Mineral Dust and Iron Solubility: Effects of Composition, Particle Size, and Surface Area



Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Sample Preparation
2.3. Scanning Electron Microscopy (SEM)
2.4. Surface Area Measurements
2.5. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) Analysis
2.6. UV-Vis Spectroscopy Analysis
3. Results and Discussion
3.1. Iron Dissolution of Minerals Extracted into Moderately Acidic and Acidic Leaching Media
3.2. Effect of Particle Size on Iron Solubility
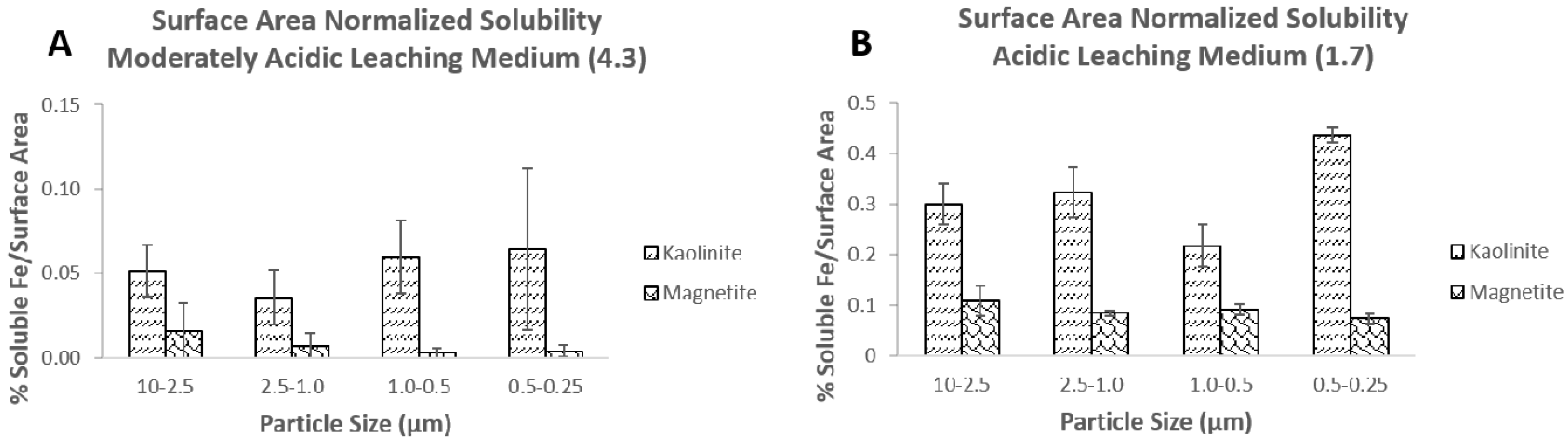
3.3. Particle Size and Surface Area Measurements
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zeebe, R.E. History of Seawater Carbonate Chemistry, Atmospheric CO2, and Ocean Acidification. Annu. Rev. Earth Planet. Sci. 2012, 40, 141–165. [Google Scholar] [CrossRef]
- Barber, R.T.; Chavez, F.P. Regulation of primary productivity rate in the equatorial Pacific. Limnol. Oceanogr. 1991, 36, 1803–1815. [Google Scholar] [CrossRef]
- Martin, J.H. Glacial-interglacial CO2 change: The iron hypothesis. Paleoceanography 1990, 5, 1–13. [Google Scholar] [CrossRef]
- Falkowski, P.G.; Barber, R.T.; Smetacek, V. Biogeochemical controls and feedbacks on ocean primary productivity. Science 1998, 281, 200–206. [Google Scholar] [CrossRef]
- Moore, J.K.; Doney, S.C.; Glover, D.M.; Fung, I.Y. Iron cycling and nutrient-limitation patterns in surface waters of the World Ocean. Deep Sea Res. Part II Trop. Stud. Oceanogr. 2001, 49, 463–507. [Google Scholar] [CrossRef]
- Ito, A.; Myriokefalitakis, S.; Kanakidou, M.; Mahowald, N.M.; Scanza, R.A.; Hamilton, D.S.; Baker, A.R.; Jickells, T.; Sarin, M.; Bikkina, S.; et al. Pyrogenic iron: The missing link to high iron solubility in aerosols. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef]
- Jickells, T.D.; An, Z.S.; Andersen, K.K.; Baker, A.R.; Bergametti, G.; Brooks, N.; Cao, J.J.; Boyd, P.W.; Duce, R.A.; Hunter, K.A.; et al. Global iron connections between desert dust, ocean biogeochemistry, and climate. Science 2005, 308, 67–71. [Google Scholar] [CrossRef]
- Boyd, P.W.; Ellwood, M.J. The biogeochemical cycle of iron in the ocean. Nat. Geosci. 2010, 3, 675–682. [Google Scholar] [CrossRef]
- Mead, C.; Herckes, P.; Majestic, B.J.; Anbar, A.D. Source apportionment of aerosol iron in the marine environment using iron isotope analysis. Geophys. Res. Lett. 2013, 40, 5722–5727. [Google Scholar] [CrossRef]
- Archer, D.E.; Johnson, K. A model of the iron cycle in the ocean. Global Biogeochem. Cycles 2000, 14, 269–279. [Google Scholar] [CrossRef]
- Chen, H.; Laskin, A.; Baltrusaitis, J.; Gorski, C.A.; Scherer, M.M.; Grassian, V.H. Coal Fly Ash as a Source of Iron in Atmospheric Dust. Environ. Sci. Technol. 2012, 46, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fan, S.M.; Sarmiento, J.L. Aeolian iron input to the ocean through precipitation scavenging: A modeling perspective and its implication for natural iron fertilization in the ocean. J. Geophys. Res. 2003, 108, 1–13. [Google Scholar] [CrossRef]
- Journet, E.; Desboeufs, K.V.; Caquineau, S.; Colin, J.-L. Mineralogy as a critical factor of dust iron solubility. Geophys. Res. Letters 2008, 35, 1–5. [Google Scholar] [CrossRef]
- Chuang, P.Y.; Duvall, R.M.; Shafer, M.M.; Schauer, J.J. The origin of water soluble particulate iron in the Asian atmospheric outflow. Geophys. Res. Lett. 2005, 32, 1–4. [Google Scholar] [CrossRef]
- Kumar, A.; Sarin, M.M.; Srinivas, B. Aerosol iron solubility over Bay of Bengal: Role of anthropogenic sources and chemical processing. Mar. Chem. 2010, 212, 167–175. [Google Scholar] [CrossRef]
- Sholkovitz, E.R.; Sedwick, P.N.; Church, T.M.; Baker, A.R.; Powell, C.F. Fractional solubility of aerosol iron: Synthesis of a global-scale data set. Geochim. Cosmochim. Acta 2012, 89, 173–189. [Google Scholar] [CrossRef]
- Oakes, M.; Weber, R.J.; Lai, B.; Russell, A.; Ingall, E.D. Characterization of iron speciation in urban and rural single particles using XANES spectroscopy and micro X-ray fluorescence measurements: Investigating the relationship between speciation and fractional iron solubility. Atmos. Chem. Phys. 2012, 12, 745–756. [Google Scholar] [CrossRef]
- Ito, A.; Xu, L. Response of acid mobilization of iron-containing mineral dust to improvement of air quality projected in the future. Atmos. Chem. Phys. 2014, 14, 3441–3459. [Google Scholar] [CrossRef]
- Raiswell, R.; Canfield, D.E. The iron biogeochemical cycle past and present. Geochem. Perspect. 2012, 1, 1–220. [Google Scholar] [CrossRef]
- Rizzolo, J.A.; Barbosa, C.G.G.; Borillo, G.C.; Godoi, A.F.L.; Souza, R.A.; Andreoli, R.V.; Manzi, A.O.; Sá, M.O.; Alves, E.G.; Pöhlker, C.; et al. Soluble iron nutrients in Saharan dust over the central Amazon rainforest. Atmos. Chem. Phys. 2017, 17, 2673–2687. [Google Scholar] [CrossRef]
- Spokes, L.J.; Jickells, T.D.; Lim, B. Solubilisation of aerosol trace metals by cloud processing: A laboratory study. Geochim. Cosmochim. Acta 1994, 58, 3281–3287. [Google Scholar] [CrossRef]
- Cwiertny, D.M.; Baltrusaitis, J.; Hunter, G.J.; Laskin, A.; Scherer, M.M.; Grassian, V.H. Characterization and acid-mobilization study of iron-containing mineral dust source minerals. J. Geophys. Res. 2008, 113, 1–18. [Google Scholar] [CrossRef]
- Wiederhold, J.G.; Kraemer, S.M.; Teutsch, N.; Borer, P.M.; Halliday, A.N.; Kretzschmar, R. Iron Isotope Fractionation during Proton-Promoted, Ligand-Controlled, and Reductive Dissolution of Goethite. Environ. Sci. Technol. 2006, 40, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.R.; Cartledge, B.T.; Haynes, J.P.; York-Marini, R.; Robinson, A.L.; Drozd, G.T.; Goldstein, A.H.; Fakra, S.C.; Majestic, B.J. Water-soluble iron emitted from vehicle exhaust is linked to primary speciated organic compounds. Atmos. Chem. Phys. 2020, 20, 1849–1860. [Google Scholar] [CrossRef]
- Schwertmann, U. Solubility and dissolution of iron oxides. Plant Soil 1991, 130, 1–25. [Google Scholar] [CrossRef]
- Fu, H.; Cwiertny, D.M.; Carmichael, G.R.; Scherer, M.M.; Grassian, V.H. Photoreductive dissolution of Fe-containing mineral dust particles in acidic media. J. Geophys. Res. 2010, 115, 1–12. [Google Scholar] [CrossRef]
- Hoffmann, P.; Dedik, A.N.; Ensling, J.; Weinbruch, S.; Weber, S.; Sinner, T.; Gutlich, P.; Ortner, H.M. Speciation of iron in atmospheric aerosol samples. J. Aerosol Sci. 1996, 2, 325–337. [Google Scholar] [CrossRef]
- Pehkonen, S.O.; Siefert, R.L.; Erel, Y.; Webb, S.; Hoffmann, M.R. Photoreduction of iron oxyhydroxides in the presence of important organic compounds. Environ. Sci. Technol. 1993, 27, 2058–2062. [Google Scholar] [CrossRef]
- Sulzberger, B.; Laubscher, H. Reactivity of various types of iron (III) (hydr)oxides towards light-induced dissolution. Marine Chem. 1995, 50, 103–115. [Google Scholar] [CrossRef]
- Paris, R.; Desboeufs, K.V.; Journet, E. Variability of dust iron solubility in atmospheric waters: Investigation of the role of oxalate organic complexation. Atmos. Environ. 2011, 45, 6510–6517. [Google Scholar] [CrossRef]
- Upadhyay, N.; Majestic, B.J.; Herckes, P. Solubility and speciation of atmospheric iron in buffer systems simulating cloud conditions. Atmos. Environ. 2011, 45, 1858–1866. [Google Scholar] [CrossRef]
- Paris, R.; Desboeufs, K.V. Effect of atmospheric organic complexation on iron-bearing dust solubility. Atmos. Chem. Phys. 2013, 13, 4895–4905. [Google Scholar] [CrossRef]
- Chen, H.; Grassian, V.H. Iron Dissolution of Dust Source Materials during Simulated Acidic Processing: The Effect of Sulfuric, Acetic, and Oxalic Acids. Environ. Sci. Technol. 2013, 47, 10312–10321. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.S.; Meskhidze, N. Atmospheric dissolved iron deposition to the global oceans: Effects of oxalate-promoted Fe dissolution, photochemical redox cycling, and dust mineralogy. Geosci. Model Dev. 2013, 6, 1137–1155. [Google Scholar] [CrossRef]
- Myriokefalitakis, S.; Daskalakis, N.; Mihalopoulos, N.; Baker, A.R.; Nenes, A.; Kanakidou, M. Changes in dissolved iron deposition to the oceans driven by human activity: A 3-D global modelling study. Biogeosciences 2015, 12, 3973–3992. [Google Scholar] [CrossRef]
- Dedik, A.N.; Hoffman, P.; Ensling, J. Chemical characterization of iron in atmospheric aerosols. Atmos. Environ. 1992, 26A, 2545–2548. [Google Scholar] [CrossRef]
- Arakaki, T.; Faust, B.C. Sources, sinks, and mechanisms of hydroxyl radical photoproduction and consumption in authentic acid continental cloud waters from Whiteface Mountain, New York: The role of the Fe (r) (r = II,III) photochemical cycle. J. Geophys. Res. 1998, 103, 3487–3504. [Google Scholar] [CrossRef]
- Baker, A.R.; Jickells, T.D. Mineral particle size as a control on aerosol iron solubility. Geophys. Res. Lett. 2006, 33, 1–4. [Google Scholar] [CrossRef]
- Ooki, A.; Nishioka, J.; Ono, T.; Noriki, S. Size dependence of iron solubility of Asian mineral dust particles. J. Geophys. Res. Atmos. 2009, 114. [Google Scholar] [CrossRef]
- Buck, C.S.; Landing, W.M.; Resing, J.A. Particle size and aerosol iron solubility: A high-resolution analysis of Atlantic aerosols. Mar. Chem. 2010, 120, 14–24. [Google Scholar] [CrossRef]
- Cartledge, B.T.; Marcotte, A.R.; Herckes, P.; Anbar, A.D.; Majestic, B.J. The Impact of Particle Size, Relative Humidity, and Sulfur Dioxide on Iron Solubility in Simulated Atmospheric Marine Aerosols. Environ. Sci. Technol. 2015, 49, 7179–7187. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; John Wiley & Sons Inc.: New York, NY, USA, 1998. [Google Scholar]
- Mahowald, N.; Albani, S.; Kok, J.F.; Engelstaeder, S.; Scanza, R.; Ward, D.S.; Fanner, M.G. The size distribution of desert dust aerosols and its impact on the Earth system. Aeolian Res. 2014, 15, 54–71. [Google Scholar] [CrossRef]
- Buseck, P.R.; Adachi, K. Nanoparticles in the atmosphere. Elements 2008, 4, 389–394. [Google Scholar] [CrossRef]
- Claquin, T.; Schulz, M.; Balkanski, Y.J. Modeling the mineralogy of atmospheric dust sources. J. Geophys. Res. 1999, 104, 22243–22256. [Google Scholar] [CrossRef]
- Glaccum, R.A.; Prospero, J.M. Saharan aerosols over the tropical North Atlantic—Mineralogy. Mar. Geol. 1980, 37, 295–321. [Google Scholar] [CrossRef]
- Avila, A.; Queralt-Mitjans, I.; Alarcón, M. Mineralogical composition of African dust delivered by red rains over northeastern Spain. J. Geophys. Res. 1997, 102, 21977–21996. [Google Scholar] [CrossRef]
- Shi, Z.; Krom, M.D.; Jickells, T.D.; Bonneville, S.; Carslaw, K.S.; Mihalopoulos, N.; Baker, A.R.; Benning, L.G. Impacts on iron solubility in the mineral dust by processes in the source region and the atmosphere: A review. Aeolian Res. 2012, 5, 21–42. [Google Scholar] [CrossRef]
- Hutchings, J.W.; Robinson, M.S.; McIlwraith, H.; Kingston, J.T.; Herckes, P. The Chemistry of Intercepted Clouds in Northern Arizona during the North American Monsoon Season. Water Air Soil Pollut. 2009, 199, 191–202. [Google Scholar] [CrossRef]
- Zhu, X.; Prospero, J.M.; Savoie, D.L.; Millero, F.J.; Zika, R.G.; Saltzman, E.S. Photoreduction of iron (III) in marine mineral aerosol solutions. J. Geophys. Res. 1993, 98, 9039–9046. [Google Scholar] [CrossRef]
- O’Dowd, C.D.; Hämeri, K.; Mäkelä, J.M.; Pirjola, L.; Kulmala, M.; Jennings, S.G.; Berresheim, H.; Hansson, H.C.; de Leeuw, G.; Kunz, G.L.; et al. A dedicated study of New Particle Formation and Fate in the Coastal Environment (PARFORCE): Overview of objectives and achievements. J. Geophys. Res. 2002, 107, 8108. [Google Scholar] [CrossRef]
- Majestic, B.J.; Schauer, J.J.; Shafer, M.M.; Turner, J.R.; Fine, P.M.; Singh, M.; Sioutas, C. Development of a Wet-Chemical Method for the Speciation of Iron in Atmospheric Aerosols. Environ. Sci. Technol. 2006, 40, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, N.; Clements, A.L.; Fraser, M.P.; Sundblom, M.; Solomon, P.; Herckes, P. Size-differentiated chemical composition of re-suspended soil dust from the desert Southwest United States. Aerosol Air Qual. Res. 2015, 15, 387–398. [Google Scholar] [CrossRef]
- Fagerlund, G. Determination of specific surface by the BET method. Matér. Constr. 1973, 6, 239–245. [Google Scholar] [CrossRef]
- Upadhyay, N.; Majestic, B.J.; Prapaipong, P.; Herckes, P. Evaluation of polyurethane foam, polypropylene, quartz fiber, and cellulose substrates for multi-element analysis of atmospheric particulate matter by ICP-MS. Anal. Bioanal. Chem. 2009, 394, 255–266. [Google Scholar] [CrossRef]
- Stookey, L.L. Ferrozine—A New Spectrophotometric Reagent for Iron. Anal. Chem. 1970, 42, 779–781. [Google Scholar] [CrossRef]
- Jeong, G.Y.; Achterberg, E.P. Chemistry and mineralogy of clay minerals in Asian and Saharan dusts and the implications for iron availability. Atmos. Chem. Phys. Discuss. 2014, 14, 15735–15770. [Google Scholar] [CrossRef]
- Longo, A.F.; Feng, Y.; Lai, B.; Landing, W.M.; Shelley, R.U.; Nenes, A.; Mihalopoulos, N.; Violaki, K.; Ingall, E.D. Influence of atmospheric processes on the solubility and composition of iron in Saharan dust. Environ. Sci. Technol. 2016, 50, 6912–6920. [Google Scholar] [CrossRef]
- Jeong, G.Y.; Nousiainen, T. TEM analysis of the internal structures and mineralogy of Asian dust particles and the implications for optical modeling. Atmos. Chem. Phys. Discuss. 2014, 14, 6619–6661. [Google Scholar] [CrossRef]
- Malden, P.J.; Meads, R.E. Substitution by iron in kaolinite. Nature 1967, 215, 844–846. [Google Scholar] [CrossRef]
- Johnston, J.H.; Cardile, C.M. Iron substitution in montmorillonite, illite, and glauconite by 57 Fe Mössbauer spectroscopy. Clays Clay Miner. 1987, 35, 170–176. [Google Scholar] [CrossRef]
- Bahranowski, K.; Serwicka, E.M.; Stock, L.; Strycharski, P. On the possibility of removal of non-structural iron from kaolinite-group minerals. Clay Miner. 1993, 28, 379–391. [Google Scholar] [CrossRef]
- Bonnet, S.; Guieu, C. Dissolution of atmospheric iron in seawater. Geophys. Res. Lett. 2004, 31, 1–4. [Google Scholar] [CrossRef]
- Rubasinghege, G.; Lentz, R.W.; Park, H.; Scherer, M.M.; Grassian, V.H. Nanorod dissolution quenched in the aggregated state. Langmuir 2010, 26, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Ito, A. Atmospheric processing of combustion aerosols as a source of bioavailable iron. Environ. Sci. Technol. Lett. 2015, 2, 70–75. [Google Scholar] [CrossRef]
- Ito, A.; Shi, Z. Delivery of anthropogenic bioavailable iron from mineral dust and combustion aerosols to the ocean. Atmos. Chem. Phys. 2016, 16, 85–99. [Google Scholar] [CrossRef]
- Zhu, X.; Prospero, J.M.; Millero, F.J.; Savoie, D.L.; Brass, G.W. The solubility of ferric iron in marine mineral aerosol solutions at ambient relative humidities. Mar. Chem. 1992, 38, 91–107. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution | Composition |
|---|---|
| Moderately acidic leaching medium (pH 4.3) | 5 mM acetate buffer *, 5 mM formate buffer **, 5 mM ammonium nitrate Ɨ |
| Acidic leaching medium (pH 1.7) | 0.1 M sulfuric acid, 0.1 M sodium chloride ƗƗ |
| Mineral (m2·g−1) | 10–2.5 µm | 2.5–1.0 µm | 1.0–0.5 µm | 0.5–0.25 µm |
|---|---|---|---|---|
| Kaolinite | 31.3 | 21.7 | 30.2 | 29.9 |
| Magnetite | 7.1 | 9.4 | 11.3 | 20.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcotte, A.R.; Anbar, A.D.; Majestic, B.J.; Herckes, P. Mineral Dust and Iron Solubility: Effects of Composition, Particle Size, and Surface Area. Atmosphere 2020, 11, 533. https://doi.org/10.3390/atmos11050533
Marcotte AR, Anbar AD, Majestic BJ, Herckes P. Mineral Dust and Iron Solubility: Effects of Composition, Particle Size, and Surface Area. Atmosphere. 2020; 11(5):533. https://doi.org/10.3390/atmos11050533
Chicago/Turabian StyleMarcotte, Aurelie R., Ariel D. Anbar, Brian J. Majestic, and Pierre Herckes. 2020. "Mineral Dust and Iron Solubility: Effects of Composition, Particle Size, and Surface Area" Atmosphere 11, no. 5: 533. https://doi.org/10.3390/atmos11050533
APA StyleMarcotte, A. R., Anbar, A. D., Majestic, B. J., & Herckes, P. (2020). Mineral Dust and Iron Solubility: Effects of Composition, Particle Size, and Surface Area. Atmosphere, 11(5), 533. https://doi.org/10.3390/atmos11050533