Development of an Understanding of Reactive Mercury in Ambient Air: A Review

 ,
, 
Abstract
1. Introduction
1.1. Discovery of GOM
1.2. Early Development of Methods
2. Early Method Intercomparisons
3. Work Pointing to Issues with the Tekran Speciation System
4. Realization RM Was Not Being Accurately Measured
4.1. Surrogate Surface Data
4.2. RAMIX
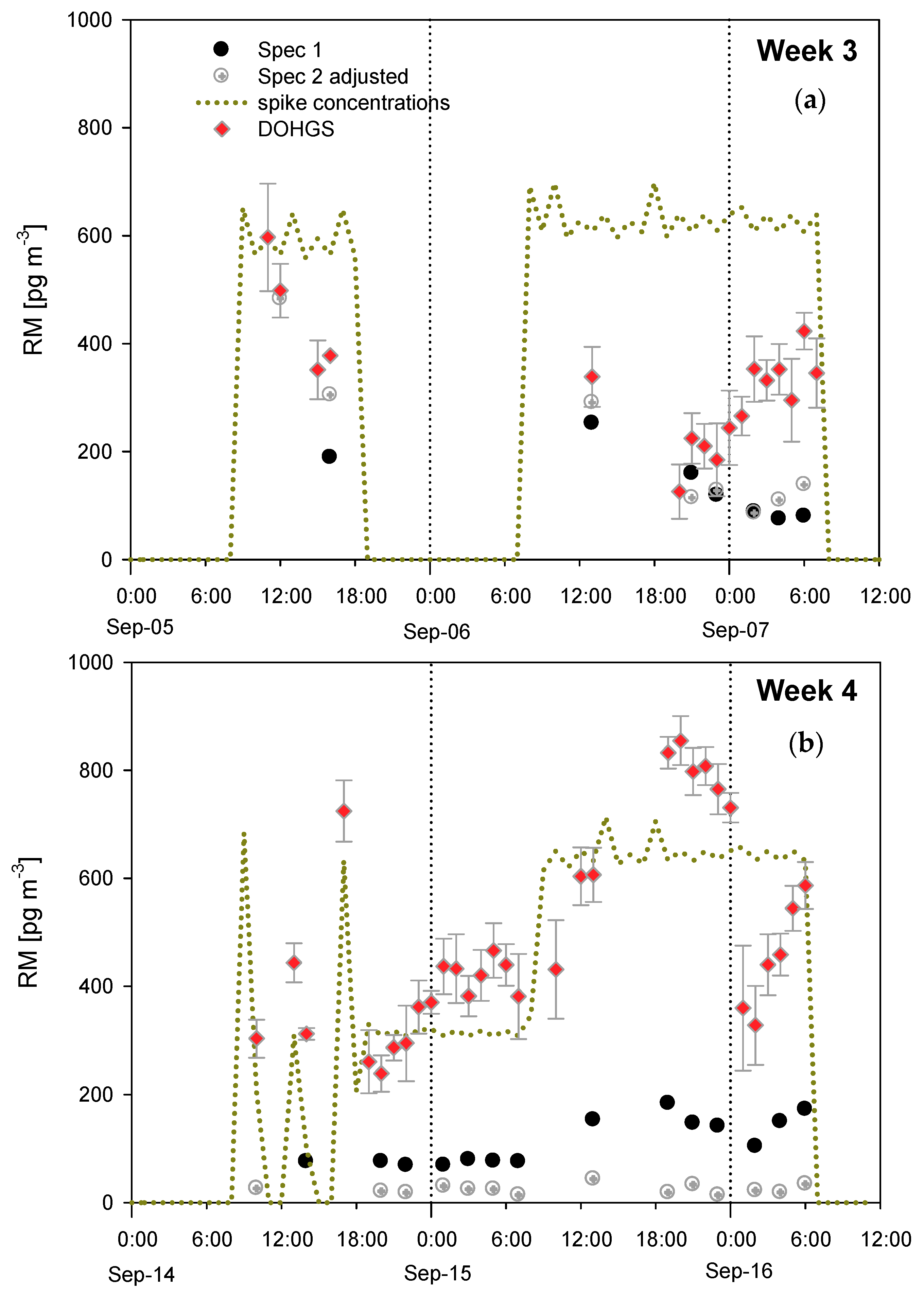
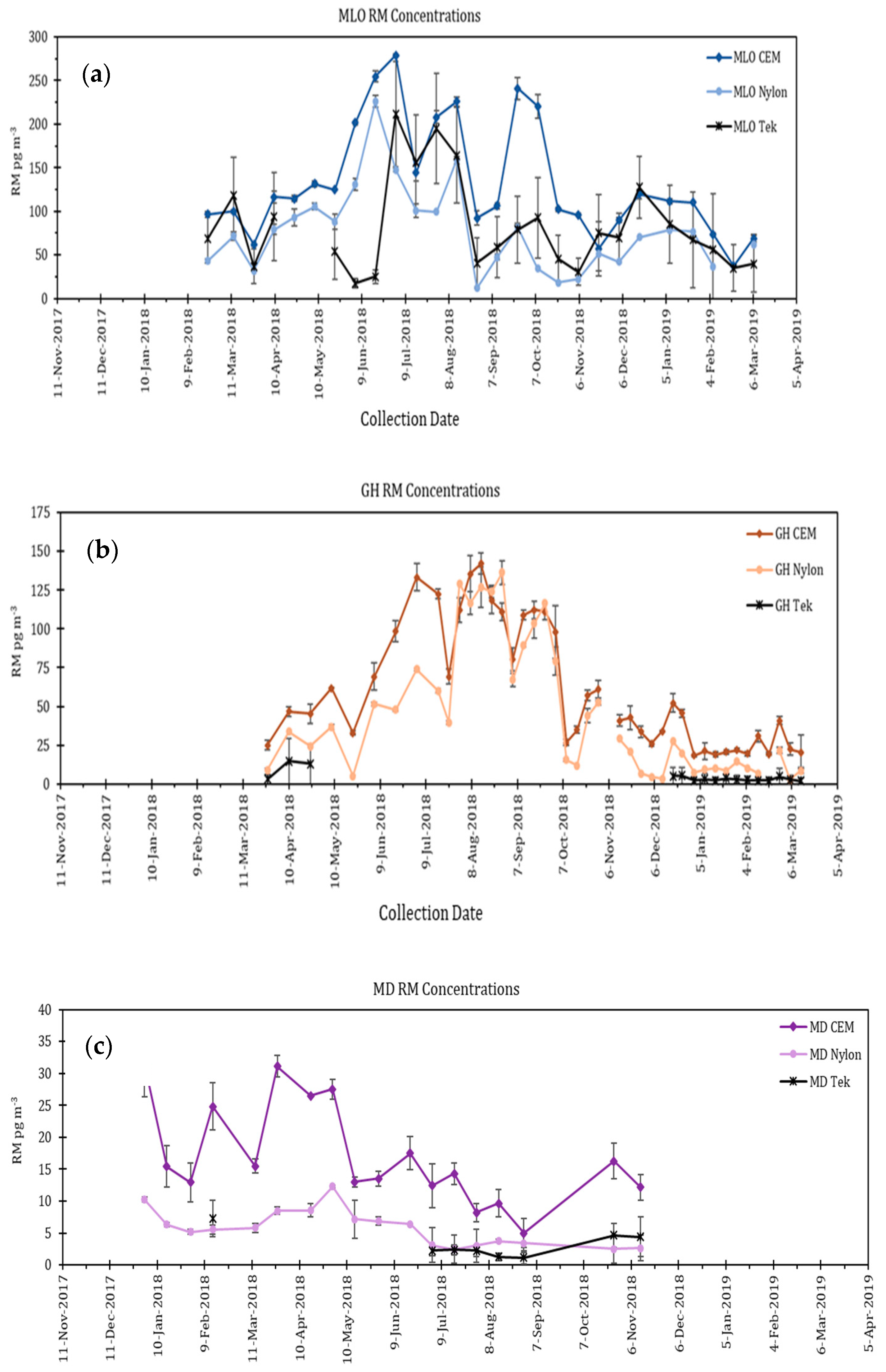
4.3. Additional Tests Following or Associated with RAMIX
5. Development of New Methods
5.1. Reactive Mercury Active System (RMAS)
5.2. Dual Channel Systems
5.3. Other Work Using CEM
5.4. Mass Spectrometric Methods
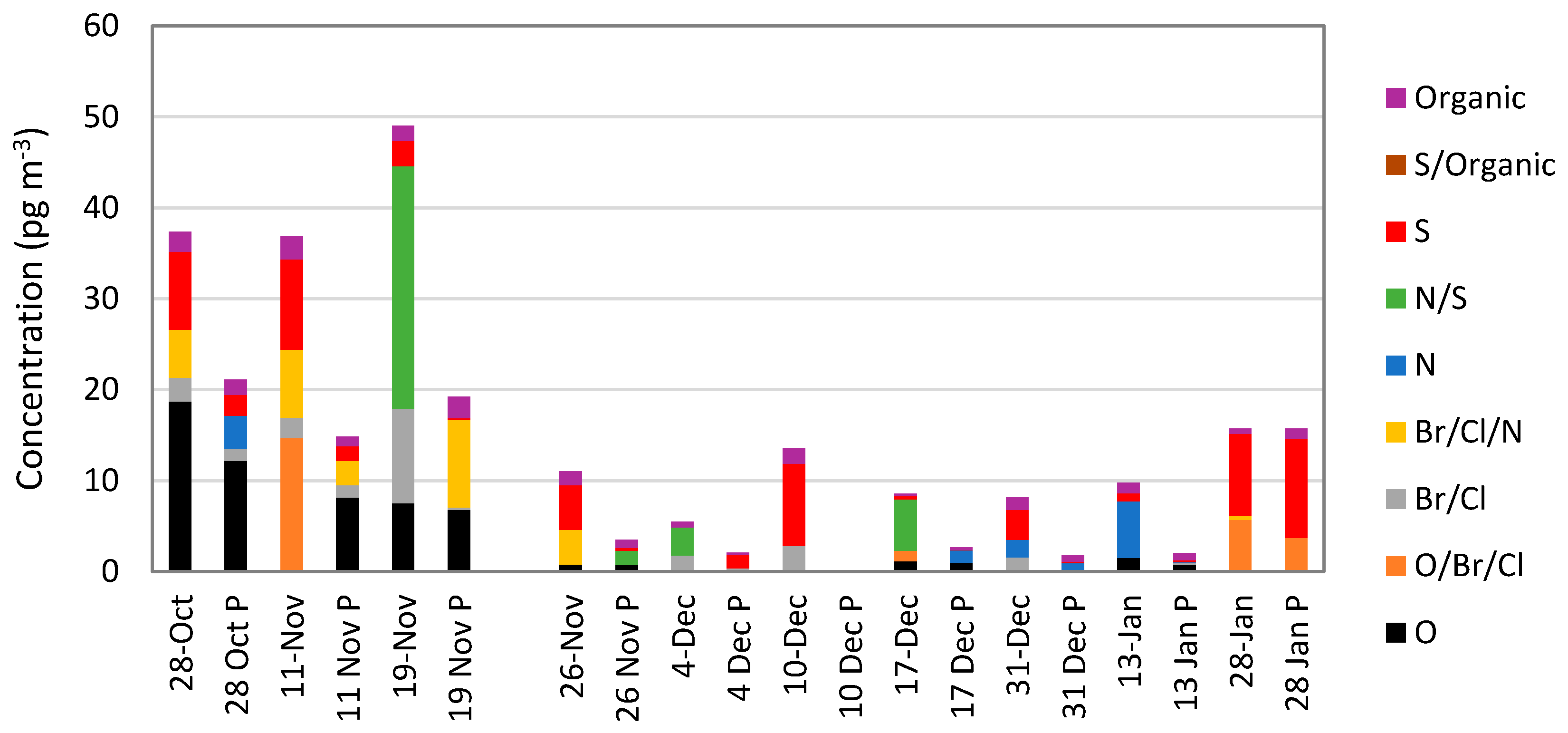
5.5. Oxidized Mercury Calibration Systems
6. What We Have Learned
7. Work Needed
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ariya, P.A.; Amyot, M.; Dastoor, A.; Deeds, D.; Feinberg, A.; Kos, G.; Poulain, A.; Ryjkov, A.; Semeniuk, K.; Subir, M.; et al. Mercury physicochemical and biogeochemical transformation in the atmosphere and at atmospheric interfaces: A review and future directions. Chem. Rev. 2015, 115, 3760–3802. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Pehkonen, S.O. Aqueous phase reactions of mercury with free radicals and chlorine: Implications for atmospheric mercury chemistry. Chemosphere 1999, 38, 1253–1263. [Google Scholar] [CrossRef]
- Saiz-Lopez, A.; Travnikov, O.; Sonke, J.E.; Thackray, C.P.; Jacob, D.J.; Carmona-García, J.; Francés-Monerris, A.; Roca-Sanjuán, R.; Acuña, A.U.; Dávalos, J.Z.; et al. Photochemistry of oxidized Hg(I) and Hg(II) species suggests missing mercury oxidation in the troposphere. Proc. Natl. Acad. Sci. USA 2020, 117, 30949–30956. [Google Scholar] [CrossRef]
- Fogg, T.R.; Fitzgerald, W.F. Mercury in southern New-England coastal rains. J. Geophys. Res. Oceans 1979, 84, 6987–6989. [Google Scholar] [CrossRef]
- Kothny, E.L. 3-phase equilibrium of mercury in nature. Adv. Chem. Ser. 1973, 123, 48–80. [Google Scholar]
- Brosset, C. Transport of airborne mercury emitted by coal burning into aquatic systems. Water Sci. Technol. 1983, 15, 59–66. [Google Scholar] [CrossRef]
- Iverfeldt, A.; Lindqvist, O. Atmospheric oxidation of elemental mercury by ozone in the aqueous phase. Atmos. Environ. 1986, 20, 1567–1573. [Google Scholar] [CrossRef]
- Xiao, Z.F.; Munthe, J.; Lindqvist, O. Sampling and determination of gaseous and particulate mercury in the atmosphere using gold-coated denuders. Water Air Soil Pollut. 1991, 56, 141–151. [Google Scholar] [CrossRef]
- Munthe, J.; Schroeder, W.H.; Xiao, Z.; Lindqvist, O. Removal of gaseous mercury from air using a gold coated denuder. Atmos. Environ. 1990, 24, 2271–2274. [Google Scholar]
- Fitzgerald, W.F.; Gill, G.A. Sub-nanogram determination of mercury by 2-stage gold amalgamation and gas-phase detection applied to atmospheric analysis. Anal. Chem. 1979, 51, 1714–1720. [Google Scholar]
- Bloom, N.; Fitzgerald, W.F. Determination of volatile mercury species at the picogram level by low-temperature gas-chromatography with cold-vapor atomic fluorescence detection. Anal. Chim. Acta. 1988, 208, 151–161. [Google Scholar] [CrossRef]
- Galbreath, K.C.; Zygarlicke, C.J. Mercury speciation in coal combustion and gasification flue gases. Environ. Sci. Technol. 1996, 30, 2421–2426. [Google Scholar] [CrossRef]
- Lindberg, S.E.; Meyers, T.P.; Taylor, G.E.; Turner, R.R.; Schroeder, W.H. Atmosphere/surface exchange of mercury in a forest: Results of modeling and gradient approaches. J. Geophys. Res. 1992, 97, 2519–2528. [Google Scholar] [CrossRef]
- Lindberg, S.E.; Turner, R.R.; Meyers, T.P.; Taylor, G.E.; Schroeder, W.H. Atmospheric concentrations and deposition of airborne mercury to Walker Branch Watershed. Water Air Soil Pollut. 1991, 56, 577–594. [Google Scholar] [CrossRef]
- Stratton, W.J.; Lindberg, S.E. Use of a refluxing mist chamber for measurement of gas-phase mercury(ii) species in the atmosphere. Water Air Soil Pollut. 1995, 80, 1269–1278. [Google Scholar] [CrossRef]
- Brosset, C.; Lord, E. Methylmercury in ambient air. Method of determination and some measurement results. Water Air Soil Pollut. 1995, 82, 739–750. [Google Scholar] [CrossRef]
- Lindberg, S.E.; Stratton, W.J. Atmospheric mercury speciation: Concentrations and behavior of reactive gaseous mercury in ambient air. Environ. Sci. Technol. 1998, 32, 49–57. [Google Scholar] [CrossRef]
- Stratton, W.J.; Lindberg, S.E.; Perry, C.J. Atmospheric mercury speciation: Laboratory and field evaluation of a mist chamber method for measuring reactive gaseous mercury. Environ. Sci. Technol. 2001, 35, 170–177. [Google Scholar] [CrossRef]
- Ebinghaus, R.; Jennings, S.G.; Schroeder, W.H.; Berg, T.; Donaghy, T.; Guentzel, J.; Kenny, C.; Kock, H.H.; Kvietkus, K.; Landing, W.; et al. International field intercomparison measurements of atmospheric mercury species at Mace Head, Ireland. Atmos. Environ. 1999, 33, 3063–3073. [Google Scholar] [CrossRef]
- Munthe, J.; Wängberg, I.; Pirrone, N.; Iverfeldt, Å.; Ferrara, R.; Ebinghaus, R.; Feng, X.; Gårdfeldt, K.; Keeler, G.; Lanzillotta, E.; et al. Intercomparison of methods for sampling and analysis of atmospheric mercury species. Atmos. Environ. 2001, 35, 3007–3017. [Google Scholar] [CrossRef]
- Feng, X.B.; Sommar, J.; Gardfeldt, K.; Lindqvist, O. Improved determination of gaseous divalent mercury in ambient air using KCl coated denuders. Fresenius J. Anal. Chem. 2000, 366, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Nacht, D.A.; Gustin, M.S.; Engle, M.A.; Zehner, R.Z.; Giglini, A.D. Quantifying total and reactive gaseous mercury at the Sulphur Banks Mercury Mine Superfund Site, Northern California. Environ. Sci. Technol. 2004, 38, 1977–1983. [Google Scholar] [CrossRef]
- Steffen, A.; Schroeder, W.; Bottenheim, J.; Narayan, J.; Fuentes, J.D. Atmospheric mercury concentrations: Measurements and profiles near snow and ice surfaces in the Canadian Arctic during Alert 2000. Atmos. Environ. 2002, 36, 2653–2661. [Google Scholar] [CrossRef]
- Sommar, J.; Feng, X.; Gardfeldt, K.; Lindqvist, O. Measurements of fractionated gaseous mercury concentrations over northwestern and central Europe, 1995–99. J. Environ. Monit. 1999, 1, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Landis, M.S.; Stevens, R.K.; Schaedlich, F.; Prestbo, E.M. Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air. Environ. Sci. Technol. 2002, 36, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Sommar, J.; Wei, S.; Lindqvist, O. Sampling and determination of gas phase divalent mercury in the air using a KCl coated denuder. Fresenius J. Anal. Chem. 1997, 358, 386–391. [Google Scholar] [CrossRef]
- Valente, R.J.; Shea, C.; Humes, K.L.; Tanner, R.L. Atmospheric mercury in the Great Smoky Mountains compared to regional and global levels. Atmos. Environ. 2007, 41, 1861–1873. [Google Scholar] [CrossRef]
- Gustin, M.S.; Huang, J.Y.; Miller, M.B.; Peterson, C.; Jaffe, D.A.; Ambrose, J.; Finley, B.D.; Lyman, S.N.; Call, K.; Talbot, R.; et al. Do we understand what the mercury speciation instruments are actually measuring? Results of RAMIX. Environ. Sci. Technol. 2013, 47, 7295–7306. [Google Scholar] [CrossRef]
- Lu, J.Y.; Schroeder, W.H.; Berg, T.; Munthe, J.; Schneeberger, D.; Schaedlich, F. A device for sampling and determination of total particulate mercury in ambient air. Anal. Chem. 1998, 70, 2403–2408. [Google Scholar] [CrossRef]
- Lynam, M.M.; Keeler, G.J. Artifacts associated with the measurement of particulate mercury in an urban environment: The influence of elevated ozone concentrations. Atmos. Environ. 2005, 39, 3081–3088. [Google Scholar] [CrossRef]
- Sheu, G.-R.; Mason, R.P. An examination of methods for the measurements of reactive gaseous mercury in the atmosphere. Environ. Sci. Technol. 2001, 35, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Lyman, S.N.; Gustin, M.S. Determinants of atmospheric mercury concentrations in Reno, Nevada, U.S.A. Sci. Total Environ. 2009, 408, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Weiss-Penzias, P.; Jaffe, D.A.; McClintick, A.; Prestbo, E.M.; Landis, M.S. Gaseous elemental mercury in the marine boundary layer: Evidence for rapid removal in anthropogenic pollution. Environ. Sci. Technol. 2003, 37, 3755–3763. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-D.; Huang, J.; Mondal, S.; Holsen, T.M. Variation in concentrations of three mercury (Hg) forms at a rural and a suburban site in New York State. Sci. Total Environ. 2013, 448, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Lyman, S.N.; Jaffe, D.A. Formation and fate of oxidized mercury in the upper troposphere and lower stratosphere. Nat. Geosci. 2012, 5, 114–117. [Google Scholar] [CrossRef]
- Swartzendruber, P.C.; Jaffe, D.A.; Finley, B. Development and first results of an aircraft-based, high time resolution technique for gaseous elemental and reactive (oxidized) gaseous mercury. Environ. Sci. Technol. 2009, 43, 7484–7489. [Google Scholar] [CrossRef]
- Lyman, S.N.; Gustin, M.S.; Prestbo, E.M.; Marsik, F.J. Estimation of dry deposition of atmospheric mercury in Nevada by direct and indirect methods. Environ. Sci. Technol. 2007, 41, 1970–1976. [Google Scholar] [CrossRef]
- Lyman, S.N.; Gustin, M.S.; Prestbo, E.M.; Kilner, P.I.; Edgerton, E.; Hartsell, B. Testing and application of surrogate surfaces for understanding potential gaseous oxidized mercury dry deposition. Environ. Sci. Technol. 2009, 43, 6235–6241. [Google Scholar] [CrossRef]
- Castro, M.S.; Moore, C.; Sherwell, J.; Brooks, S.B. Dry deposition of gaseous oxidized mercury in western Maryland. Sci. Total Environ. 2012, 417, 232–240. [Google Scholar] [CrossRef]
- Fang, G.C.; Lin, Y.H.; Chang, C.Y. Use of mercury dry deposition samplers to quantify dry deposition of particulate-bound mercury and reactive gaseous mercury at a traffic sampling site. Environ. Forensics 2013, 14, 182–186. [Google Scholar] [CrossRef]
- Sather, M.E.; Mukerjee, S.; Allen, K.L.; Smith, L.; Mathew, J.; Jackson, C.; Callison, R.; Scrapper, L.; Hathcoat, A.; Adam, J.; et al. Gaseous oxidized mercury dry deposition measurements in the southwestern USA: A comparison between Texas, eastern Oklahoma, and the Four Corners area. Sci. World J. 2014, 2014, 580723. [Google Scholar] [CrossRef] [PubMed]
- Sather, M.E.; Mukerjee, S.; Smith, L.; Mathew, J.; Jackson, C.; Callison, R.; Scrapper, L.; Hathcoat, A.; Adam, J.; Keese, D.; et al. Gaseous oxidized mercury dry deposition measurements in the Four Corners area and eastern Oklahoma, U.S.A. Atmos. Pollut. Res. 2013, 4, 168–180. [Google Scholar] [CrossRef]
- Peterson, C.; Alishahi, M.; Gustin, M.S. Testing the use of passive sampling systems for understanding air mercury concentrations and dry deposition across Florida, USA. Sci. Total Environ. 2012, 424, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Finley, B.D.; Jaffe, D.A.; Call, K.; Lyman, S.; Gustin, M.S.; Peterson, C.; Miller, M.; Lyman, T. Development, testing, and deployment of an air sampling manifold for spiking elemental and oxidized mercury during the Reno Atmospheric Mercury Intercomparison Experiment (RAMIX). Environ. Sci. Technol. 2013, 47, 7277–7284. [Google Scholar] [CrossRef]
- Ambrose, J.L.; Lyman, S.N.; Huang, J.Y.; Gustin, M.S.; Jaffe, D.A. Fast time resolution oxidized mercury measurements during the Reno Atmospheric Mercury Intercomparison Experiment (RAMIX). Environ. Sci. Technol. 2013, 47, 7285–7294. [Google Scholar] [CrossRef]
- Hynes, A.J.; Everhart, S.; Bauer, D.; Remeika, J.; Ernest, C.T. In situ and denuder-based measurements of elemental and reactive gaseous mercury with analysis by laser-induced fluorescence results from the Reno Atmospheric Mercury Intercomparison Experiment. Atmos. Chem. Phys. 2017, 17, 465–483. [Google Scholar] [CrossRef]
- Ambrose, J.L.; Gratz, L.E.; Jaffe, D.A.; Campos, T.; Flocke, F.M.; Knapp, D.J.; Stechman, D.M.; Stell, M.; Weinheimer, A.J.; Cantrell, C.A.; et al. Mercury emission ratios from coal-fired power plants in the southeastern United States during NOMADSS. Environ. Sci. Technol. 2015, 49, 10389–10397. [Google Scholar] [CrossRef]
- Luippold, A.; Gustin, M.S.; Dunham-Cheatham, S.M.; Castro, M.; Luke, W.; Lyman, S.; Zhang, L. Use of multiple lines of evidence to understand reactive mercury concentrations and chemistry in Hawai’i, Nevada, Maryland, and Utah, USA. Environ. Sci. Technol. 2020, 54, 7922–7931. [Google Scholar] [CrossRef]
- Huang, J.Y.; Miller, M.B.; Weiss-Penzias, P.; Gustin, M.S. Comparison of gaseous oxidized Hg measured by KCl-coated denuders, and nylon and cation exchange membranes. Environ. Sci. Technol. 2013, 47, 7307–7316. [Google Scholar] [CrossRef]
- Rutter, A.P.; Schauer, J.J. The impact of aerosol composition on the particle to gas partitioning of reactive mercury. Environ. Sci. Technol. 2007, 41, 3934–3939. [Google Scholar] [CrossRef]
- Rutter, A.P.; Schauer, J.J. The effect of temperature on the gas-particle partitioning of reactive mercury in atmospheric aerosols. Atmos. Environ. 2007, 41, 8647–8657. [Google Scholar] [CrossRef]
- Talbot, R.; Mao, H.; Feddersen, D.; Smith, M.; Kim, S.Y.; Sive, B.; Haase, K.; Ambrose, J.; Zhou, Y.; Russo, R. Assessment of particulate mercury measured with the manual and automated methods. Atmosphere 2010, 2, 1–20. [Google Scholar] [CrossRef]
- Gustin, M.S.; Amos, H.M.; Huang, J.; Miller, M.B.; Heidecorn, K. Measuring and modeling mercury in the atmosphere: A critical review. Atmos. Chem. Phys. 2015, 15, 5697–5713. [Google Scholar] [CrossRef]
- Lyman, S.N.; Cheng, I.; Gratz, L.E.; Weiss-Penzias, P.; Zhang, L. An updated review of atmospheric mercury. Sci. Total Environ. 2020, 707, 135575. [Google Scholar] [CrossRef] [PubMed]
- Gustin, M.S.; Dunham-Cheatham, S.M.; Zhang, L.; Lyman, S.; Choma, N.; Castro, M. Use of membranes and detailed HYSPLIT analyses to understand atmospheric particulate, gaseous oxidized, and reactive mercury chemistry. Environ. Sci. Technol. 2020. [Google Scholar] [CrossRef]
- Huang, J.Y.; Gustin, M.S. Uncertainties of gaseous oxidized mercury measurements using KCl-coated denuders, cation-exchange membranes, and nylon membranes: Humidity influences. Environ. Sci. Technol. 2015, 49, 6102–6108. [Google Scholar] [CrossRef] [PubMed]
- McClure, C.D.; Jaffe, D.A.; Edgerton, E.S. Evaluation of the KCI denuder method for gaseous oxidized mercury using HgBr2 at an in-service AMNet site. Environ. Sci. Technol. 2014, 48, 11437–11444. [Google Scholar] [CrossRef]
- Huang, J.Y.; Miller, M.B.; Edgerton, E.; Gustin, M.S. Deciphering potential chemical compounds of gaseous oxidized mercury in Florida, USA. Atmos. Chem. Phys. 2017, 17, 1689–1698. [Google Scholar] [CrossRef]
- Gustin, M.S.; Pierce, A.M.; Huang, J.Y.; Miller, M.B.; Holmes, H.A.; Loria-Salazar, S.M. Evidence for different reactive Hg sources and chemical compounds at adjacent valley and high elevation locations. Environ. Sci. Technol. 2016, 50, 12225–12231. [Google Scholar] [CrossRef]
- He, Y.; Mason, R.P. Comparison of reactive gaseous mercury measured by KCl-coated denuders andcation exchange membranes during the Pacific GEOTRACES GP15 expedition. Atmos. Environ. 2020, 241, 117973. [Google Scholar] [CrossRef]
- Miller, M.B.; Howard, D.A.; Pierce, A.M.; Cook, K.; Keywood, M.; Powell, J.; Gustin, M.S.; Edwards, G.C. Atmospheric reactive mercury concentrations in coastal Australia and the Southern Ocean. Sci. Total Environ. 2021, 751, 141681. [Google Scholar] [CrossRef] [PubMed]
- Luippold, A.; Gustin, M.S.; Dunham-Cheatham, S.M.; Zhang, L. Improvement of quantification and identification of atmospheric reactive mercury. Atmos. Environ. 2020, 224, 117307. [Google Scholar] [CrossRef]
- Lyman, S.N.; Jaffe, D.A.; Gustin, M.S. Release of mercury halides from KCl denuders in the presence of ozone. Atmos. Chem. Phys. 2010, 10, 8197–8204. [Google Scholar] [CrossRef]
- Gratz, L.E.; Ambrose, J.L.; Jaffe, D.A.; Shah, V.; Jaeglé, L.; Stutz, J.; Festa, J.; Spolaor, M.; Tsai, C.; Selin, N.E.; et al. Oxidation of mercury by bromine in the subtropical Pacific free troposphere. Geophys. Res. Lett. 2015, 42, 10–494. [Google Scholar] [CrossRef]
- Gustin, M.S.; Dunham-Cheatham, S.M.; Zhang, L. Comparison of 4 methods for measurement of reactive, gaseous oxidized, and particulate bound mercury. Environ. Sci. Technol. 2019, 53, 14489–14495. [Google Scholar] [CrossRef]
- Lyman, S.N.; Gratz, L.; Dunham-Cheatham, S.M.; Gustin, M.S.; Luippold, A. Improvements to the accuracy of atmospheric oxidized mercury measurements. Environ. Sci. Technol. 2020, 54, 13379–13388. [Google Scholar] [CrossRef]
- Miller, M.B.; Gustin, M.S.; Edwards, G.C. Reactive mercury flux measurements using cation exchange membranes. Atmos. Meas. Tech. Discuss. 2018. [Google Scholar] [CrossRef]
- Zhang, L.; Wright, L.P.; Blanchard, P. A review of current knowledge concerning dry deposition of atmospheric mercury. Atmos. Environ. 2009, 43, 5853–5864. [Google Scholar] [CrossRef]
- Marusczak, N.; Sonke, J.E.; Fu, X.W.; Jiskra, M. Tropospheric GOM at the Pic du Midi Observatory-Correcting bias in denuder based observations. Environ. Sci. Technol. 2017, 51, 863–869. [Google Scholar] [CrossRef]
- Dunham-Cheatham, S.M.; Lyman, S.; Gustin, M.S. Evaluation of sorption surface materials for reactive mercury compounds. Atmos. Environ. 2020, 242, 117836. [Google Scholar] [CrossRef]
- Deeds, D.A.; Ghoshdastidar, A.; Raofie, F.; Guérette, E.A.; Tessier, A.; Ariya, P.A. Development of a particle-trap preconcentration-soft ionization mass spectrometric technique for the quantification of mercury halides in air. Anal. Chem. 2015, 87, 5109–5116. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.P.; Lyman, S.N.; Jaffe, D.A.; Allen, T.; O’Neil, T.L. Detection and quantification of gas-phase oxidized mercury compounds by GC/MS. Atmos. Meas. Tech. 2016, 9, 2195–2205. [Google Scholar] [CrossRef]
- Khalizov, A.F.; Guzman, F.J.; Cooper, M.; Mao, N.; Antley, J.; Bozzelli, J. Direct detection of gas-phase mercuric chloride by ion drift-chemical ionization mass spectrometry. Atmos. Environ. 2020, 238, 117687. [Google Scholar] [CrossRef]
- Gustin, M.S.; Jaffe, D. Reducing the uncertainty in measurement and understanding of mercury in the atmosphere. Environ. Sci. Technol. 2010, 44, 2222–2227. [Google Scholar] [CrossRef]
- Jaffe, D.A.; Lyman, S.; Amos, H.M.; Gustin, M.S.; Huang, J.; Selin, N.E.; Levin, L.; Ter Schure, A.; Mason, R.P.; Talbot, R.; et al. Progress on understanding atmospheric mercury hampered by uncertain measurements. Environ. Sci. Technol. 2014, 48, 7204–7206. [Google Scholar] [CrossRef]
- Lyman, S.N.; Jones, C.; O’Neil, T.; Allen, T.; Miller, M.; Gustin, M.S.; Pierce, A.M.; Luke, W.; Ren, X.; Kelley, P. Automated calibration of atmospheric oxidized mercury measurements. Environ. Sci. Technol. 2016, 50, 12921–12927. [Google Scholar] [CrossRef]
- Sari, S.; Timo, R.; Jussi, H.; Panu, H. Dynamic calibration method for reactive gases. Meas. Sci. Technol. 2019, 31, 034001. [Google Scholar] [CrossRef]
- González, R.O.; Díaz-Somoano, M.; Antón, M.L.; Martínez-Tarazona, M.R. Effect of adding aluminum salts to wet FGD systems upon the stabilization of mercury. Fuel 2012, 96, 568–571. [Google Scholar] [CrossRef]
- ISO/IEC Guide 98-3:2008. Available online: http://www.iso.org/sites/JCGM/JCGM-introduction.htm (accessed on 4 January 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | HgCl2 | HgBr2 | HgO | Hg(NO3)2 | HgSO4 |
|---|---|---|---|---|---|
| KCl denuder (x) vs. nylon membrane (y) | y = 1.6x + 0.002 r2 = 0.97, n = 12 | y = 1.7x + 0.01 r2 = 0.99, n = 10 | y = 1.8x + 0.02 r2 = 0.99, n = 8 | y = 1.4x + 0.04 r2 = 0.90, n = 12 | y = 1.9x − 0.1 r2 = 0.6, n = 12 |
| KCl denuder vs. CEM (y) | y = 2.4x + 0.1 r2 = 0.58, n = 9 | y = 1.6x + 0.2 r2 = 0.86, n = 5 | y = 3.7x + 0.1 r2 = 0.99, n = 6 | y = 12.6x − 0.02 r2 = 0.50, n = 6 | y = 2.3x + 0.01 r2 = 0.95, n = 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustin, M.S.; Dunham-Cheatham, S.M.; Huang, J.; Lindberg, S.; Lyman, S.N. Development of an Understanding of Reactive Mercury in Ambient Air: A Review. Atmosphere 2021, 12, 73. https://doi.org/10.3390/atmos12010073
Gustin MS, Dunham-Cheatham SM, Huang J, Lindberg S, Lyman SN. Development of an Understanding of Reactive Mercury in Ambient Air: A Review. Atmosphere. 2021; 12(1):73. https://doi.org/10.3390/atmos12010073
Chicago/Turabian StyleGustin, Mae Sexauer, Sarrah M. Dunham-Cheatham, Jiaoyan Huang, Steve Lindberg, and Seth N. Lyman. 2021. "Development of an Understanding of Reactive Mercury in Ambient Air: A Review" Atmosphere 12, no. 1: 73. https://doi.org/10.3390/atmos12010073
APA StyleGustin, M. S., Dunham-Cheatham, S. M., Huang, J., Lindberg, S., & Lyman, S. N. (2021). Development of an Understanding of Reactive Mercury in Ambient Air: A Review. Atmosphere, 12(1), 73. https://doi.org/10.3390/atmos12010073