Atmospheric Trace Metal Deposition from Natural and Anthropogenic Sources in Western Australia

 , , , , and
, , , , and
Abstract
1. Introduction
2. Experiments
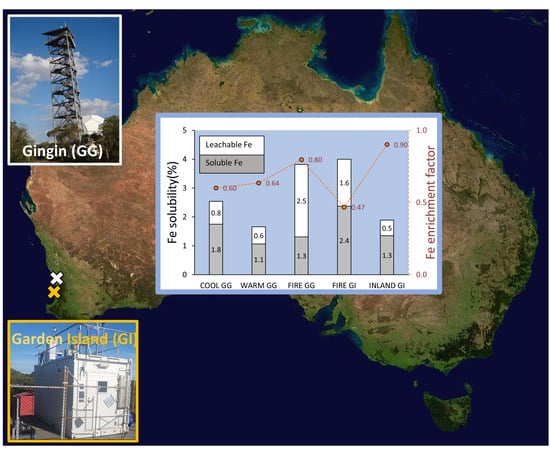
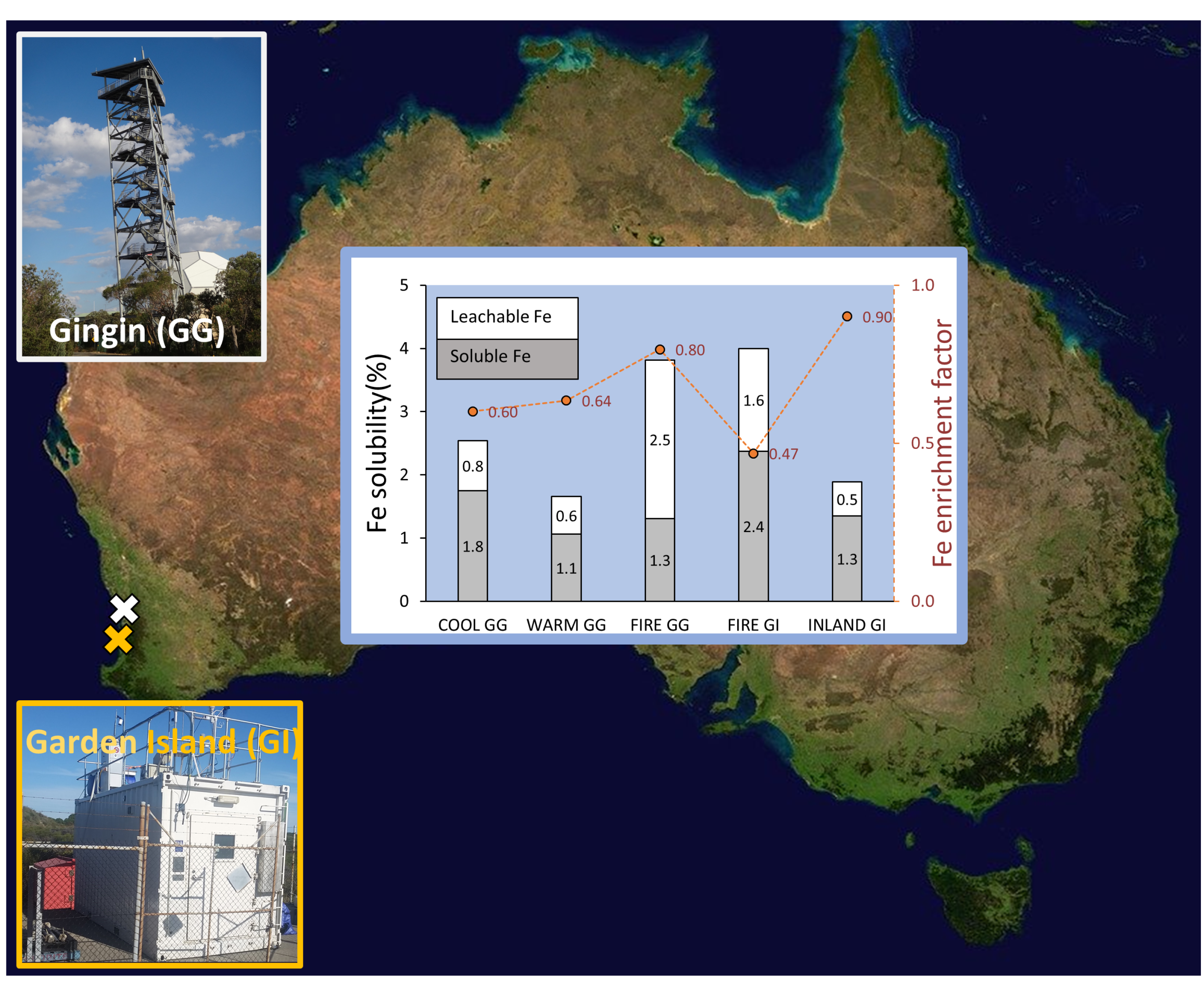
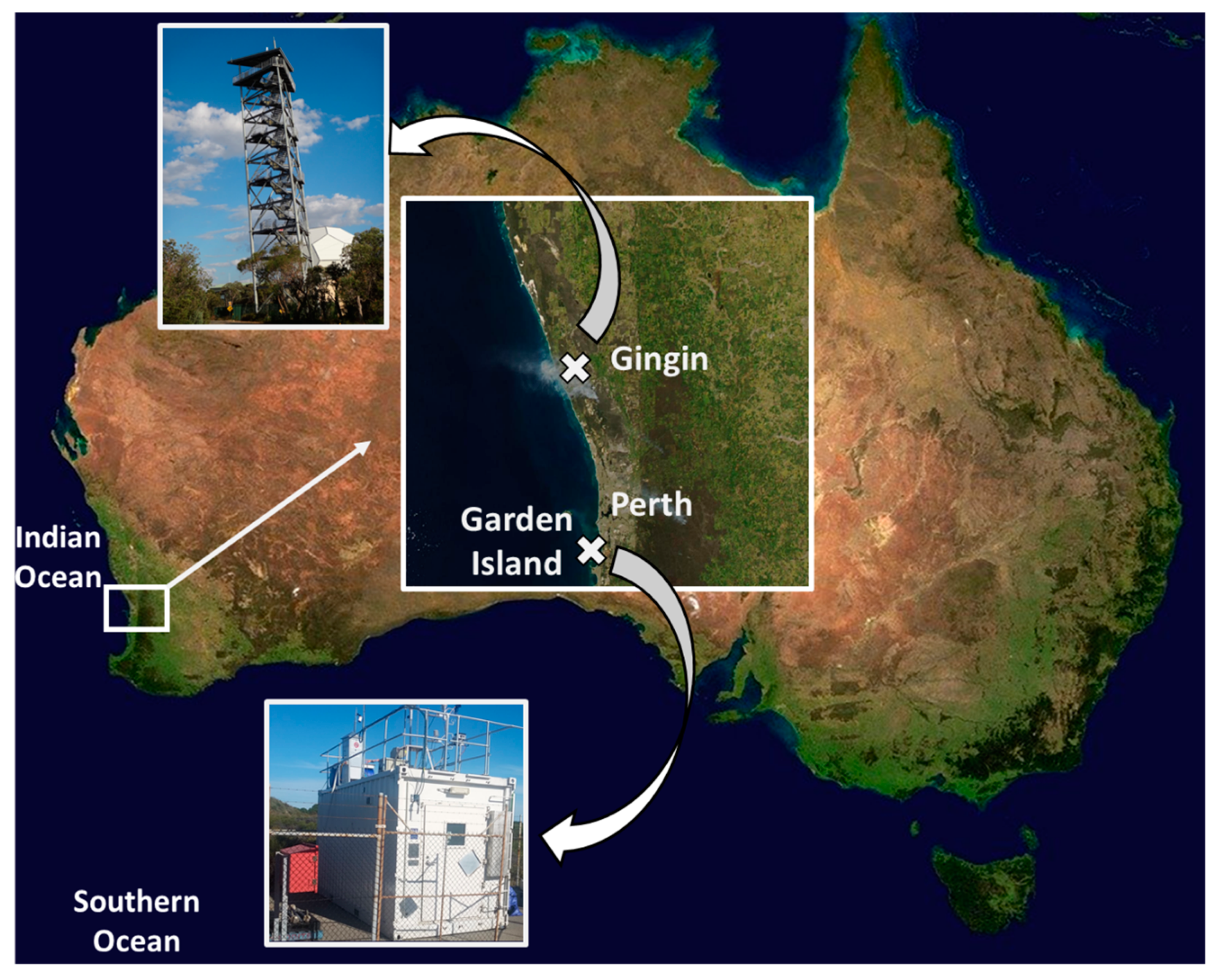
2.1. Study Site and Sample Collection
2.2. Trace Metal Analysis
2.3. Major Ion Analysis
2.4. Back Trajectories and Burned Area Analysis
2.5. Aerosols Sources by Positive Matrix Factorisation
2.6. Other Calculations
3. Results and Discussion
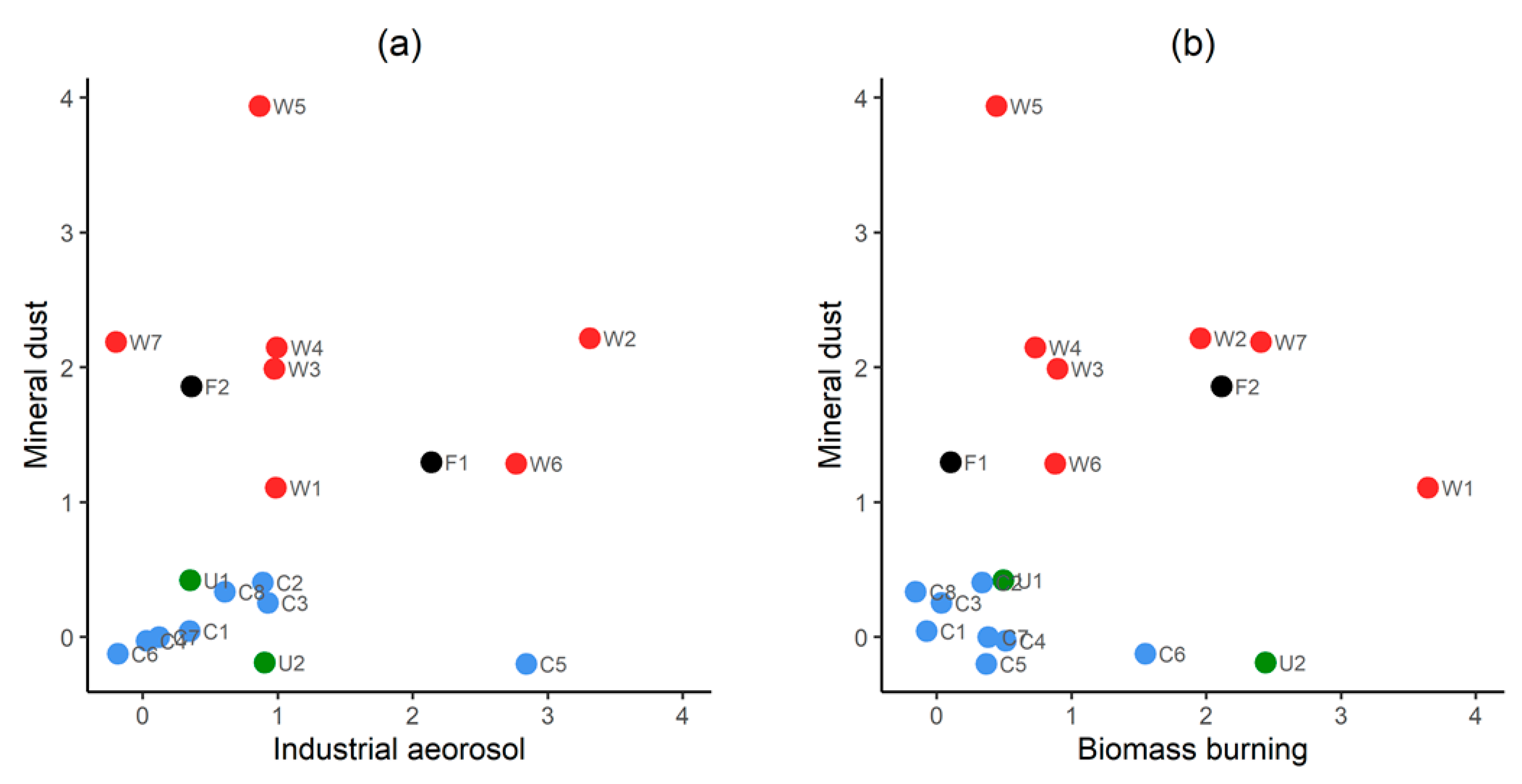
3.1. Seasonality in Aerosol Provenance: Sample Classification
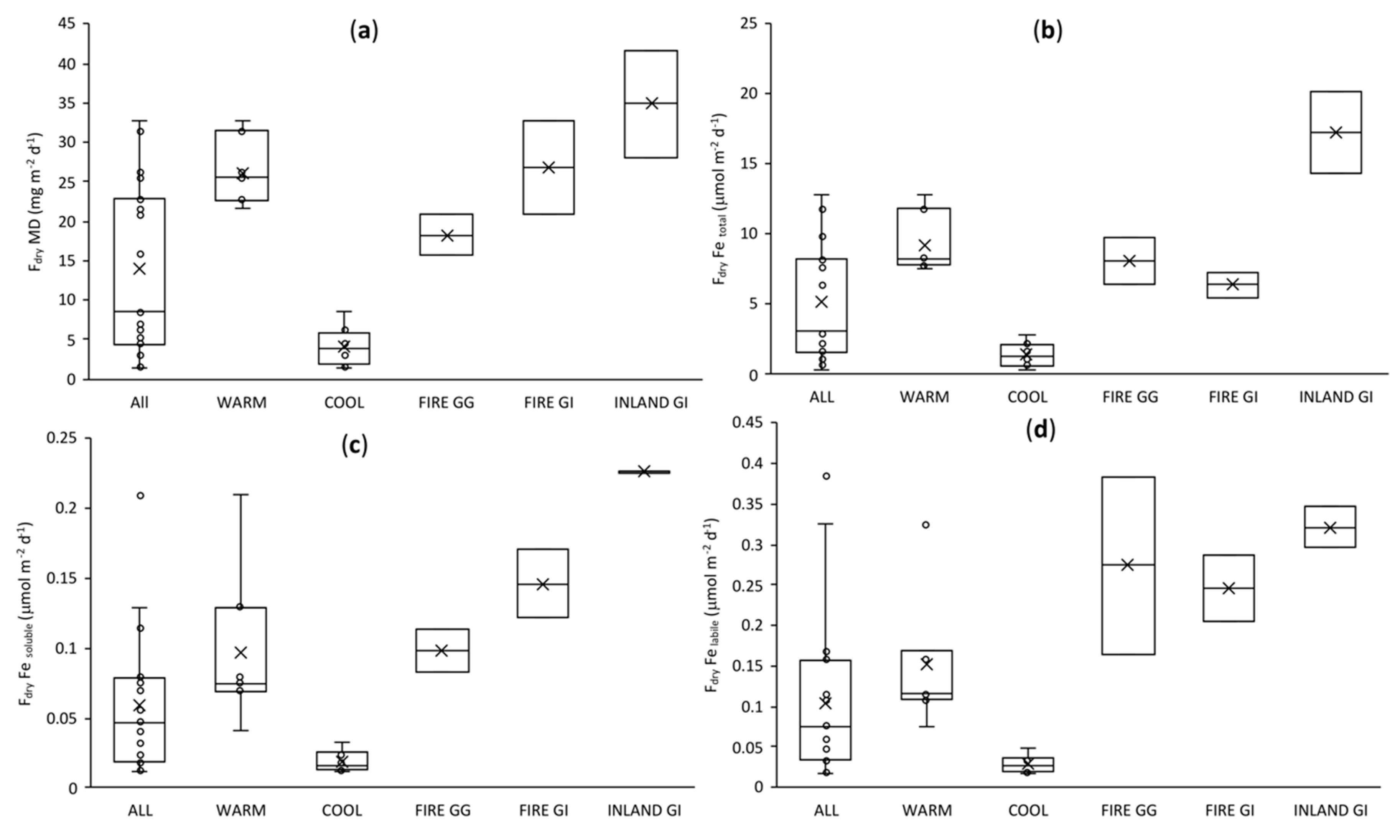
3.2. Mineral Dust Contribution to Atmospheric Trace Metal Deposition
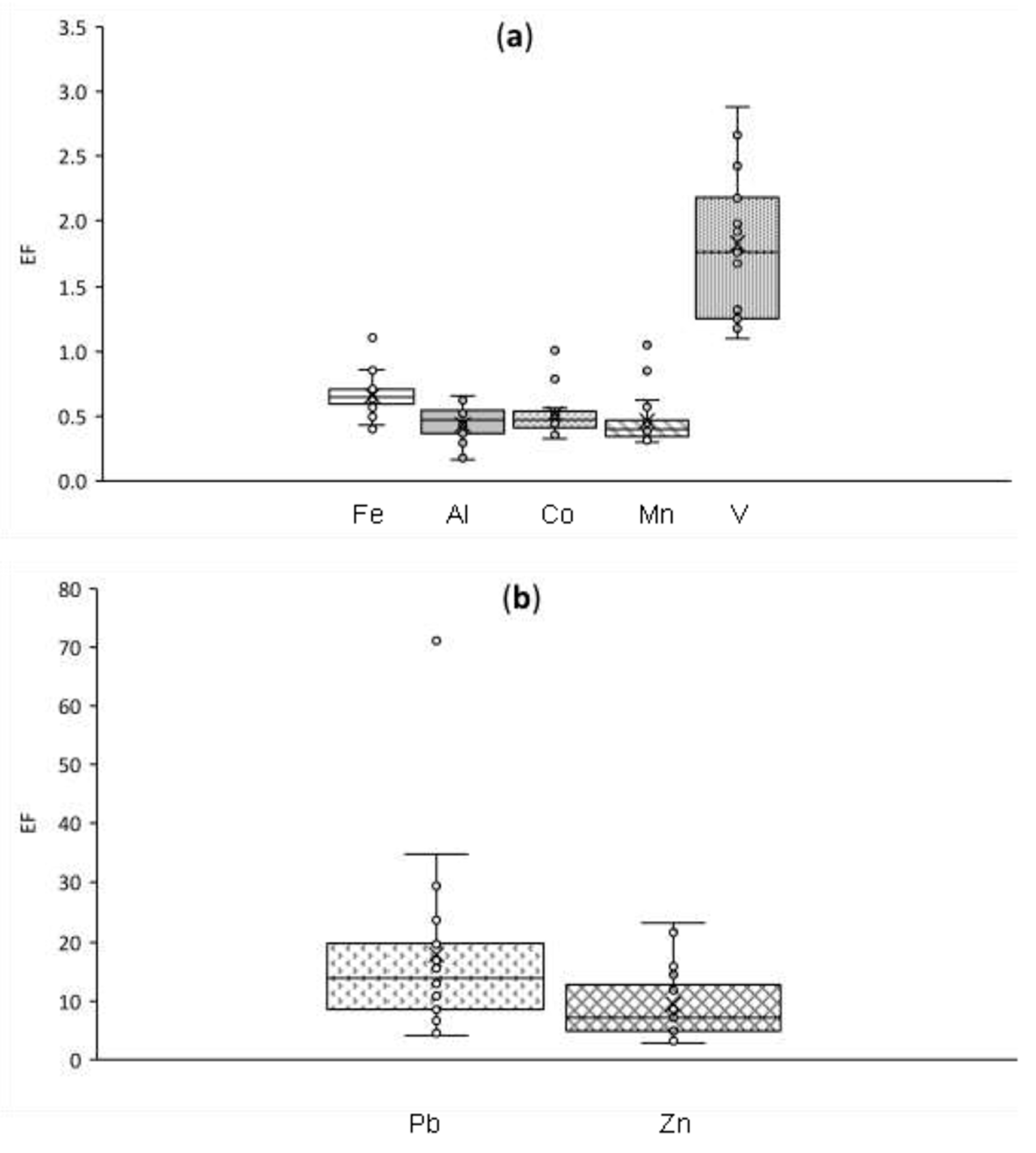
3.2.1. Enrichment Factors
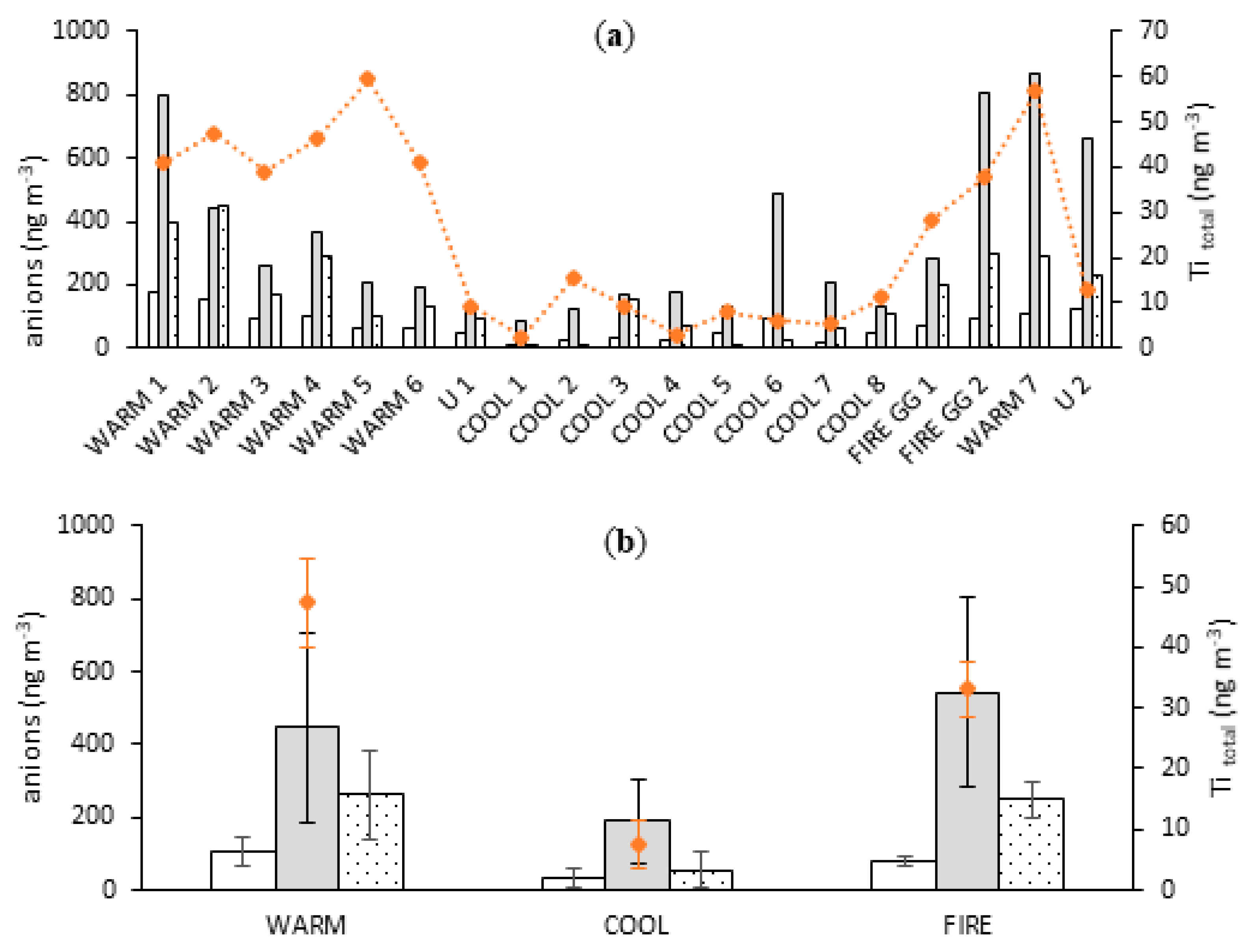
3.2.2. Correlation with Mineral Dust Markers
3.2.3. Iron Enrichment of Australian Aerosols and Soils
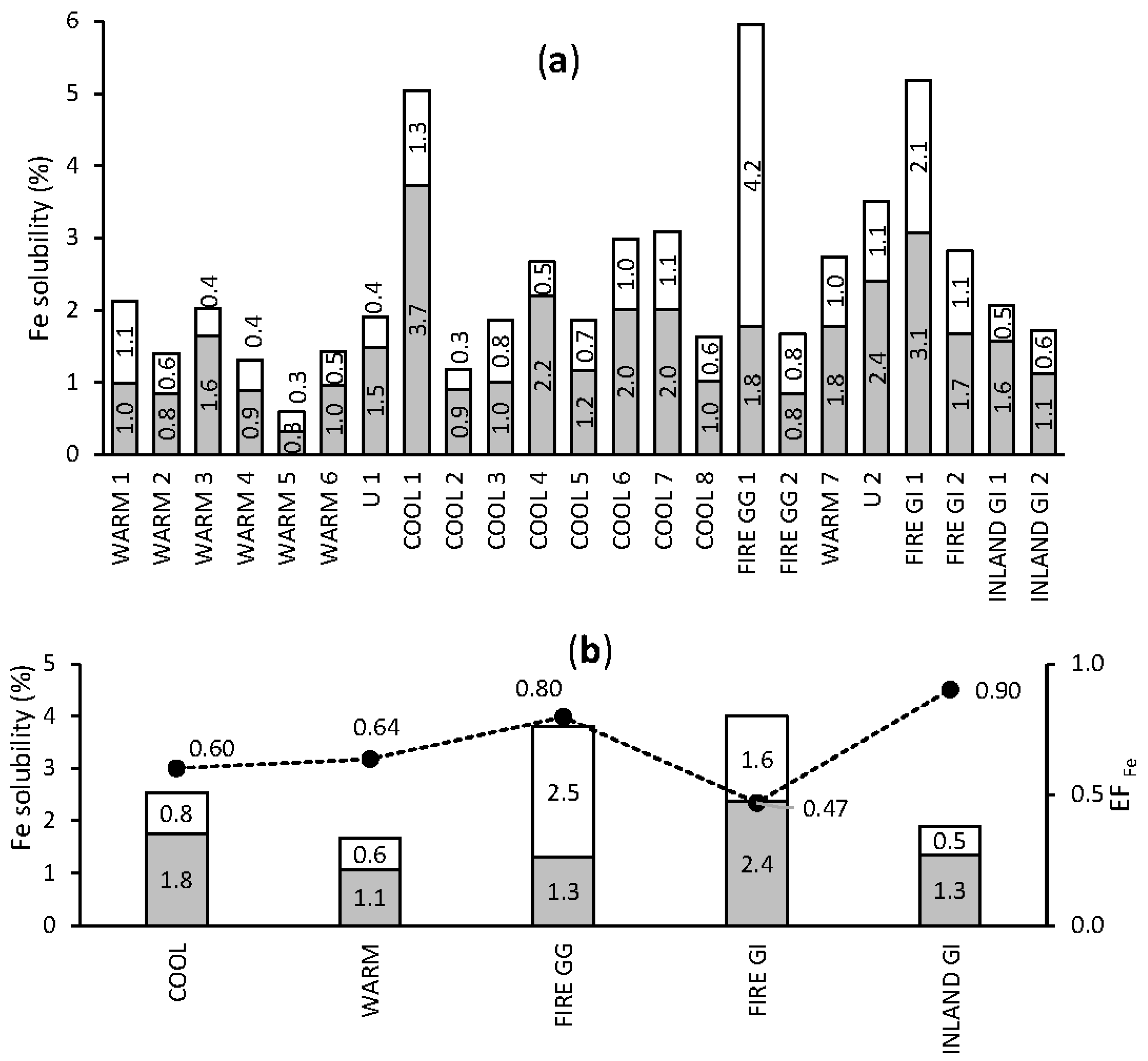
3.3. Seasonality in Bioactive Trace Metal Solubilities and Enrichment Factors
3.3.1. Iron
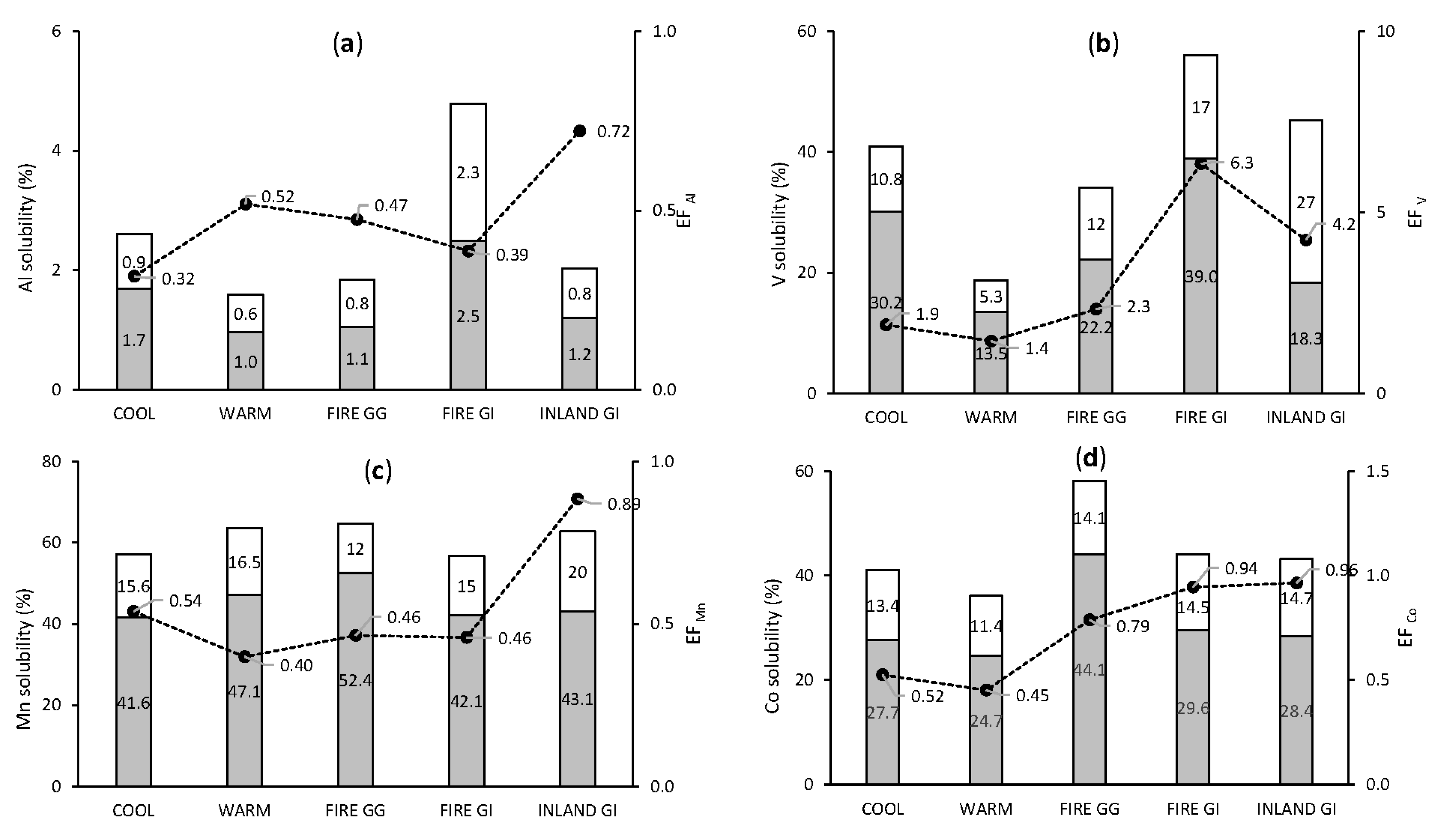
3.3.2. Crustal and Mixed Origin Elements: Al, V, Mn, and Co
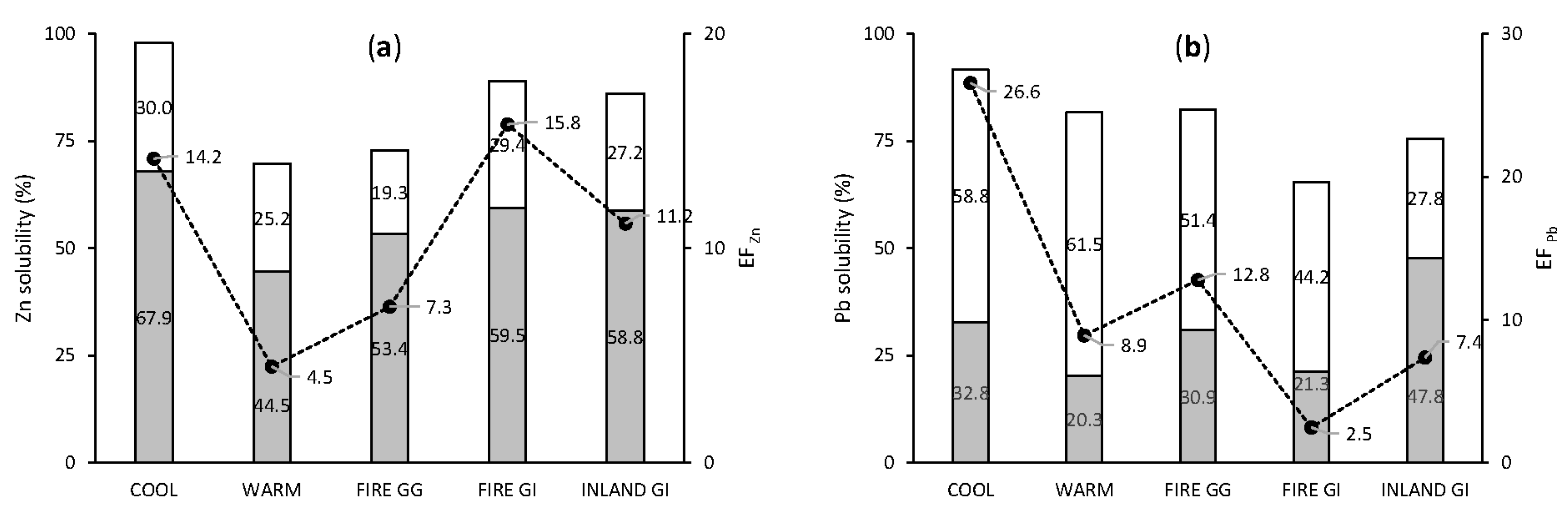
3.3.3. Anthropogenic Elements: Pb and Zn
3.4. Sources of Soluble and Leachable Bioactive Trace Metals at Gingin Site
3.4.1. Solubility as a Function of EFs
3.4.2. Fractional Iron Solubility as a Function of Soluble Major Ions
3.4.3. Fractional Iron Solubility as a Function of Mineral Dust Tracer Solubility
3.5. Sources Identification and Estimation of Their Contribution in Total Collected Aerosols: PMF Analysis
3.6. Dry Deposition Fluxes of Fe: Comparison with Models and Data from Other Regions of Australia
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Quality Control of the Leaching Protocol
Appendix A.1. Contribution of TM’s in Procedural Blank to Aerosols

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| W1 | W2 | W3 | W4 | W5 | W6 | U1 | C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 | F1 | F2 | W7 | U2 | |
| soluble | |||||||||||||||||||
| Al | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 16 | 3 | 4 | 9 | 6 | 2 | 9 | 2 | 1 | 0 | 1 | 1 |
| Ti | 2 | 1 | 1 | 1 | 6 | 3 | 13 | 10 | 5 | 4 | 23 | 4 | 4 | 6 | 2 | 2 | 1 | 1 | 2 |
| V | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mn | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fe | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 3 | 1 | 1 | 3 | 2 | 1 | 3 | 1 | 0 | 0 | 0 | 0 |
| Co | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 9 | 1 | 1 | 4 | 1 | 1 | 3 | 0 | 0 | 0 | 0 | 1 |
| Zn | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 0 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Pb | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| leachable | |||||||||||||||||||
| Al | 1 | 1 | 2 | 1 | 1 | 2 | 6 | 24 | 7 | 5 | 26 | 10 | 6 | 13 | 5 | 1 | 1 | 1 | 3 |
| Ti | 3 | 4 | 22 | 5 | 10 | 6 | 31 | 35 | 31 | 10 | 48 | 15 | 7 | 19 | 8 | 4 | 2 | 2 | 3 |
| V | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mn | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Fe | 1 | 1 | 3 | 1 | 2 | 2 | 4 | 16 | 7 | 3 | 22 | 5 | 3 | 9 | 2 | 0 | 0 | 1 | 2 |
| Co | 0 | 0 | 1 | 0 | 0 | 1 | 2 | 10 | 2 | 1 | 8 | 2 | 2 | 1 | 1 | 0 | 0 | 1 | 2 |
| Zn | 4 | 3 | 7 | 2 | 3 | 2 | 5 | 6 | 5 | 3 | 18 | 5 | 4 | 8 | 6 | 2 | 3 | 3 | 7 |
| Pb | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| refractory | |||||||||||||||||||
| Al | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | 1 | 1 | 2 | 2 | 1 | 2 | 0 | 0 | 0 | 0 | 0 |
| Ti | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 1 | 0 | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| V | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 10 | 1 | 1 | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Mn | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 5 | 1 | 1 | 2 | 2 | 1 | 2 | 0 | 0 | 0 | 0 | 1 |
| Fe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 1 | 0 | 2 | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 |
| Co | 1 | 1 | 1 | 1 | 1 | 1 | 3 | 36 | 5 | 2 | 12 | 8 | 4 | 4 | 2 | 1 | 1 | 1 | 2 |
| Zn | 5 | 3 | 10 | 4 | 4 | 4 | 15 | 78 | 26 | 7 | 35 | 11 | 35 | 20 | 7 | 2 | 4 | 8 | 9 |
| Pb | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 7 | 1 | 0 | 2 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 0 |
| total | |||||||||||||||||||
| Al | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | 1 | 1 | 2 | 2 | 1 | 2 | 0 | 0 | 0 | 0 | 0 |
| Ti | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 1 | 0 | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| V | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 1 | 0 | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Cr | 2 | 1 | 3 | 1 | 2 | 2 | 5 | 40 | 11 | 4 | 27 | 11 | 11 | 15 | 2 | 0 | 1 | 2 | 4 |
| Mn | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Fe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 1 | 0 | 2 | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 |
| Co | 1 | 1 | 1 | 0 | 1 | 1 | 3 | 28 | 4 | 2 | 11 | 4 | 3 | 3 | 1 | 0 | 0 | 1 | 2 |
| Cu | 8 | 4 | 20 | 5 | 7 | 6 | 18 | 53 | 24 | 10 | 52 | 39 | 14 | 39 | 9 | 3 | 3 | 6 | 16 |
| Zn | 4 | 2 | 7 | 2 | 3 | 2 | 6 | 13 | 7 | 3 | 21 | 5 | 6 | 11 | 4 | 2 | 2 | 4 | 5 |
| Ba | 1 | 0 | 1 | 1 | 0 | 1 | 3 | 13 | 5 | 2 | 12 | 8 | 3 | 8 | 2 | 0 | 0 | 1 | 2 |
| La | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Ce | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| U | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 1 | 1 | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Pb | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
- Correlations with mineral dust tracers in Section 3.2.2: %PBZ,∑l < 25% and outlier values from box and whisker plots of EF (Figure A1).
- Seasonality in bioactive trace metal solubilities and enrichment factors (Section 3.3): %PBAl, refractory of the sample COOL 4 was 26% for leachable Al. However, this result was included because the %PBAl, labile was below 25% and improved seasonal statistical robustness.
- Solubility as a function of EFs (Section 3.4.1): Fe and Co values for outlier sample FIRE GG 1 were excluded.
- Correlation between Fe solubility with Ti-normalised Pb, V, and Zn in Section 3.4.1: sample FIRE GG 1 was excluded from samples collected at Gingin because of the outstanding EF of these elements.

Appendix A.2. Precision of Single Sample Analysis
| Soluble | Leachable | Soluble * [46] | Refractory | Total | Total [46] | |
|---|---|---|---|---|---|---|
| Al | 1.9 | 7.0 | 0.6–31.7 | 10.3 | 10.2 | 0.0–20.2 |
| Ti | 12.3 | 10.5 | 1.7–2.1 | 9.3 | 9.3 | 0.3–20.0 |
| V | 1.4 | 5.9 | 0.5–31.2 | 9.4 | 8.8 | 0.0–21.2 |
| Mn | 5.7 | 4.9 | - | 8.7 | 6.2 | - |
| Fe | 1.9 | 8.4 | 1.0–36.1 | 9.5 | 9.5 | 0.0–20.1 |
| Co | 5.0 | 12.9 | - | 11.9 | 10.9 | - |
| Zn | 2.3 | 4.0 | 1.9–21.9 | 5.4 | 2.5 | 0.1–21.2 |
| Pb | 2.0 | 1.0 | 0.7–45.2 | 8.4 | 3.5 | 0.6–21.2 |
References
- Siegenthaler, U.; Sarmiento, J.L. Atmospheric carbon dioxide and the ocean. Nature 1993, 365, 119–125. [Google Scholar] [CrossRef]
- Martin, J.H.; Fitzwater, S.E. Iron deficiency limits phytoplankton growth in the north-east Pacific subarctic. Nature 1988, 331, 341–343. [Google Scholar] [CrossRef]
- Martin, J.H.; Coale, K.H.; Johnson, K.S.; Fitzwater, S.E.; Gordon, R.M.; Tanner, S.J.; Hunter, C.N.; Elrod, V.A.; Nowicki, J.L.; Coley, T.L.; et al. Testing the iron hypothesis in ecosystems of the equatorial Pacific Ocean. Nature 1994, 371, 123–129. [Google Scholar] [CrossRef]
- Boyd, P.W.; Jickells, T.; Law, C.S.; Blain, S.; Boyle, E.A.; Buesseler, K.O.; Coale, K.H.; Cullen, J.J.; de Baar, H.J.W.; Follows, M.; et al. Mesoscale Iron Enrichment Experiments 1993–2005: Synthesis and Future Directions. Science 2007, 315, 612–617. [Google Scholar] [CrossRef]
- Moore, C.M.; Mills, M.M.; Arrigo, K.R.; Berman-Frank, I.; Bopp, L.; Boyd, P.W.; Galbraith, E.D.; Geider, R.J.; Guieu, C.; Jaccard, S.L.; et al. Processes and patterns of oceanic nutrient limitation. Nat. Geosci. 2013, 6, 701–710. [Google Scholar] [CrossRef]
- Duce, R.A. The atmospheric input of trace species to the world ocean. Glob. Biogeochem. Cycles 1991, 5, 193–259. [Google Scholar] [CrossRef]
- Duce, R.A.; Tindale, N.W. Atmospheric transport of iron and its deposition in the ocean. Limnol. Oceanogr. 1991, 36, 1715–1726. [Google Scholar] [CrossRef]
- Ussher, S.J.; Achterberg, E.P.; Worsfold, P.J. Marine Biogeochemistry of Iron. Environ. Chem. 2004, 1, 67–80. [Google Scholar] [CrossRef]
- Jickells, T.D.; An, Z.S.; Andersen, K.K.; Baker, A.R.; Bergametti, G.; Brooks, N.; Cao, J.J.; Boyd, P.W.; Duce, R.A.; Hunter, K.A.; et al. Global Iron Connections Between Desert Dust, Ocean Biogeochemistry, and Climate. Science 2005, 308, 67–71. [Google Scholar] [CrossRef]
- Mahowald, N.M.; Engelstaedter, S.; Luo, C.; Sealy, A.; Artaxo, P.; Benitez-Nelson, C.; Bonnet, S.; Chen, Y.; Chuang, P.Y.; Cohen, D.; et al. Atmospheric iron deposition: Global distribution, variability, and human perturbations. Annu. Rev. Mar. Sci. 2009, 1, 245–278. [Google Scholar] [CrossRef]
- Baker, A.R.; Croot, P.L. Atmospheric and marine controls on aerosol iron solubility in seawater. Mar. Chem. 2010, 120, 4–13. [Google Scholar] [CrossRef]
- Mahowald, N.; Ward, D.S.; Kloster, S.; Flanner, M.G.; Heald, C.L.; Heavens, N.G.; Hess, P.G.; Lamarque, J.-F.; Chuang, P.Y. Aerosol Impacts on Climate and Biogeochemistry. Annu. Rev. Environ. Resour. 2011, 36, 45–74. [Google Scholar] [CrossRef]
- Sholkovitz, E.R.; Sedwick, P.N.; Church, T.M.; Baker, A.R.; Powell, C.F. Fractional solubility of aerosol iron: Synthesis of a global-scale data set. Geochim. Cosmochim. Acta 2012, 89, 173–189. [Google Scholar] [CrossRef]
- Scanza, R.A. The Impact of Resolving Mineralogy of Dust on Climate and Biogeochemistry. Ph.D. Thesis, Cornell University, Ann Arbor, MI, USA, 2016. [Google Scholar]
- Perron, M.M.G.; Strzelec, M.; Gault-Ringold, M.; Proemse, B.C.; Boyd, P.W.; Bowie, A.R. Assessment of leaching protocols to determine the solubility of trace metals in aerosols. Talanta 2020, 208, 120377. [Google Scholar] [CrossRef] [PubMed]
- Sedwick, P.N.; Sholkovitz, E.R.; Church, T.M. Impact of anthropogenic combustion emissions on the fractional solubility of aerosol iron: Evidence from the Sargasso Sea. Geochem. Geophys. Geosyst. 2007, 8, Q10Q06. [Google Scholar] [CrossRef]
- Schroth, A.W.; Crusius, J.; Sholkovitz, E.R.; Bostick, B.C. Iron solubility driven by speciation in dust sources to the ocean. Nat. Geosci. 2009, 2, 337–340. [Google Scholar] [CrossRef]
- Takahashi, Y.; Higashi, M.; Furukawa, T.; Mitsunobu, S. Change of iron species and iron solubility in Asian dust during the long-range transport from western China to Japan. Atmos. Chem. Phys. 2011, 11, 11237–11252. [Google Scholar] [CrossRef]
- Ito, A. Atmospheric Processing of Combustion Aerosols as a Source of Bioavailable Iron. Environ. Sci. Technol. Lett. 2015, 2, 70–75. [Google Scholar] [CrossRef]
- Ito, A.; Penner, J.E. Global estimates of biomass burning emissions based on satellite imagery for the year 2000. J. Geophys. Res. D Atmos. 2004, 109, D14S05. [Google Scholar] [CrossRef]
- Guieu, C.; Bonnet, S.; Wagener, T.; Loÿe-Pilot, M.D. Biomass burning as a source of dissolved iron to the open ocean? Geophys. Res. Lett. 2005, 32, L19608. [Google Scholar] [CrossRef]
- Paris, R.; Desboeufs, K.V.; Formenti, P.; Nava, S.; Chou, C. Chemical characterisation of iron in dust and biomass burning aerosols during AMMA-SOP0/DABEX: Implication for iron solubility. Atmos. Chem. Phys. 2010, 10, 4273–4282. [Google Scholar] [CrossRef]
- Luo, C.; Mahowald, N.; Bond, T.; Chuang, P.Y.; Artaxo, P.; Siefert, R.; Chen, Y.; Schauer, J. Combustion iron distribution and deposition. Glob. Biogeochem. Cycles 2008, 22, GB1012. [Google Scholar] [CrossRef]
- Desboeufs, K.V.; Sofikitis, A.; Losno, R.; Colin, J.L.; Ausset, P. Dissolution and solubility of trace metals from natural and anthropogenic aerosol particulate matter. Chemosphere 2005, 58, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Chance, R.; Jickells, T.D.; Baker, A.R. Atmospheric trace metal concentrations, solubility and deposition fluxes in remote marine air over the south-east Atlantic. Mar. Chem. 2015, 177, 45–56. [Google Scholar] [CrossRef]
- Tanaka, T.Y.; Chiba, M. A numerical study of the contributions of dust source regions to the global dust budget. Glob. Planet. Chang. 2006, 52, 88–104. [Google Scholar] [CrossRef]
- Bowler, J.M. Aridity in Australia: Age, origins and expression in aeolian landforms and sediments. Earth Sci. Rev. 1976, 12, 279–310. [Google Scholar] [CrossRef]
- Hesse, P.P.; McTainsh, G.H. Australian dust deposits: Modern processes and the Quaternary record. Quat. Sci. Rev. 2003, 22, 2007–2035. [Google Scholar] [CrossRef]
- Kiefert, L.; McTainsh, G.H. Oxygen isotope abundance in the quartz fraction of aeolian dust: Implications for soil and ocean sediment formation in the Australasian region. Soil Res. 1996, 34, 467–473. [Google Scholar] [CrossRef]
- Mahowald, N.M.; Baker, A.R.; Bergametti, G.; Brooks, N.; Duce, R.A.; Jickells, T.D.; Kubilay, N.; Prospero, J.M.; Tegen, I. Atmospheric global dust cycle and iron inputs to the ocean. Glob. Biogeochem. Cycles 2005, 19, GB4025. [Google Scholar] [CrossRef]
- Radhi, M.; Box, M.A.; Box, G.P.; Gupta, P.; Christopher, S.A. Evolution of the optical properties of biomass-burning aerosol during the 2003 southeast Australian bushfires. Appl. Opt. 2009, 48, 1764–1773. [Google Scholar] [CrossRef]
- Yusiharni, E.; Gilkes, R.J. Changes in the mineralogy and chemistry of a lateritic soil due to a bushfire at Wundowie, Darling Range, Western Australia. Geoderma 2012, 191, 140–150. [Google Scholar] [CrossRef]
- Duc, H.N.; Chang, L.T.-C.; Azzi, M.; Jiang, N. Smoke aerosols dispersion and transport from the 2013 New South Wales (Australia) bushfires. Environ. Monit. Assess. 2018, 190, 428. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.D.; Crawford, J.; Stelcer, E.; Atanacio, A.J. Application of positive matrix factorization, multi-linear engine and back trajectory techniques to the quantification of coal-fired power station pollution in metropolitan Sydney. Atmos. Environ. 2012, 61, 204–211. [Google Scholar] [CrossRef]
- Cohen, D.D.; Stelcer, E.; Atanacio, A.; Crawford, J. The application of IBA techniques to air pollution source fingerprinting and source apportionment. Nucl. Instrum. Methods Phys. Res. Sect. B 2014, 318, 113–118. [Google Scholar] [CrossRef]
- Cohen, D.D.; Stelcer, E.; Garton, D.; Crawford, J. Fine particle characterisation, source apportionment and long-range dust transport into the Sydney Basin: A long term study between 1998 and 2009. Atmos. Pollut. Res. 2011, 2, 182–189. [Google Scholar] [CrossRef]
- Taylor, M.P. Atmospherically deposited trace metals from bulk mineral concentrate port operations. Sci. Total Environ. 2015, 515–516, 143–152. [Google Scholar] [CrossRef]
- Rate, A.W. Multielement geochemistry identifies the spatial pattern of soil and sediment contamination in an urban parkland, Western Australia. Sci. Total Environ. 2018, 627, 1106–1120. [Google Scholar] [CrossRef]
- Bureau of Meteorology. Available online: http://www.bom.gov.au/ (accessed on 4 October 2019).
- Driscoll, D.; Milkovits, G.; Freudenberger, D. Impact and Use of Firewood in Australia; CSIRO Sustainable Ecosystems: Canberra, Australia, 2000. [Google Scholar]
- Murdoch University. Available online: http://www.see.murdoch.edu.au/resources/info/Res/wood/ (accessed on 24 October 2019).
- Cropp, R.A.; Gabric, A.J.; Levasseur, M.; McTainsh, G.H.; Bowie, A.; Hassler, C.S.; Law, C.S.; McGowan, H.; Tindale, N.; Viscarra Rossel, R. The likelihood of observing dust-stimulated phytoplankton growth in waters proximal to the Australian continent. J. Mar. Syst. 2013, 117–118, 43–52. [Google Scholar] [CrossRef]
- de Baar, H.J.W.; de Jong, J.T.M.; Bakker, D.C.E.; Löscher, B.M.; Veth, C.; Bathmann, U.; Smetacek, V. Importance of iron for plankton blooms and carbon dioxide drawdown in the Southern Ocean. Nature 1995, 373, 412–415. [Google Scholar] [CrossRef]
- Boyd, P.; LaRoche, J.; Gall, M.; Frew, R.; McKay, R.M.L. Role of iron, light, and silicate in controlling algal biomass in subantarctic waters SE of New Zealand. J. Geophys. Res. Ocean. 1999, 104, 13395–13408. [Google Scholar] [CrossRef]
- Thompson, P.A.; Bonham, P.; Waite, A.M.; Clementson, L.A.; Cherukuru, N.; Hassler, C.; Doblin, M.A. Contrasting oceanographic conditions and phytoplankton communities on the east and west coasts of Australia. Deep Sea Res. Part II Top. Stud. Oceanogr. 2011, 58, 645–663. [Google Scholar] [CrossRef]
- Morton, P.L.; Landing, W.M.; Hsu, S.C.; Milne, A.; Aguilar-Islas, A.M.; Baker, A.R.; Bowie, A.R.; Buck, C.S.; Gao, Y.; Gichuki, S.; et al. Methods for the sampling and analysis of marine aerosols: Results from the 2008 GEOTRACES aerosol intercalibration experiment. Limnol. Oceanogr. Methods 2013, 11, 62–78. [Google Scholar] [CrossRef]
- World Atlas of Desertification, 3rd ed.; Cherlet, M., Hutchinson, C., Reynolds, J., Hill, J., Sommer, S., von Maltitz, G., Eds.; Publication Office of the European Union: Luxembourg, 2018. [Google Scholar] [CrossRef]
- Keene, W.C.; Pszenny, A.A.P.; Galloway, J.N.; Hawley, M.E. Sea-salt corrections and interpretation of constituent ratios in marine precipitation. J. Geophys. Res. Atmos. 1986, 91, 6647–6658. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-time Environmental Applications and Display sYstem: READY. Environ. Model. Softw. 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Landgate Fire Watch. Available online: http://firewatch-pro.landgate.wa.gov.au/home.php (accessed on 4 October 2019).
- NASA. Available online: https://zoom.earth/ (accessed on 4 October 2019).
- Paatero, P.; Tapper, U. Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Norris, G.; Duvall, R.; Brown, S.; Bai, S. EPA Positive Matrix Factorization (PMF) 5.0 Fundamentals and User Guide. In Guide (User Guide No. EPA/600/R-14/108); U.S. Environmental Protection Agency: Washington, DC, USA, 2014. [Google Scholar]
- Evans, J.D. Straightforward Statistics for the Behavioral Sciences; Thomson Brooks/Cole Publishing Co.: Belmont, CA, USA, 1996. [Google Scholar]
- Wedepohl, H.K. The composition of the continental crust. Geochim. Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Cullis, C.F.; Hirschler, M.M. Atmospheric sulphur: Natural and man-made sources. Atmos. Environ. (1967) 1980, 14, 1263–1278. [Google Scholar] [CrossRef]
- Galbally, I.; Roy, C.R. The Fate of Nitrogen Compounds in the Stmosphere; Springer: Dordrecht, The Netherlands, 1983; pp. 265–284. [Google Scholar] [CrossRef]
- Kawamura, K.; Kaplan, I.R. Motor exhaust emissions as a primary source for dicarboxylic acids in Los Angeles ambient air. Environ. Sci. Technol. 1987, 21, 105–110. [Google Scholar] [CrossRef]
- Chebbi, A.; Carlier, P. Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review. Atmos. Environ. 1996, 30, 4233–4249. [Google Scholar] [CrossRef]
- Gillett, W.R.; Galbally, I.; Ayers, G.P.; Selleck, P.W.; Jennifer, P.; Meyer, C.P.; Keywood, M.; Fedele, R. Oxalic acid and oxalate in the atmosphere. In Proceedings of the 4th IUAPPA World Congress Clean Air Partnerships: Coming Together for Clean Air, Brisbane, Australia, 9–13 September 2007. [Google Scholar]
- Warneck, P. In-cloud chemistry opens pathway to the formation of oxalic acid in the marine atmosphere. Atmos. Environ. Atmos. Environ. 2003, 37, 2423–2427. [Google Scholar] [CrossRef]
- Winton, V.H.L.; Edwards, R.; Bowie, A.R.; Keywood, M.; Williams, A.G.; Chambers, S.; Selleck, P.W.; Desservettaz, M.; Mallet, M.; Paton-Walsh, C. Dry season aerosol iron solubility in tropical northern Australia. Atmos. Chem. Phys. Discuss. 2016, 16, 12829–12848. [Google Scholar] [CrossRef]
- Buck, C.S.; Aguilar-Islas, A.; Marsay, C.; Kadko, D.; Landing, W.M. Trace element concentrations, elemental ratios, and enrichment factors observed in aerosol samples collected during the US GEOTRACES eastern Pacific Ocean transect (GP16). Chem. Geol. 2019, 511, 212–224. [Google Scholar] [CrossRef]
- Viana, M.; Amato, F.; Alastuey, A.; Querol, X.; Moreno, T.; García Dos Santos, S.; Herce, M.D.; Fernández-Patier, R. Chemical Tracers of Particulate Emissions from Commercial Shipping. Environ. Sci. Technol. 2009, 43, 7472–7477. [Google Scholar] [CrossRef]
- Radhi, M.; Box, M.A.; Box, G.P.; Keywood, M.D.; Cohen, D.D.; Stelcer, E.; Mitchell, R.M. Size-resolved chemical composition of Australian dust aerosol during winter. Environ. Chem. 2011, 8, 248–262. [Google Scholar] [CrossRef]
- Radhi, M.; Box, M.A.; Box, G.P.; Mitchell, R.M.; Cohen, D.D.; Stelcer, E.; Keywood, M.D. Size-resolved mass and chemical properties of dust aerosols from Australia’s Lake Eyre Basin. Atmos. Environ. 2010, 44, 3519–3528. [Google Scholar] [CrossRef]
- Radhi, M.; Box, M.A.; Box, G.P.; Mitchell, R.M.; Cohen, D.D.; Stelcer, E.; Keywood, M.D. Optical, physical and chemical characteristics of Australian continental aerosols: Results from a field experiment. Atmos. Chem. Phys. 2010, 10, 5925–5942. [Google Scholar] [CrossRef]
- Perron, M.M.G.; Proemse, B.C.; Strzelec, M.; Gault-Ringold, M.; Boyd, P.W.; Sanz Rodriguez, E.; Paull, B.; Bowie, A.R. Origin, transport and deposition of aerosol iron to Australian coastal waters. Atmos. Environ. 2020, 228, 117432. [Google Scholar] [CrossRef]
- Mahowald, N.M.; Hamilton, D.S.; Mackey, K.R.M.; Moore, J.K.; Baker, A.R.; Scanza, R.A.; Zhang, Y. Aerosol trace metal leaching and impacts on marine microorganisms. Nat. Commun. 2018, 9, 2614. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, X.H.H.; Bian, Q.; Griffith, S.M.; Louie, P.K.K.; Yu, J. Sources and atmospheric processes impacting oxalate at a suburban coastal site in Hong Kong: Insights inferred from 1 year hourly measurements. J. Geophys. Res. Atmos. 2015, 120, 9772–9788. [Google Scholar] [CrossRef]
- Park, S.-S.; Sim, S.Y.; Bae, M.-S.; Schauer, J.J. Size distribution of water-soluble components in particulate matter emitted from biomass burning. Atmos. Environ. 2013, 73, 62–72. [Google Scholar] [CrossRef]
- Takahashi, Y.; Furukawa, T.; Kanai, Y.; Uematsu, M.; Zheng, G.; Marcus, M.A. Seasonal changes in Fe species and soluble Fe concentration in the atmosphere in the Northwest Pacific region based on the analysis of aerosols collected in Tsukuba, Japan. Atmos. Chem. Phys. 2013, 13, 7695–7710. [Google Scholar] [CrossRef]
- Kong, L.; Yang, Y.; Zhang, S.; Zhao, X.; Du, H.; Fu, H.; Zhang, S.; Cheng, T.; Yang, X.; Chen, J.; et al. Observations of linear dependence between sulfate and nitrate in atmospheric particles. J. Geophys. Res. Atmos. 2014, 119, 341–361. [Google Scholar] [CrossRef]
- Block, C.; Dams, R. Lead contents of coalL, coal ash and fly ash. Water Air Soil Pollut. 1975, 5, 207–211. [Google Scholar] [CrossRef]
- Chaudhary, S.; Banerjee, D.K. Speciation of some heavy metals in coal fly ash. Chem. Speciat. Bioavailab. 2007, 19, 95–102. [Google Scholar] [CrossRef][Green Version]
- Maresca, A.; Hyks, J.; Astrup, T.F. Recirculation of biomass ashes onto forest soils: Ash composition, mineralogy and leaching properties. Waste Manag. 2017, 70, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, S.V.; Baxter, D.; Andersen, L.K.; Vassileva, C.G. An overview of the composition and application of biomass ash. Part 1. Phase–mineral and chemical composition and classification. Fuel 2013, 105, 40–76. [Google Scholar] [CrossRef]
- Vassilev, S.V.; Vassileva, C.G.; Baxter, D. Trace element concentrations and associations in some biomass ashes. Fuel 2014, 129, 292–313. [Google Scholar] [CrossRef]
- Ketris, M.P.; Yudovich, Y.E. Estimations of Clarkes for Carbonaceous biolithes: World averages for trace element contents in black shales and coals. Int. J. Coal Geol. 2009, 78, 135–148. [Google Scholar] [CrossRef]
- National Pollution Inventory. Available online: http://www.npi.gov.au/ (accessed on 21 September 2019).
- Strzelec, M.; Proemse, B.C.; Gault-Ringold, M.; Boyd, P.W.; Perron, M.M.G.; Schofield, R.; Ryan, R.G.; Ristovski, Z.D.; Alroe, J.; Humphries, R.S.; et al. Atmospheric trace metal deposition near the Great Barrier Reef, Australia. Atmosphere 2020, 11, 390. [Google Scholar] [CrossRef]








| Element | N | rAl | rTi | EFaerosols | EFsoil 1 | EFsoil 2 |
|---|---|---|---|---|---|---|
| Al | 19 | 1.00 | 0.98 | 0.5 | 0.9 | 0.6 |
| Ti | 19 | 0.98 | 1.00 | 1 | 1.0 | 1.0 |
| V | 17 | 0.91 | 0.92 | 1.8 | 3.1 | 0.6 |
| Mn | 17 | 0.93 | 0.91 | 0.4 | 0.1 | 0.1 |
| Fe | 17 | 0.98 | 0.98 | 0.6 | 2.9 | 0.4 |
| Co | 16 | 0.98 | 0.98 | 0.5 | 0.1 | 0.1 |
| Zn | 19 | 0.67 | 0.77 | 7.2 | 0.4 | 1.6 |
| Pb | 18 | 0.52 | 0.62 | 14.0 | 0.6 | 1.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strzelec, M.; Proemse, B.C.; Barmuta, L.A.; Gault-Ringold, M.; Desservettaz, M.; Boyd, P.W.; Perron, M.M.G.; Schofield, R.; Bowie, A.R. Atmospheric Trace Metal Deposition from Natural and Anthropogenic Sources in Western Australia. Atmosphere 2020, 11, 474. https://doi.org/10.3390/atmos11050474
Strzelec M, Proemse BC, Barmuta LA, Gault-Ringold M, Desservettaz M, Boyd PW, Perron MMG, Schofield R, Bowie AR. Atmospheric Trace Metal Deposition from Natural and Anthropogenic Sources in Western Australia. Atmosphere. 2020; 11(5):474. https://doi.org/10.3390/atmos11050474
Chicago/Turabian StyleStrzelec, Michal, Bernadette C. Proemse, Leon A. Barmuta, Melanie Gault-Ringold, Maximilien Desservettaz, Philip W. Boyd, Morgane M. G. Perron, Robyn Schofield, and Andrew R. Bowie. 2020. "Atmospheric Trace Metal Deposition from Natural and Anthropogenic Sources in Western Australia" Atmosphere 11, no. 5: 474. https://doi.org/10.3390/atmos11050474
APA StyleStrzelec, M., Proemse, B. C., Barmuta, L. A., Gault-Ringold, M., Desservettaz, M., Boyd, P. W., Perron, M. M. G., Schofield, R., & Bowie, A. R. (2020). Atmospheric Trace Metal Deposition from Natural and Anthropogenic Sources in Western Australia. Atmosphere, 11(5), 474. https://doi.org/10.3390/atmos11050474