lyndin, M.; Kravtsova, O.; Sikora, K.; Lyndina, Y.; Kuzenko, Y.; Awuah, W.A.; Abdul-Rahman, T.; Hyriavenko, N.; Sikora, V.; Romaniuk, A. COX2 Effects on endometrial carcinomas progression. Pathol. Res. Pract. 2022, 238, 154082. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Siminska, D.; Gassowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-kappaB Activation: A Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 10701. [Google Scholar] [CrossRef] [PubMed]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- He, S.; Moutaoufik, M.T.; Islam, S.; Persad, A.; Wu, A.; Aly, K.A.; Fonge, H.; Babu, M.; Cayabyab, F.S. HERG channel and cancer: A mechanistic review of carcinogenic processes and therapeutic potential. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188355. [Google Scholar] [CrossRef]
- Arcangeli, A.; Becchetti, A. hERG Channels: From Antitargets to Novel Targets for Cancer Therapy. Clin. Cancer Res. 2017, 23, 3–5. [Google Scholar] [CrossRef]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef]
- Leskela, S.; Perez-Mies, B.; Rosa-Rosa, J.M.; Cristobal, E.; Biscuola, M.; Palacios-Berraquero, M.L.; Ong, S.; Matias-Guiu Guia, X.; Palacios, J. Molecular Basis of Tumor Heterogeneity in Endometrial Carcinosarcoma. Cancers 2019, 11, 964. [Google Scholar] [CrossRef]
- Fagundes, R.; Teixeira, L.K. Cyclin E/CDK2: DNA Replication, Replication Stress and Genomic Instability. Front. Cell Dev. Biol. 2021, 9, 774845. [Google Scholar] [CrossRef]
- Bogani, G.; Ray-Coquard, I.; Concin, N.; Ngoi, N.Y.L.; Morice, P.; Enomoto, T.; Takehara, K.; Denys, H.; Nout, R.A.; Lorusso, D.; et al. Uterine serous carcinoma. Gynecol. Oncol. 2021, 162, 226–234. [Google Scholar] [CrossRef]
- Xu, M.; Wang, Y.; Xia, R.; Wei, Y.; Wei, X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. 2021, 54, e13115. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, T.; Warr, G.; Loy, J.; Bravo, R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 1997, 186, 1757–1762. [Google Scholar] [CrossRef]
- Fei, L.; Ren, X.; Yu, H.; Zhan, Y. Targeting the CCL2/CCR2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Front. Immunol. 2021, 12, 771210. [Google Scholar] [CrossRef]
- Chen, W.; Fang, Y.; Wang, H.; Tan, X.; Zhu, X.; Xu, Z.; Jiang, H.; Wu, X.; Hong, W.; Wang, X.; et al. Role of chemokine receptor 2 in rheumatoid arthritis: A research update. Int. Immunopharmacol. 2023, 116, 109755. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.H.; Ence, Z.E.; Felsted, A.M.; Parker, A.C.; Piccolo, S.R.; Robison, R.A.; O’Neill, K.L. Potential new biomarkers for endometrial cancer. Cancer Cell Int. 2019, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, R.; Zhang, H.; Sun, X.; Li, X.; Yang, M.; Zhang, X. Prognostic significance and immunological role of HPRT1 in human cancers. Biomol. Biomed. 2024, 24, 262–291. [Google Scholar] [CrossRef]
- Sun, D.; Zhang, A.; Gao, B.; Zou, L.; Huang, H.; Zhao, X.; Xu, D. Identification of Alternative Splicing-Related Genes CYB561 and FOLH1 in the Tumor-Immune Microenvironment for Endometrial Cancer Based on TCGA Data Analysis. Front. Genet. 2022, 13, 770569. [Google Scholar] [CrossRef]
- Mhawech-Fauceglia, P.; Smiraglia, D.J.; Bshara, W.; Andrews, C.; Schwaller, J.; South, S.; Higgs, D.; Lele, S.; Herrmann, F.; Odunsi, K. Prostate-specific membrane antigen expression is a potential prognostic marker in endometrial adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 571–577. [Google Scholar] [CrossRef]
- Wang, R.; He, G.; Nelman-Gonzalez, M.; Ashorn, C.L.; Gallick, G.E.; Stukenberg, P.T.; Kirschner, M.W.; Kuang, J. Regulation of Cdc25C by ERK-MAP kinases during the G2/M transition. Cell 2007, 128, 1119–1132. [Google Scholar] [CrossRef]
- Vassileva, V.; Millar, A.; Briollais, L.; Chapman, W.; Bapat, B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res. 2002, 62, 4095–4099. [Google Scholar]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Karlsson-Rosenthal, C.; Millar, J.B. Cdc25: Mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006, 16, 285–292. [Google Scholar] [CrossRef]
- Vizan, P.; Di Croce, L.; Aranda, S. Functional and Pathological Roles of AHCY. Front. Cell Dev. Biol. 2021, 9, 654344. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.C.; Esteve, P.O.; Ruse, C.I.; Pradhan, S. S-adenosylhomocysteine Hydrolase Participates in DNA Methylation Inheritance. J. Mol. Biol. 2018, 430, 2051–2065. [Google Scholar] [CrossRef]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D—Many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Kallstrom, H.; Lundgren, A.; Barsoum, E.; Rosenthal, C.K. Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J. Cell Biol. 2005, 171, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Galaktionov, K.; Lee, A.K.; Eckstein, J.; Draetta, G.; Meckler, J.; Loda, M.; Beach, D. CDC25 phosphatases as potential human oncogenes. Science 1995, 269, 1575–1577. [Google Scholar] [CrossRef]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef]
- Wolosowicz, M.; Prokopiuk, S.; Kaminski, T.W. The Complex Role of Matrix Metalloproteinase-2 (MMP-2) in Health and Disease. Int. J. Mol. Sci. 2024, 25, 13691. [Google Scholar] [CrossRef]
- Fernandez-Patron, C.; Kassiri, Z.; Leung, D. Modulation of Systemic Metabolism by MMP-2: From MMP-2 Deficiency in Mice to MMP-2 Deficiency in Patients. Compr. Physiol. 2016, 6, 1935–1949. [Google Scholar] [CrossRef]
- de Almeida, L.G.N.; Thode, H.; Eslambolchi, Y.; Chopra, S.; Young, D.; Gill, S.; Devel, L.; Dufour, A. Matrix Metalloproteinases: From Molecular Mechanisms to Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 2022, 74, 712–768. [Google Scholar] [CrossRef] [PubMed]
- Alaseem, A.; Alhazzani, K.; Dondapati, P.; Alobid, S.; Bishayee, A.; Rathinavelu, A. Matrix Metalloproteinases: A challenging paradigm of cancer management. Semin. Cancer Biol. 2019, 56, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Ran, S. The Role of TLR4 in Chemotherapy-Driven Metastasis. Cancer Res. 2015, 75, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Yamamoto, H.; Kawakami, T. Maintenance of homeostasis by TLR4 ligands. Front. Immunol. 2024, 15, 1286270. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Lupi, L.A.; Cucielo, M.S.; Silveira, H.S.; Gaiotte, L.B.; Cesario, R.C.; Seiva, F.R.F.; de Almeida Chuffa, L.G. The role of Toll-like receptor 4 signaling pathway in ovarian, cervical, and endometrial cancers. Life Sci. 2020, 247, 117435. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.; Lee, J.H.; Hwangbo, C. Toll-like receptor 4 (TLR4): New insight immune and aging. Immun. Ageing 2023, 20, 67. [Google Scholar] [CrossRef]
- Lopez-Serra, P.; Marcilla, M.; Villanueva, A.; Ramos-Fernandez, A.; Palau, A.; Leal, L.; Wahi, J.E.; Setien-Baranda, F.; Szczesna, K.; Moutinho, C.; et al. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat. Commun. 2014, 5, 3608. [Google Scholar] [CrossRef]
- Ooi, A.T.; Gomperts, B.N. Molecular pathways: Targeting cellular energy metabolism in cancer via inhibition of SLC2A1 and LDHA. Clin. Cancer Res. 2015, 21, 2440–2444. [Google Scholar] [CrossRef]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism 2016, 65, 124–139. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Formaldehyde, 2-butoxyethanol and 1-tert-butoxypropan-2-ol. IARC Monogr. Eval. Carcinog. Risks Hum. 2006, 88, 1–478. [Google Scholar]
- Loomis, D.; Guyton, K.Z.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Vilahur, N.; Mattock, H.; Straif, K.; et al. Carcinogenicity of benzene. Lancet Oncol. 2017, 18, 1574–1575. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Agents Classified by the IARC Monographs. Volumes 1–123. IARC. 2018. Available online: https://monographs.iarc.who.int/wp-content/uploads/2018/09/ClassificationsAlphaOrder.pdf (accessed on 7 April 2025).
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Outdoor Air Pollution. IARC Monogr. Eval. Carcinog. Risks Hum. 2016, 109, 9–444. [Google Scholar]
- Blanc-Lapierre, A.; Sauve, J.F.; Parent, M.E. Occupational exposure to benzene, toluene, xylene and styrene and risk of prostate cancer in a population-based study. Occup. Environ. Med. 2018, 75, 562–572. [Google Scholar] [CrossRef]
- El-Zaemey, S.; Anand, T.N.; Heyworth, J.S.; Boyle, T.; van Tongeren, M.; Fritschi, L. Case-control study to assess the association between colorectal cancer and selected occupational agents using INTEROCC job exposure matrix. Occup. Environ. Med. 2018, 75, 290–295. [Google Scholar] [CrossRef]
- Kim, S.; Park, E.; Song, S.H.; Lee, C.W.; Kwon, J.T.; Park, E.Y.; Kim, B. Toluene concentrations in the blood and risk of thyroid cancer among residents living near national industrial complexes in South Korea: A population-based cohort study. Environ. Int. 2021, 146, 106304. [Google Scholar] [CrossRef]
- Warden, H.; Richardson, H.; Richardson, L.; Siemiatycki, J.; Ho, V. Associations between occupational exposure to benzene, toluene and xylene and risk of lung cancer in Montreal. Occup. Environ. Med. 2018, 75, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Kong, D.; Ci, M.; Guan, Y.; Luo, C.; Zhang, X.; Gao, F.; Li, M.; Deng, G. Oxidative Stress Effects of Multiple Pollutants in an Indoor Environment on Human Bronchial Epithelial Cells. Toxics 2023, 11, 251. [Google Scholar] [CrossRef]
- Dees, C.; Askari, M.; Henley, D. Carcinogenic potential of benzene and toluene when evaluated using cyclin-dependent kinase activation and p53-DNA binding. Environ. Health Perspect. 1996, 104 (Suppl. S6), 1289–1292. [Google Scholar] [CrossRef]
- Dees, C.; Travis, C. Hyperphosphorylation of p53 induced by benzene, toluene, and chloroform. Cancer Lett. 1994, 84, 117–123. [Google Scholar] [CrossRef]
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Bolden, A.L.; Schultz, K.; Pelch, K.E.; Kwiatkowski, C.F. Exploring the endocrine activity of air pollutants associated with unconventional oil and gas extraction. Environ. Health 2018, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Kassotis, C.D.; Klemp, K.C.; Vu, D.C.; Lin, C.H.; Meng, C.X.; Besch-Williford, C.L.; Pinatti, L.; Zoeller, R.T.; Drobnis, E.Z.; Balise, V.D.; et al. Endocrine-Disrupting Activity of Hydraulic Fracturing Chemicals and Adverse Health Outcomes After Prenatal Exposure in Male Mice. Endocrinology 2015, 156, 4458–4473. [Google Scholar] [CrossRef]
- Brown-Woodman, P.D.; Webster, W.S.; Picker, K.; Huq, F. In vitro assessment of individual and interactive effects of aromatic hydrocarbons on embryonic development of the rat. Reprod. Toxicol. 1994, 8, 121–135. [Google Scholar] [CrossRef]
- Ono, A.; Sekita, K.; Ogawa, Y.; Hirose, A.; Suzuki, S.; Saito, M.; Naito, K.; Kaneko, T.; Furuya, T.; Kawashima, K.; et al. Reproductive and developmental toxicity studies of toluene. II. Effects of inhalation exposure on fertility in rats. J. Environ. Pathol. Toxicol. Oncol. 1996, 15, 9–20. [Google Scholar] [PubMed]
- Ungvary, G.; Tatrai, E. On the embryotoxic effects of benzene and its alkyl derivatives in mice, rats and rabbits. Arch. Toxicol. Suppl. 1985, 8, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Cho, S.I.; Sammel, M.; You, L.; Cui, S.; Huang, Y.; Ma, G.; Padungtod, C.; Pothier, L.; Niu, T.; et al. Association of petrochemical exposure with spontaneous abortion. Occup. Environ. Med. 1998, 55, 31–36. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Glaviano, A.; Singh, S.K.; Lee, E.H.C.; Okina, E.; Lam, H.Y.; Carbone, D.; Reddy, E.P.; O’Connor, M.J.; Koff, A.; Singh, G.; et al. Cell cycle dysregulation in cancer. Pharmacol. Rev. 2025, 77, 100030. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Das, S.K.; Lewis, B.A.; Levens, D. MYC: A complex problem. Trends Cell Biol. 2023, 33, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, J.; Liao, M.; Yang, Y.; Wang, Y.; Yuan, Y.; Ouyang, L. Folate-mediated one-carbon metabolism: A targeting strategy in cancer therapy. Drug Discov. Today 2021, 26, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- An, J.; Li, F.; Qin, Y.; Zhang, H.; Ding, S. Low concentrations of FA exhibits the Hormesis effect by affecting cell division and the Warburg effect. Ecotoxicol. Environ. Saf. 2019, 183, 109576. [Google Scholar] [CrossRef]
- Chen, Z.; Salam, M.T.; Toledo-Corral, C.; Watanabe, R.M.; Xiang, A.H.; Buchanan, T.A.; Habre, R.; Bastain, T.M.; Lurmann, F.; Wilson, J.P.; et al. Ambient Air Pollutants Have Adverse Effects on Insulin and Glucose Homeostasis in Mexican Americans. Diabetes Care 2016, 39, 547–554. [Google Scholar] [CrossRef]
- Miller, D.B.; Karoly, E.D.; Jones, J.C.; Ward, W.O.; Vallanat, B.D.; Andrews, D.L.; Schladweiler, M.C.; Snow, S.J.; Bass, V.L.; Richards, J.E.; et al. Inhaled ozone (O3)-induces changes in serum metabolomic and liver transcriptomic profiles in rats. Toxicol. Appl. Pharmacol. 2015, 286, 65–79. [Google Scholar] [CrossRef]
- Bollati, V.; Baccarelli, A.; Hou, L.; Bonzini, M.; Fustinoni, S.; Cavallo, D.; Byun, H.M.; Jiang, J.; Marinelli, B.; Pesatori, A.C.; et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007, 67, 876–880. [Google Scholar] [CrossRef]
- Feng, J.; Liu, C.W.; Peng, J.; Hsiao, Y.C.; Chen, D.; Jin, C.; Lu, K. Formaldehyde Exposure Induces Systemic Epigenetic Alterations in Histone Methylation and Acetylation. bioRxiv 2025. [Google Scholar] [CrossRef]
- Poursafa, P.; Kamali, Z.; Fraszczyk, E.; Boezen, H.M.; Vaez, A.; Snieder, H. DNA methylation: A potential mediator between air pollution and metabolic syndrome. Clin. Epigenetics 2022, 14, 82. [Google Scholar] [CrossRef]
- Rider, C.F.; Carlsten, C. Air pollution and DNA methylation: Effects of exposure in humans. Clin. Epigenetics 2019, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, R.S.; Bevan, G.H.; Palanivel, R.; Das, L.; Rajagopalan, S. Oxidative stress pathways of air pollution mediated toxicity: Recent insights. Redox Biol. 2020, 34, 101545. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Hseu, Y.C.; Liang, S.H.; Kuo, J.Y.; Chen, S.C. Assessment of genotoxicity of methyl-tert-butyl ether, benzene, toluene, ethylbenzene, and xylene to human lymphocytes using comet assay. J. Hazard. Mater. 2008, 153, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, R.H., Jr.; Bender, R.P.; Osheroff, N. Effects of benzene metabolites on DNA cleavage mediated by human topoisomerase II alpha: 1,4-hydroquinone is a topoisomerase II poison. Chem. Res. Toxicol. 2005, 18, 761–770. [Google Scholar] [CrossRef]
- Monks, T.J.; Butterworth, M.; Lau, S.S. The fate of benzene-oxide. Chem. Biol. Interact. 2010, 184, 201–206. [Google Scholar] [CrossRef]
- Chen, H.; Eastmond, D.A. Topoisomerase inhibition by phenolic metabolites: A potential mechanism for benzene’s clastogenic effects. Carcinogenesis 1995, 16, 2301–2307. [Google Scholar] [CrossRef]
- Nadalutti, C.A.; Prasad, R.; Wilson, S.H. Perspectives on formaldehyde dysregulation: Mitochondrial DNA damage and repair in mammalian cells. DNA Repair 2021, 105, 103134. [Google Scholar] [CrossRef]
- Stucki, D.; Stahl, W. Carbon monoxide—Beyond toxicity? Toxicol. Lett. 2020, 333, 251–260. [Google Scholar] [CrossRef]
- Jayakumar, R.; Sasikala, K. Evaluation of DNA damage in jewellery workers occupationally exposed to nitric oxide. Environ. Toxicol. Pharmacol. 2008, 26, 259–261. [Google Scholar] [CrossRef]
- Wagner, J.R.; Madugundu, G.S.; Cadet, J. Ozone-Induced DNA Damage: A Pandora’s Box of Oxidatively Modified DNA Bases. Chem. Res. Toxicol. 2021, 34, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Sandalio, L.M.; Pelaez-Vico, M.A.; Molina-Moya, E.; Romero-Puertas, M.C. Peroxisomes as redox-signaling nodes in intracellular communication and stress responses. Plant Physiol. 2021, 186, 22–35. [Google Scholar] [CrossRef]
- Walker, C.L.; Pomatto, L.C.D.; Tripathi, D.N.; Davies, K.J.A. Redox Regulation of Homeostasis and Proteostasis in Peroxisomes. Physiol. Rev. 2018, 98, 89–115. [Google Scholar] [CrossRef]
- Ahn, C.; Jeung, E.B. Endocrine-Disrupting Chemicals and Disease Endpoints. Int. J. Mol. Sci. 2023, 24, 5342. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Jeung, E.B.; Cho, M.H.; Kim, T.H.; Leung, P.C.; Choi, K.C. Molecular mechanism(s) of endocrine-disrupting chemicals and their potent oestrogenicity in diverse cells and tissues that express oestrogen receptors. J. Cell Mol. Med. 2013, 17, 1–11. [Google Scholar] [CrossRef]
- Yilmaz, B.; Terekeci, H.; Sandal, S.; Kelestimur, F. Endocrine disrupting chemicals: Exposure, effects on human health, mechanism of action, models for testing and strategies for prevention. Rev. Endocr. Metab. Disord. 2020, 21, 127–147. [Google Scholar] [CrossRef]
- Bulog, A.; Karaconji, I.B.; Sutic, I.; Micovic, V. Immunomodulation of cell-mediated cytotoxicity after chronic exposure to vapors. Coll. Antropol. 2011, 35 (Suppl. S2), 61–64. [Google Scholar] [PubMed]
- De Celis, R.; Feria-Velasco, A.; Bravo-Cuellar, A.; Hicks-Gomez, J.J.; Garcia-Iglesias, T.; Preciado-Martinez, V.; Munoz-Islas, L.; Gonzalez-Unzaga, M. Expression of NK cells activation receptors after occupational exposure to toxics: A preliminary study. Immunol. Lett. 2008, 118, 125–131. [Google Scholar] [CrossRef]
- Park, J.; Kang, G.H.; Kim, Y.; Lee, J.Y.; Song, J.A.; Hwang, J.H. Formaldehyde exposure induces differentiation of regulatory T cells via the NFAT-mediated T cell receptor signalling pathway in Yucatan minipigs. Sci. Rep. 2022, 12, 8149. [Google Scholar] [CrossRef]
- Park, J.; Yang, H.S.; Song, M.K.; Kim, D.I.; Lee, K. Formaldehyde exposure induces regulatory T cell-mediated immunosuppression via calcineurin-NFAT signalling pathway. Sci. Rep. 2020, 10, 17023. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Yuan, L.; Wei, C.; Zhao, Y.; Qian, Y.; Ma, P.; Ding, S.; Yang, X.; Wang, X. Effects of combined exposure to formaldehyde and benzene on immune cells in the blood and spleen in Balb/c mice. Environ. Toxicol. Pharmacol. 2016, 45, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Shu, Q.; Ma, H.; Wang, T.; Wang, P.; Xu, H. Formaldehyde promotes tumor-associated macrophage polarizations and functions through induction of HIF-1alpha-mediated glycolysis. Toxicol. Lett. 2023, 390, 5–14. [Google Scholar] [CrossRef]
- Ye, S.; Ma, Y.; Li, S.; Luo, S.; Wei, L.; Hu, D.; Xiao, F. Ambient NO(2) hinders neutrophil extracellular trap formation in rats: Assessment of the role of neutrophil autophagy. J. Hazard. Mater. 2023, 457, 131755. [Google Scholar] [CrossRef]
- Fritsche, K.L. The science of fatty acids and inflammation. Adv. Nutr. 2015, 6, 293S–301S. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Bushrow, A.E.; Burmeister, C.; Birbeck, J.; Chen, Y.; Lamerato, L.; Lemke, L.D.; Li, J.; Mor, G.; O’Leary, B.F.; Peters, R.M.; et al. Ambient BTEX exposure and mid-pregnancy inflammatory biomarkers in pregnant African American women. J. Reprod. Immunol. 2021, 145, 103305. [Google Scholar] [CrossRef]
- Guo, H.; Ahn, S.; Zhang, L. Benzene-associated immunosuppression and chronic inflammation in humans: A systematic review. Occup. Environ. Med. 2020, 78, 377–384. [Google Scholar] [CrossRef]
- Liu, K.; Cao, H.; Li, B.; Guo, C.; Zhao, W.; Han, X.; Zhang, H.; Wang, Z.; Tang, N.; Niu, K.; et al. Long-term exposure to ambient nitrogen dioxide and ozone modifies systematic low-grade inflammation: The CHCN-BTH study. Int. J. Hyg. Environ. Health 2022, 239, 113875. [Google Scholar] [CrossRef]
- Rappazzo, K.M.; Nichols, J.L.; Rice, R.B.; Luben, T.J. Ozone exposure during early pregnancy and preterm birth: A systematic review and meta-analysis. Environ. Res. 2021, 198, 111317. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, W.; Liu, Q.; Li, Z.; Lei, L.; Ren, L.; Deng, F.; Guo, X.; Wu, S. Association between gaseous air pollutants and biomarkers of systemic inflammation: A systematic review and meta-analysis. Environ. Pollut. 2022, 292, 118336. [Google Scholar] [CrossRef]
- Ziegler, K.; Kunert, A.T.; Reinmuth-Selzle, K.; Leifke, A.L.; Widera, D.; Weller, M.G.; Schuppan, D.; Frohlich-Nowoisky, J.; Lucas, K.; Poschl, U. Chemical modification of pro-inflammatory proteins by peroxynitrite increases activation of TLR4 and NF-kappaB: Implications for the health effects of air pollution and oxidative stress. Redox Biol. 2020, 37, 101581. [Google Scholar] [CrossRef] [PubMed]
- Faloppa, C.C.; Baiocchi, G.; Cunha, I.W.; Fregnani, J.H.; Osorio, C.A.; Fukazawa, E.M.; Kumagai, L.Y.; Badiglian-Filho, L.; Pinto, G.L.; Soares, F.A. NF-kappaB and COX-2 expression in nonmalignant endometrial lesions and cancer. Am. J. Clin. Pathol. 2014, 141, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.Z.; Yang, H.L.; Ha, S.Y.; Chang, K.K.; Mei, J.; Zhou, W.J.; Qiu, X.M.; Wang, X.Q.; Zhu, R.; Li, D.J.; et al. Cyclooxygenase-2 in Endometriosis. Int. J. Biol. Sci. 2019, 15, 2783–2797. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Molecular Weight | Molecular Formula | SMILES Structure | Carcinogenicity | ||
|---|---|---|---|---|---|---|
| ADMETLAB | ProTox | admetSAR | ||||
| Benzene | 78.11 g/mol | C6H6 | C1=CC=CC=C1 | 0.969 | 0.92 | 0.5524 |
| Toluene | 92.14 g/mol | C7H8|C6H5CH3 | CC1=CC=CC=C1 | 0.957 | 0.88 | 0.5357 |
| Sulfur Dioxide | 64.07 g/mol | O2S|SO2 | O=S=O | 0.786 | 0.6 | 0.6371 |
| Nitric Oxide | 30.006 g/mol | NO | [N]=O | 0.635 | 0.59 | 0.4813 |
| Nitrogen Dioxide | 46.006 g/mol | NO2 | N(=O) [O] | 0.976 | 0.51 | 0.4606 |
| Carbon Monoxide | 28.010 g/mol | CO | [C−] # [O+] | 1 | 0.51 | 0.5716 |
| Ozone | 47.998 g/mol | O3 | [O−] [O+]=O | 0.981 | 0.56 | 0.4759 |
| Ethylbenzene | 106.16 g/mol | C8H10 | CCC1=CC=CC=C1 | 0.736 | 0.89 | 0.5524 |
| Formaldehyde | 30.026 g/mol | CH2O|H2CO | C=O | 0.654 | 0.78 | 0.7138 |
| Gene 1 | Functional Role in EC/Cancer | Associated Pathways | Stably Bound Pollutants 2 | References |
|---|---|---|---|---|
| TNF | Tumor Necrosis Factor: Encodes a pro-inflammatory cytokine regulating immune responses, apoptosis, and cell survival. Exerts context-dependent dual roles—promotes tumor growth, angiogenesis, invasion, and immune evasion via NF-κB activation; alternatively induces apoptosis and anti-tumor immunity through TNFR1-mediated death receptor signaling and caspase activation. Outcome influenced by receptor subtype, cell type, and microenvironment. | NF-κB signaling, MAPK signaling, cytokine–cytokine receptor interaction, apoptosis, etc. | Ethylbenzene, Toluene, Benzene | [25,26] |
| ESR1 | Estrogen Receptor 1: Encodes a nuclear hormone receptor that binds estrogen response elements to regulate genes involved in proliferation, survival, and angiogenesis. Promotes tumor growth by activating pathways such as PI3K/AKT, MAPK, and E2F in type I EC (hormone-dependent); high expression typically correlates with better differentiation and prognosis. Downregulation—commonly due to promoter methylation or mutation—is frequent in high-risk or advanced-stage EC, contributing to hormone resistance, poor differentiation, and invasiveness. This shift may promote a transition to hormone-independent growth, resembling the aggressive, poorly differentiated characteristics of type II EC. Loss also disrupts immune surveillance by reducing CD8+ T-cell infiltration and enhancing pro-tumorigenic signaling (e.g., VEGF, TGF-β), creating an immunosuppressive microenvironment. Thus, ESR1 plays a context-dependent role in EC, with its presence supporting controlled proliferation and its loss linked to progression and immune evasion. | Estrogen signaling, PI3K/Akt signaling, MAPK signaling, Wnt/β-catenin, etc. | Ethylbenzene, Toluene | [27,28,29,30,31,32,33,34] |
| IL1B | Interleukin-1 Beta: Pro-inflammatory cytokine produced by immune cells, fibroblasts, and tumor cells upon inflammasome activation. Promotes cancer progression by enhancing angiogenesis, proliferation, invasion, metastasis, and by driving chronic inflammation and immunosuppressive cell recruitment. In some contexts, supports anti-tumor immunity via dendritic cell and Th1 activation. | NF-κB signaling, cytokine–cytokine receptor interaction, PI3K/Akt signaling, MAPK signaling, STAT3 signaling, etc. | Ethylbenzene | [35,36,37,38,39] |
| NFKB1 | Nuclear Factor Kappa B Subunit 1: Encodes p105/p50, a transcription factor regulating immunity, inflammation, apoptosis, and survival. Promotes tumor progression via p50:p65 heterodimers that activate pro-inflammatory and pro-survival genes; p50:p50 homodimers with Bcl-3 may also drive oncogenesis. Conversely, p50 homodimers lacking transactivation domains can suppress inflammation, inhibit immune evasion, and block survival of DNA-damaged cells, acting as context-dependent tumor suppressors. | NF-κB signaling, apoptosis, PI3K/Akt signaling, MAPK signaling, DNA damage response (DDR) Pathway, etc. | Ethylbenzene, Toluene | [40,41] |
| PTGS2 | Prostaglandin–Endoperoxide Synthase 2: Encodes COX-2, a key enzyme in prostaglandin biosynthesis and inflammation. Often upregulated in cancer, promoting angiogenesis, proliferation, invasion, immune evasion, and resistance to apoptosis and therapy. Reduced expression may disrupt inflammatory balance and immune regulation, favoring immune escape and aggressive tumor behavior. | NF-κB signaling, HIF-1 signaling, PI3K/Akt signaling, MAPK signaling, prostaglandin synthesis pathway, etc. | Ethylbenzene, Toluene, Benzene | [42,43,44] |
| KCNH2 | Voltage-gated potassium channel involved in membrane repolarization and electrical excitability. Limited direct evidence in EC; overexpression in other cancers linked to enhanced proliferation and migration. | Ion channel signaling, potential role in PI3K/Akt signaling, etc. | - | [45,46] |
| CCNE1 | Cyclin E1: Promotes G1/S transition by forming a complex with CDK2. Frequently amplified in EC, especially serous-like or copy-number high subtypes. Overexpression associated with genomic instability and poor prognosis. | Cell cycle, p53 signaling, PI3K/Akt signaling, etc. | Ethylbenzene, Toluene, Benzene | [47,48,49,50] |
| CCR2 | C-C Chemokine Receptor Type 2: Mediates monocyte/macrophage chemotaxis and shapes the tumor microenvironment. Promotes cancer progression by recruiting immunosuppressive TAMs and Tregs, enhancing tumor survival, invasion, and metastasis. Reduced signaling may impair immune surveillance, facilitating chronic inflammation or immune escape depending on context. | CCL2/CCR2 signaling axis, PI3K/Akt signaling, MAPK/ERK signaling, NF-κB signaling, etc. | - | [51,52,53,54] |
| HPRT1 | Hypoxanthine Phosphoribosyltransferase: Catalyzes purine salvage supporting nucleotide biosynthesis. Commonly overexpressed in EC and other cancers, facilitating rapid proliferation by supplying purine precursors for DNA replication and growth. | Purine metabolism, nucleotide biosynthesis, etc. | - | [55,56] |
| FOLH1 | Folate Hydrolase 1: Involved in one-carbon metabolism. Downregulated in advanced-stage EC, associated with poor prognosis and reduced immune infiltration (CD8+ T cells, dendritic cells). Epigenetic silencing via promoter methylation may promote immune evasion and metabolic dysregulation in the tumor microenvironment. | Glutamate metabolism, folate biosynthesis, etc. | Ethylbenzene | [57,58] |
| CDC25C | Cell Cycle Phosphatase: Promotes G2/M transition by activating CDK1. Overexpression accelerates mitosis, driving uncontrolled proliferation and genomic instability. In EC, especially MSI-high subtypes, CDC25C mutations or dysregulation may enhance tumor progression. | Cell cycle, G2/M checkpoint, p53 signaling, etc. | - | [59,60,61,62] |
| AHCY | S-adenosylhomocysteine Hydrolase (AHCY): Regulates methylation potential by hydrolyzing SAH, maintaining SAM:SAH balance essential for DNA, RNA, and protein methylation. Supports chromatin regulation and nucleotide synthesis. Limited direct evidence in EC but overexpression may promote epigenetic reprogramming, nucleotide synthesis, and oxidative stress adaptation, contributing to tumor progression. | Methionine metabolism, epigenetic regulation, DNA methylation, oxidative stress response, etc. | Ethylbenzene, Toluene | [63,64] |
| CTSD | Cathepsin D: Secreted lysosomal protease acting as an autocrine/paracrine mitogen. Promotes tumor proliferation, invasion, metastasis, and angiogenesis. | Lysosomal pathway, extracellular matrix degradation, MAPK signaling, apoptosis, etc. | Ethylbenzene | [65] |
| CDC25B | Cell cycle phosphatase promoting G2/M transition via CDK1–cyclin B1 activation, facilitating mitotic entry and proliferation. Frequently overexpressed in cancers, enabling checkpoint bypass, chromosomal instability, and tumor aggressiveness. | Cell cycle, G2/M checkpoint, p53 signaling, etc. | - | [62,66,67,68] |
| MMP2 | Zinc-Dependent Matrix Metalloproteinase: Degrades extracellular matrix (ECM) components. Although direct evidence in EC is limited, it is overexpressed in aggressive tumors, promoting angiogenesis, migration, and pro-tumor signaling (e.g., VEGF, TGF-β). Deficiency impairs ECM remodeling, causing accumulation of pro-inflammatory mediators (e.g., MCP-3, TNF-α), which may foster metabolic and inflammatory dysregulation, a tumor-promoting microenvironment, and genomic instability. | ECM remodeling, NF-κB signaling, TGF-β signaling, VEGF pathway, etc. | Ethylbenzene, Toluene | [69,70,71,72] |
| TLR4 | Pattern recognition receptor detecting pathogen- and damage-associated molecular patterns (PAMPs/DAMPs), triggering pro-inflammatory and immune responses via NF-κB and IRF3 pathways. Limited direct evidence in EC; overexpressed in several cancers, promoting tumor growth, inflammation, and chemoresistance. Reduced expression may impair dendritic cell activation, antigen presentation, and cytotoxic T-cell recruitment, weakening anti-tumor immunity. Also involved in tissue integrity and epithelial repair; downregulation may hinder these processes and attenuate type I interferon responses, fostering an immunosuppressive microenvironment. | NF-κB signaling, IRF3 pathway, Toll-like receptor signaling, immune cell recruitment, epithelial repair, etc. | Ethylbenzene, Toluene | [73,74,75,76,77] |
| SLC2A1 | Glucose Transporter 1: Facilitates glucose uptake and supports metabolic reprogramming via the Warburg effect. Frequently overexpressed in cancers, promoting proliferation, survival, and chemoresistance. | HIF-1 signaling, glucose metabolism, PI3K-Akt-mTOR, Ras-MAPK, c-MYC signaling, p53 suppression, etc. | Ethylbenzene, Toluene | [78,79,80] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Zou, Y. From Target Prediction to Mechanistic Insights: Revealing Air Pollution-Driven Mechanisms in Endometrial Cancer via Interpretable Machine Learning and Molecular Docking. Atmosphere 2025, 16, 841. https://doi.org/10.3390/atmos16070841
Liu H, Zou Y. From Target Prediction to Mechanistic Insights: Revealing Air Pollution-Driven Mechanisms in Endometrial Cancer via Interpretable Machine Learning and Molecular Docking. Atmosphere. 2025; 16(7):841. https://doi.org/10.3390/atmos16070841
Chicago/Turabian StyleLiu, Hongyao, and Yueqing Zou. 2025. "From Target Prediction to Mechanistic Insights: Revealing Air Pollution-Driven Mechanisms in Endometrial Cancer via Interpretable Machine Learning and Molecular Docking" Atmosphere 16, no. 7: 841. https://doi.org/10.3390/atmos16070841
APA StyleLiu, H., & Zou, Y. (2025). From Target Prediction to Mechanistic Insights: Revealing Air Pollution-Driven Mechanisms in Endometrial Cancer via Interpretable Machine Learning and Molecular Docking. Atmosphere, 16(7), 841. https://doi.org/10.3390/atmos16070841