Traceable Determination of Atmospheric Mercury Using Iodinated Activated Carbon Traps


Abstract
:1. Introduction
2. Experiments
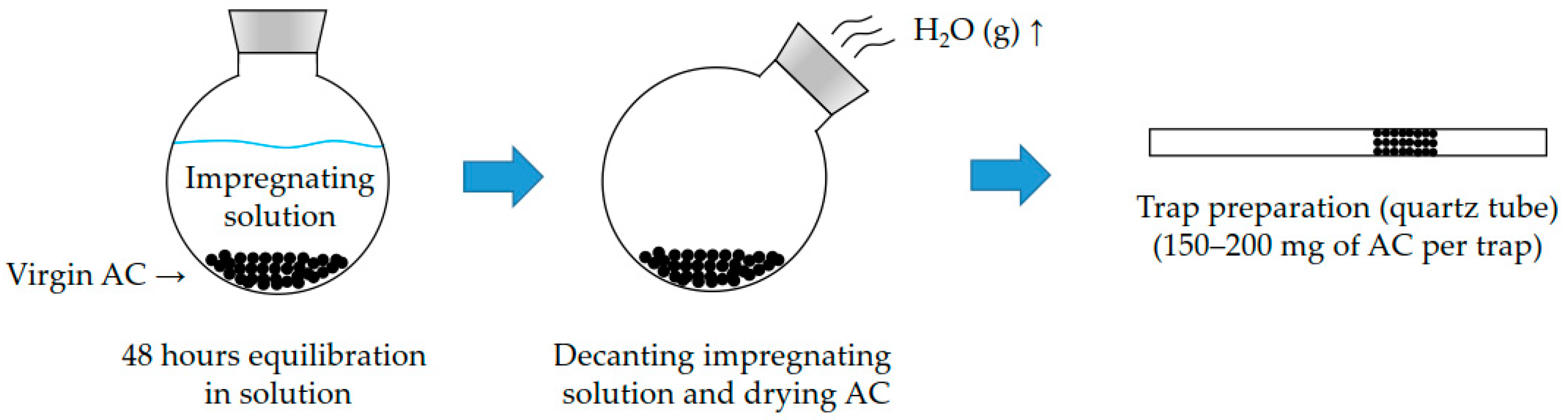
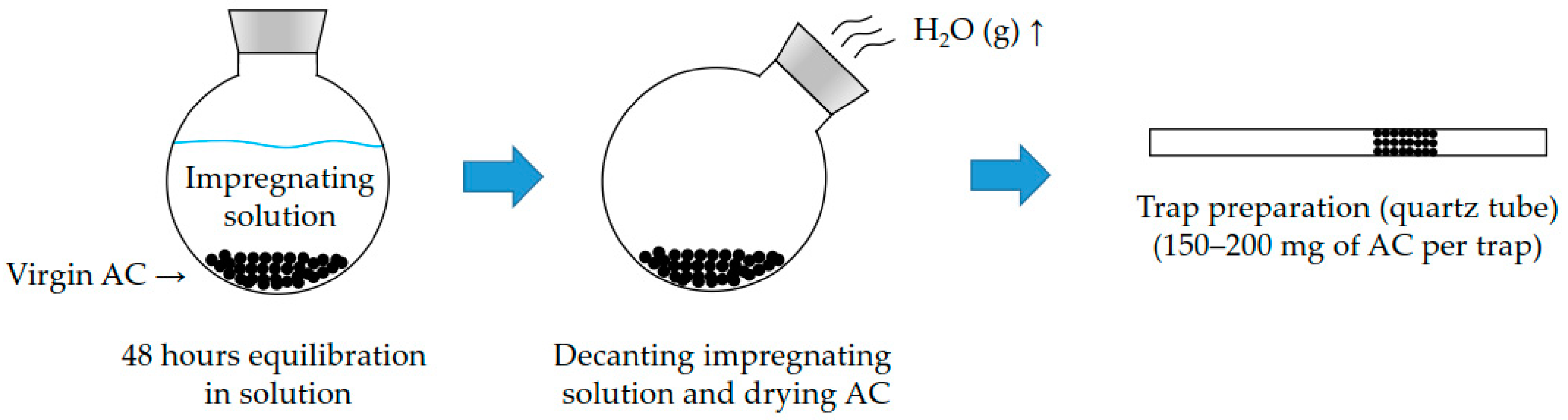
2.1. Preparation of In-House Impregnated Activated Carbon
2.2. Preparation of Activated Carbon Traps
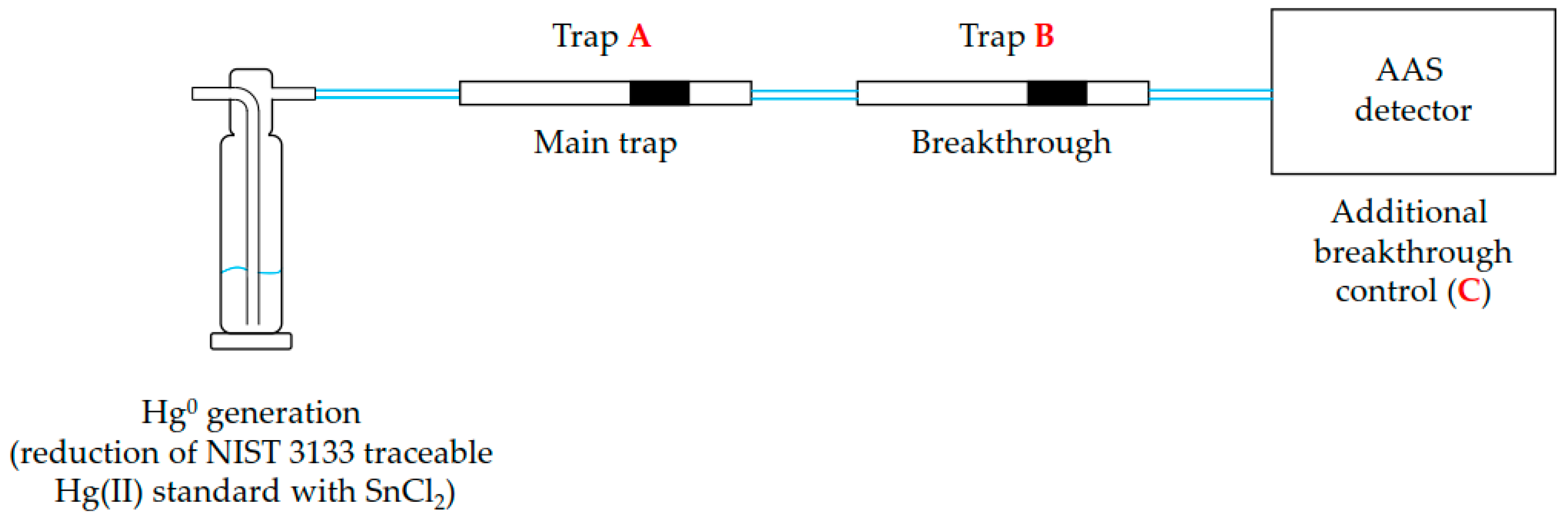
2.3. Loading of Mercury on Activated Carbon Traps
2.4. Determination of Mercury on Activated Carbon Traps
2.5. Determination of Mercury in Real Sample Using Impregnated Activated Carbon Traps—Proof of Concept
2.6. Thermal Fractionation of Mercury on Activated Carbon Traps
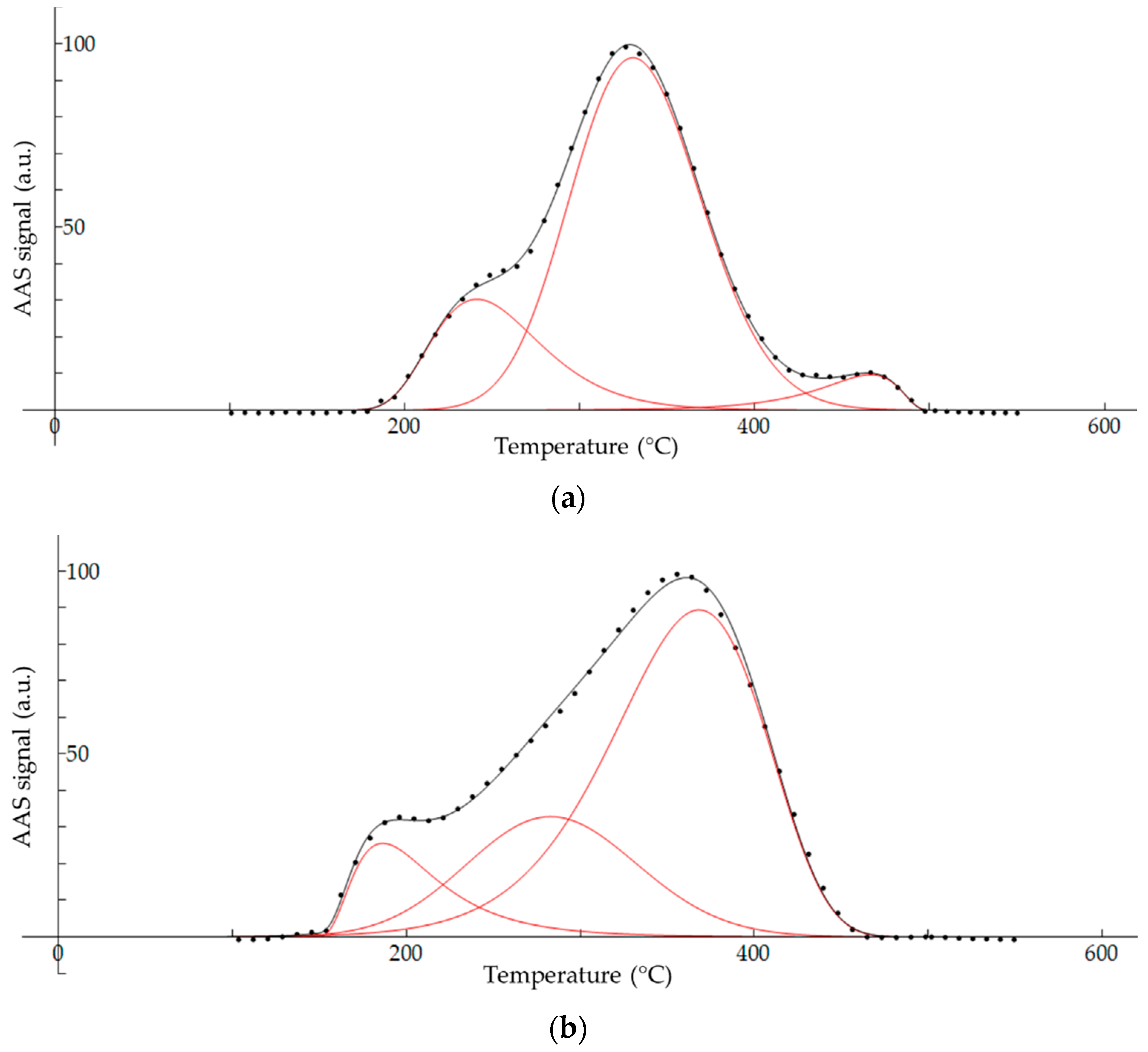
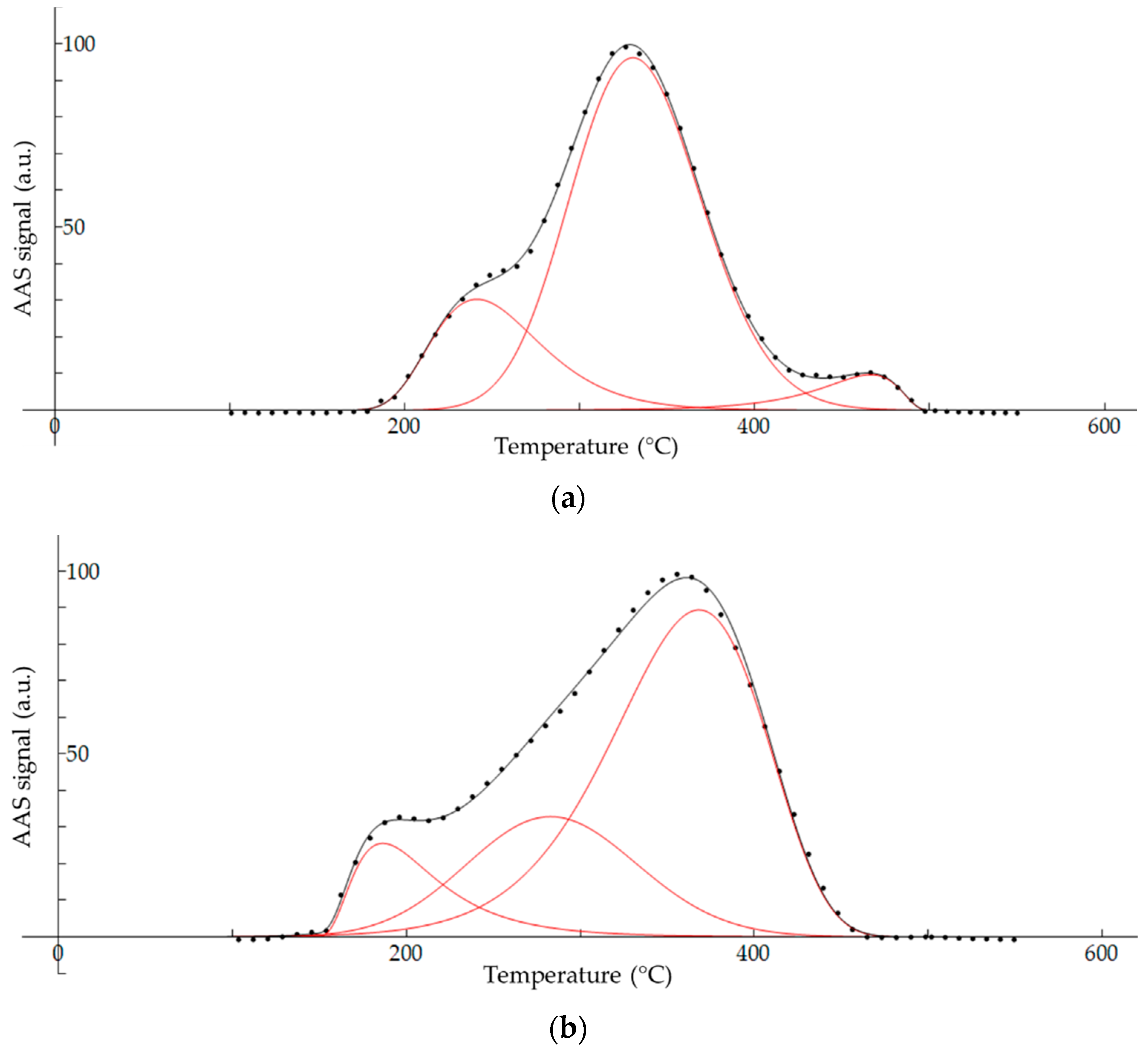
2.7. Peak Deconvulation of Fractionation Thermograms
3. Results and Discussion
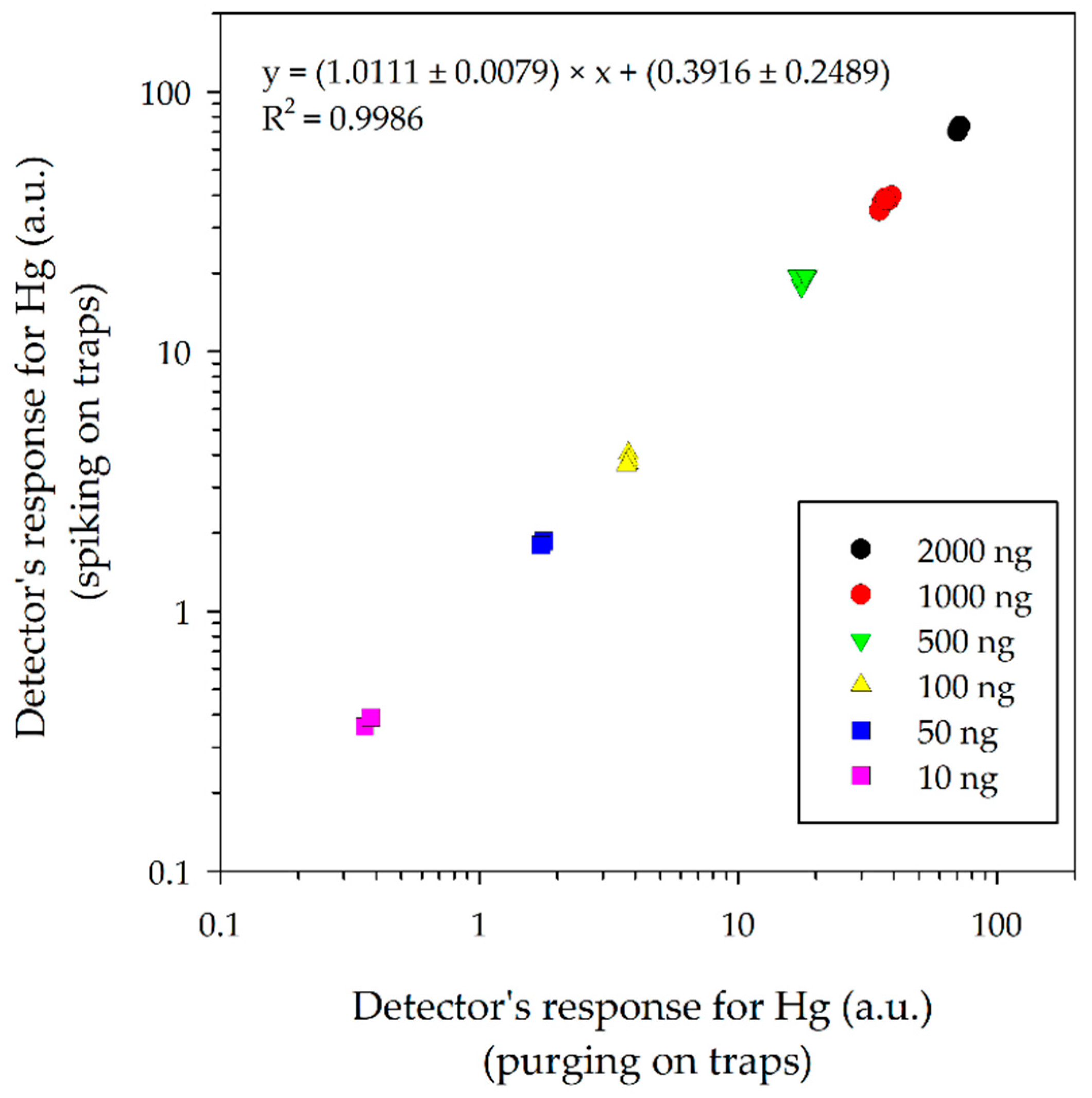
3.1. Comparison of the Detectors Response for Mercury on Activated Carbon Traps Using Two Methods
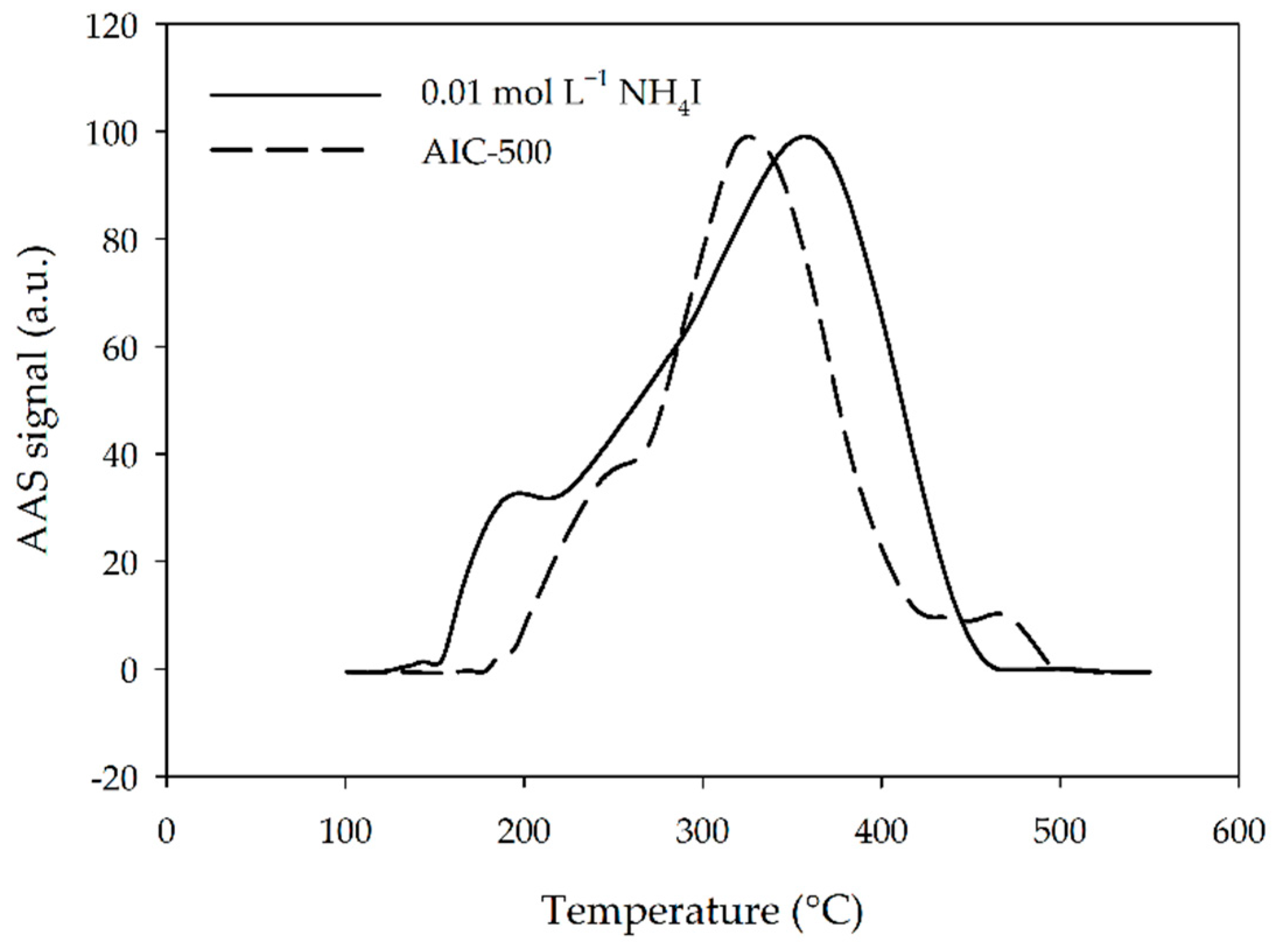
3.2. Comparison of Hg Adsorption on Different Impregnated Activated Carbon Traps
3.3. Determination of Mercury in Real Atmospheric Sample—Proof of Concept
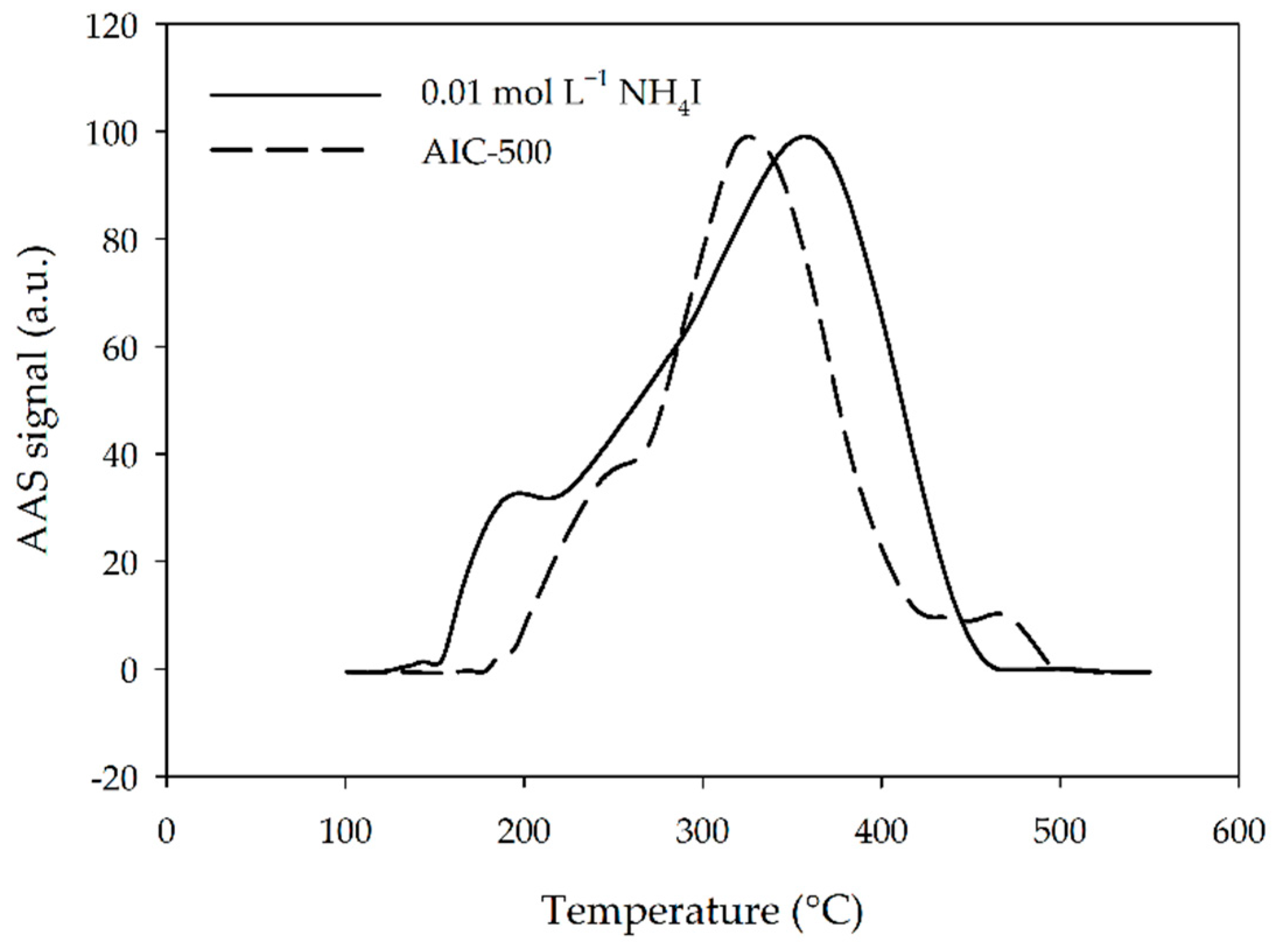
3.4. Thermal Fractionation of Hg on Impregnated Activated Carbon
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, J.-D.; Zheng, W. Human Exposure and Health Effects of Inorganic and Elemental Mercury. J. Prev. Med. Public Health 2012, 45, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Fernandes Azevedo, B.; Barros Furieri, L.; Peçanha, F.M.; Wiggers, G.A.; Frizera Vassallo, P.; Ronacher Simões, M.; Fiorim, J.; Rossi de Batista, P.; Fioresi, M.; Rossoni, L.; et al. Toxic Effects of Mercury on the Cardiovascular and Central Nervous Systems. J. Biomed. Biotechnol. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Amos, H.M.; Jacob, D.J.; Streets, D.G.; Sunderland, E.M. Legacy impacts of all-time anthropogenic emissions on the global mercury cycle. Biogeochem. Cycles 2013, 27, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.P.; Choi, A.L.; Fitzgerald, W.F.; Hammerschmidt, C.R.; Lamborg, C.H.; Soerensen, A.L.; Sunderland, E.M. Mercury biogeochemical cycling in the ocean and policy implications. Environ. Res. 2012, 119, 101–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariya, P.A.; Amyot, M.; Dastoor, A.; Deeds, D.; Feinberg, A.; Kos, G.; Poulain, A.; Ryjkov, A.; Semeniuk, K.; Subir, M.; et al. Mercury Physicochemical and Biogeochemical Transformation in the Atmosphere and at Atmospheric Interfaces: A Review and Future Directions. Chem. Rev. 2015, 115, 3760–3802. [Google Scholar] [CrossRef] [PubMed]
- Gustin, M.S.; Evers, D.C.; Bank, M.S.; Hammerschmidt, C.R.; Pierce, A.; Basu, N.; Blum, J.; Bustamante, P.; Chen, C.; Driscoll, C.T.; et al. Importance of Integration and Implementation of Emerging and Future Mercury Research into the Minamata Convention. Environ. Sci. Technol. 2016, 50, 2767–2770. [Google Scholar] [CrossRef]
- Obrist, D.; Kirk, J.L.; Zhang, L.; Sunderland, E.M.; Jiskra, M.; Selin, N.E. A review of global environmental mercury processes in response to human and natural perturbations: Changes of emissions, climate, and land use. Ambio 2018, 47, 116–140. [Google Scholar] [CrossRef] [Green Version]
- Bank, M.S. Mercury in the Environment: Pattern and Process; University of California Press: Los Angeles, CA, USA, 2012; ISBN 9780520271630. [Google Scholar]
- Ren, X.; Luke, W.T.; Kelley, P.; Cohen, M.D.; Olson, M.L.; Walker, J.; Cole, R.; Archer, M.; Artz, R.; Stein, A.A. Long-Term Observations of Atmospheric Speciated Mercury at a Coastal Site in the Northern Gulf of Mexico during 2007–2018. Atmosphere 2020, 11, 268. [Google Scholar] [CrossRef] [Green Version]
- Sprovieri, F.; Hedgecock, I.M.; Pirrone, N. An investigation of the origins of reactive gaseous mercury in the Mediterranean marine boundary layer. Atmos. Chem. Phys. 2010, 10, 3985–3997. [Google Scholar] [CrossRef] [Green Version]
- Sholupov, S.; Pogarev, S.; Ryzhov, V.; Mashyanov, N.; Stroganov, A. Zeeman atomic absorption spectrometer RA-915+ for direct determination of mercury in air and complex matrix samples. Fuel Process. Technol. 2004, 85, 473–485. [Google Scholar] [CrossRef]
- Fu, X.; Heimbürger, L.E.; Sonke, J.E. Collection of atmospheric gaseous mercury for stable isotope analysis using iodine-and chlorine-impregnated activated carbon traps. J. Anal. At. Spectrom. 2014, 29, 841–852. [Google Scholar] [CrossRef]
- Bérail, S.; Cavalheiro, J.; Tessier, E.; Barre, J.P.G.; Pedrero, Z.; Donard, O.F.X.; Amouroux, D. Determination of total Hg isotopic composition at ultra-trace levels by on line cold vapor generation and dual gold-amalgamation coupled to MC-ICP-MS. J. Anal. At. Spectrom. 2017, 32, 373–384. [Google Scholar] [CrossRef]
- Horvat, M. Determination of Mercury and its Compounds in Water, Sediment, Soil and Biological Samples. In Dynamics of Mercury Pollution on Regional and Global Scales; Springer: New York, NY, USA, 2005; pp. 153–190. [Google Scholar]
- Huang, J.; Lyman, S.N.; Hartman, J.S.; Gustin, M.S. A review of passive sampling systems for ambient air mercury measurements. Environ. Sci. Process. Impacts 2014, 16, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Granite, E.J.; Pennline, H.W.; Hargis, R.A. Novel Sorbents for Mercury Removal from Flue Gas. Ind. Eng. Chem. Res. 2000, 39, 1020–1029. [Google Scholar] [CrossRef]
- Lee, S.J.; Seo, Y.-C.; Jurng, J.; Lee, T.G. Removal of gas-phase elemental mercury by iodine- and chlorine-impregnated activated carbons. Atmos. Environ. 2004, 38, 4887–4893. [Google Scholar] [CrossRef]
- Musmarra, D.; Karatza, D.; Lancia, A.; Prisciandaro, M.; Di Celso, G.M. A comparison among different sorbents for mercury adsorption from flue gas. Chem. Eng. Trans. 2015, 43, 2461–2466. [Google Scholar] [CrossRef]
- Yu, J.-G.; Yue, B.-Y.; Wu, X.-W.; Liu, Q.; Jiao, F.-P.; Jiang, X.-Y.; Chen, X.-Q. Removal of mercury by adsorption: A review. Environ. Sci. Pollut. Res. 2016, 23, 5056–5076. [Google Scholar] [CrossRef]
- US EPA. Method 30B—Mercury Sorbent Trap Procedure. Available online: https://www.epa.gov/sites/production/files/2017-08/documents/method_30b.pdf (accessed on 3 May 2018).
- Saxholm, S.; Rajamäki, T.; Hämäläinen, J.; Hildén, P. Dynamic calibration method for reactive gases. Meas. Sci. Technol. 2019, 31, 1–14. [Google Scholar] [CrossRef]
- Dumarey, R.; Temmerman, E.; Adams, R.; Hoste, J. The accuracy of the vapour-injection calibration method for the determination of mercury by amalgamation/cold-vapour atomic absorption spectrometry. Anal. Chim. Acta 1985, 170, 337–340. [Google Scholar] [CrossRef]
- Dumarey, R.; Brown, R.J.C.; Corns, W.T.; Brown, A.S.; Stockwell, P.B. Elemental mercury vapour in air: The origins and validation of the ‘Dumarey equation’ describing the mass concentration at saturation. Accredit. Qual. Assur. 2010, 15, 409–414. [Google Scholar] [CrossRef]
- Huber, M.L.; Laesecke, A.; Friend, D.G. Correlation for the Vapor Pressure of Mercury. Ind. Eng. Chem. Res. 2006, 45, 7351–7361. [Google Scholar] [CrossRef]
- Gustin, M.S.; Huang, J.; Miller, M.B.; Peterson, C.; Jaffe, D.A.; Ambrose, J.; Finley, B.D.; Lyman, S.N.; Call, K.; Talbot, R.; et al. Do we understand what the mercury speciation instruments are actually measuring? Results of RAMIX. Environ. Sci. Technol. 2013, 47, 7295–7306. [Google Scholar] [CrossRef] [PubMed]
- Quétel, C.R.; Zampella, M.; Brown, R.J.C. Temperature dependence of Hg vapour mass concentration at saturation in air: New SI traceable results between 15 and 30°C. TrAC—Trends Anal. Chem. 2016, 85, 81–88. [Google Scholar] [CrossRef]
- US EPA. Method 1631, Revision E: Mercury in Water by Oxidation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectrometry. Available online: https://www.epa.gov/sites/production/files/2015-08/documents/method_1631e_2002.pdf (accessed on 2 February 2020).
- Berisha, S.; Živković, I.; Kotnik, J.; Mlakar, T.L.; Horvat, M. Quantification of total mercury in samples from cement production processing with thermal decomposition coupled with AAS. Accredit. Qual. Assur. 2020, 25, 233–242. [Google Scholar] [CrossRef]
- Takaku, Y.; Shimamura, T.; Masuda, K.; Igarashi, Y. Iodine Determination in Natural and Tap Water Using Inductively Coupled Plasma Mass Spectrometry. Anal. Sci. 1995, 11, 823–827. [Google Scholar] [CrossRef] [Green Version]
- Jerše, A.; Jaćimović, R.; Maršić, N.K.; Germ, M.; Šircelj, H.; Stibilj, V. Determination of iodine in plants by ICP-MS after alkaline microwave extraction. Microchem. J. 2018, 137, 355–362. [Google Scholar] [CrossRef]
- ERM-EF412 Brown Coal (GCV, Ash, Volatile Matter, Elements). Available online: https://crm.jrc.ec.europa.eu/p/40455/40467/By-material-matrix/Fuels/ERM-EF412-BROWN-COAL-GCV-Ash-Volatile-Matter-Elements/ERM-EF412 (accessed on 1 May 2020).
- Sedlar, M.; Pavlin, M.; Popovič, A.; Horvat, M. Temperature stability of mercury compounds in solid substrates. Open Chem. 2014, 13, 404–419. [Google Scholar] [CrossRef] [Green Version]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Barnett, N.W.; Kirkbright, G.F. Electrothermal vaporisation sample introduction into an atmospheric pressure helium microwave-induced plasma for the determination of iodine in hydrochloric acid. J. Anal. At. Spectrom. 1986, 1, 337. [Google Scholar] [CrossRef]
- Cizdziel, J.; Jiang, Y.; Nallamothu, D.; Brewer, J.; Gao, Z. Air/Surface Exchange of Gaseous Elemental Mercury at Different Landscapes in Mississippi, USA. Atmosphere 2019, 10, 538. [Google Scholar] [CrossRef] [Green Version]
- Gratz, L.; Eckley, C.; Schwantes, S.; Mattson, E. Ambient Mercury Observations near a Coal-Fired Power Plant in a Western U.S. Urban Area. Atmosphere 2019, 10, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slemr, F.; Brunke, E.-G.; Ebinghaus, R.; Temme, C.; Munthe, J.; Wängberg, I.; Schroeder, W.; Steffen, A.; Berg, T. Worldwide trend of atmospheric mercury since 1977. Geophys. Res. Lett. 2003, 30, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Sprovieri, F.; Pirrone, N.; Ebinghaus, R.; Kock, H.; Dommergue, A. A review of worldwide atmospheric mercury measurements. Atmos. Chem. Phys. 2010, 10, 8245–8265. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yu, B.; Yang, L.; Wang, L.; Fu, J.; Liang, Y.; Bu, D.; Yin, Y.; Hu, L.; Shi, J.; et al. Terrestrial mercury transformation in the Tibetan Plateau: New evidence from stable isotopes in upland buzzards. J. Hazard. Mater. 2020, 400, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Reinfelder, J.R.; Fu, P.; Huang, W. Variation in the mercury concentration and stable isotope composition of atmospheric total suspended particles in Beijing, China. J. Hazard. Mater. 2020, 383, 1–7. [Google Scholar] [CrossRef]
- Weiser, H.B. The Luminescence of the Iodide of Millon’s Base. J. Phys. Chem. 1917, 21, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Urben, P.G. Bretherick’s Handbook of Reactive Chemical Hazards, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Lund, H.; Oeckler, O.; Schröder, T.; Schulz, A.; Villinger, A. Mercury Azides and the Azide of Millon’s Base. Angew. Chemie Int. Ed. 2013, 52, 10900–10904. [Google Scholar] [CrossRef]
- Tornieporth-Oetting, I.C.; Klapötke, T.M. Covalent Inorganic Azides. Angew. Chemie Int. Ed. 1995, 34, 511–520. [Google Scholar] [CrossRef]
- Nockemann, P.; Meyer, G. Formation of NH4[Hg3(NH)2](NO3)3 and Transformation to [Hg2N](NO3). Z. Anorg. Allg. Chem. 2002, 628, 2709–2714. [Google Scholar] [CrossRef]
- Matsumura, Y. Adsorption of mercury vapor on the surface of activated carbons modified by oxidation or iodization. Atmos. Environ. 1974, 8, 1321–1327. [Google Scholar] [CrossRef]
- Sasmaz, E.; Kirchofer, A.; Jew, A.D.; Saha, A.; Abram, D.; Jaramillo, T.F.; Wilcox, J. Mercury chemistry on brominated activated carbon. Fuel 2012, 99, 188–196. [Google Scholar] [CrossRef]
- Granite, E.J.; Pennline, H.W.; Hargis, R.A. Sorbents for Mercury Removal from Flue Gas; U.S. Department of Energy: Pittsburgh, PA, USA, 1998.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activated Carbon/Impregnating Solution | Trap A (%) 1 | Trap B (%) 1 |
|---|---|---|
| AIC-500 (n = 9) | 100 ± 1.78 | <LOD 2 − 0.10 |
| 0.10 mol L−1 NH4I (n = 7) | 75.7 ± 6.32 | 0.71 ± 0.22 |
| 0.03 mol L−1 NH4I (n = 14) | 101 ± 3.25 | 0.74 ± 0.89 |
| 0.01 mol L−1 NH4I (n = 15) | 99.3 ± 2.52 | <LOD 2 − 0.27 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Živković, I.; Berisha, S.; Kotnik, J.; Jagodic, M.; Horvat, M. Traceable Determination of Atmospheric Mercury Using Iodinated Activated Carbon Traps. Atmosphere 2020, 11, 780. https://doi.org/10.3390/atmos11080780
Živković I, Berisha S, Kotnik J, Jagodic M, Horvat M. Traceable Determination of Atmospheric Mercury Using Iodinated Activated Carbon Traps. Atmosphere. 2020; 11(8):780. https://doi.org/10.3390/atmos11080780
Chicago/Turabian StyleŽivković, Igor, Sabina Berisha, Jože Kotnik, Marta Jagodic, and Milena Horvat. 2020. "Traceable Determination of Atmospheric Mercury Using Iodinated Activated Carbon Traps" Atmosphere 11, no. 8: 780. https://doi.org/10.3390/atmos11080780
APA StyleŽivković, I., Berisha, S., Kotnik, J., Jagodic, M., & Horvat, M. (2020). Traceable Determination of Atmospheric Mercury Using Iodinated Activated Carbon Traps. Atmosphere, 11(8), 780. https://doi.org/10.3390/atmos11080780