Atmospheric Trace Metal Deposition near the Great Barrier Reef, Australia

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Experiments
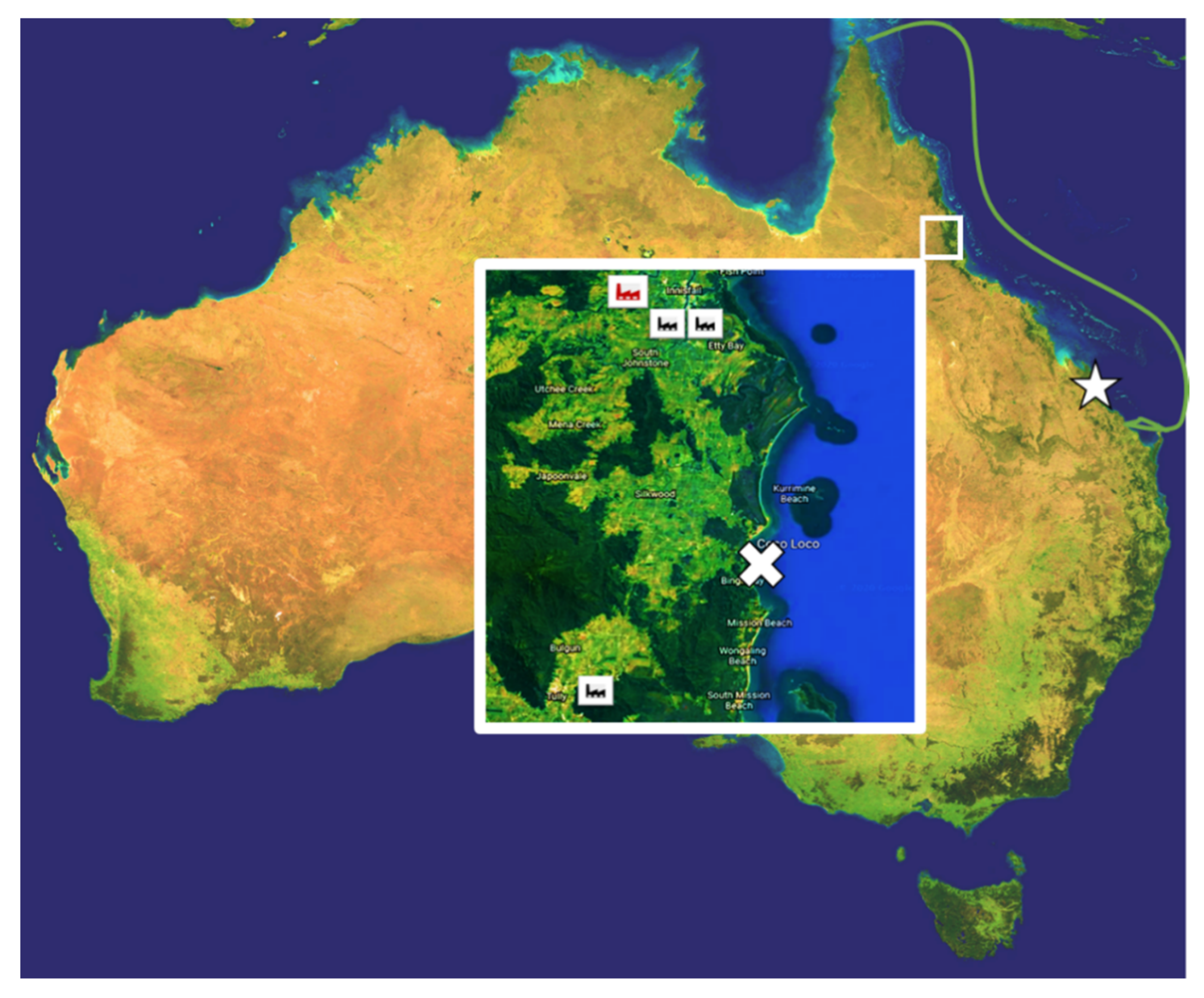
2.1. Sample Collection
2.2. Back Trajectories Analysis
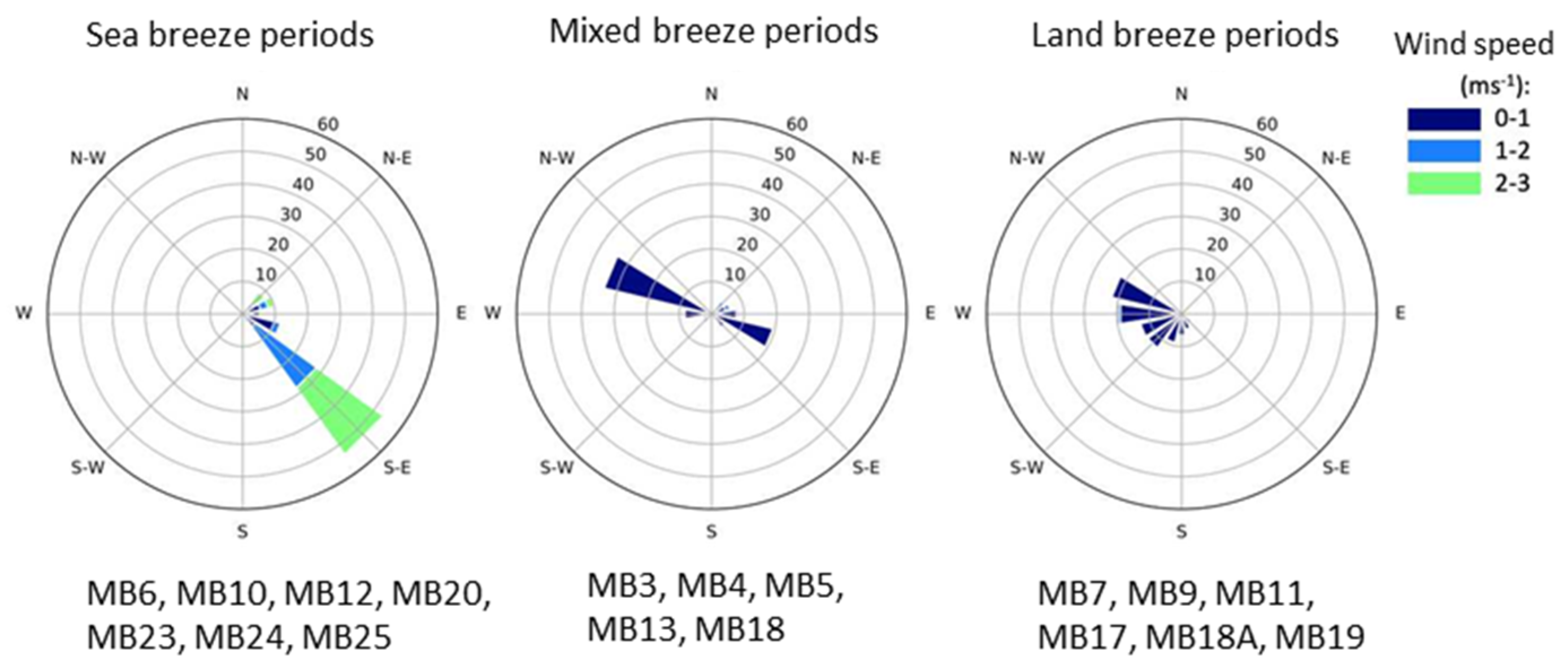
2.3. Wind Direction Analysis
2.4. Aerosol Sample Preparation
2.5. Rainwater Sample Preparation
2.6. Trace Metal Analysis by ICP-MS
2.7. Major Ion Analysis
2.7.1. Aerosol Sample Preparation
2.7.2. Major Ion Analysis by IC
2.8. Black Carbon, Organic Carbon, and Levoglucosan Derivatives
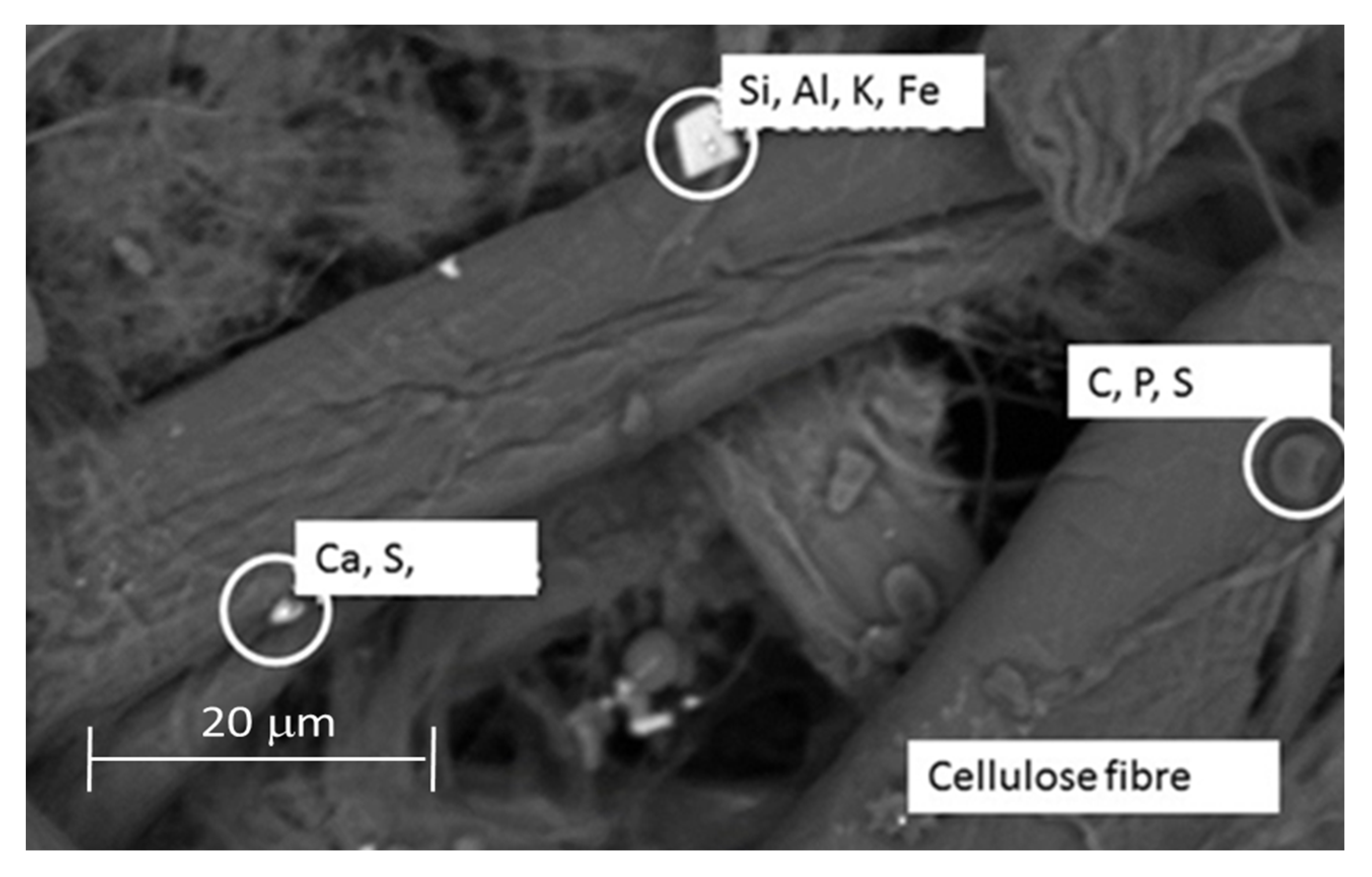
2.9. Scanning Electron Microscopy
2.10. Enrichment Factor
2.11. Other Calculations
3. Results and Discussion
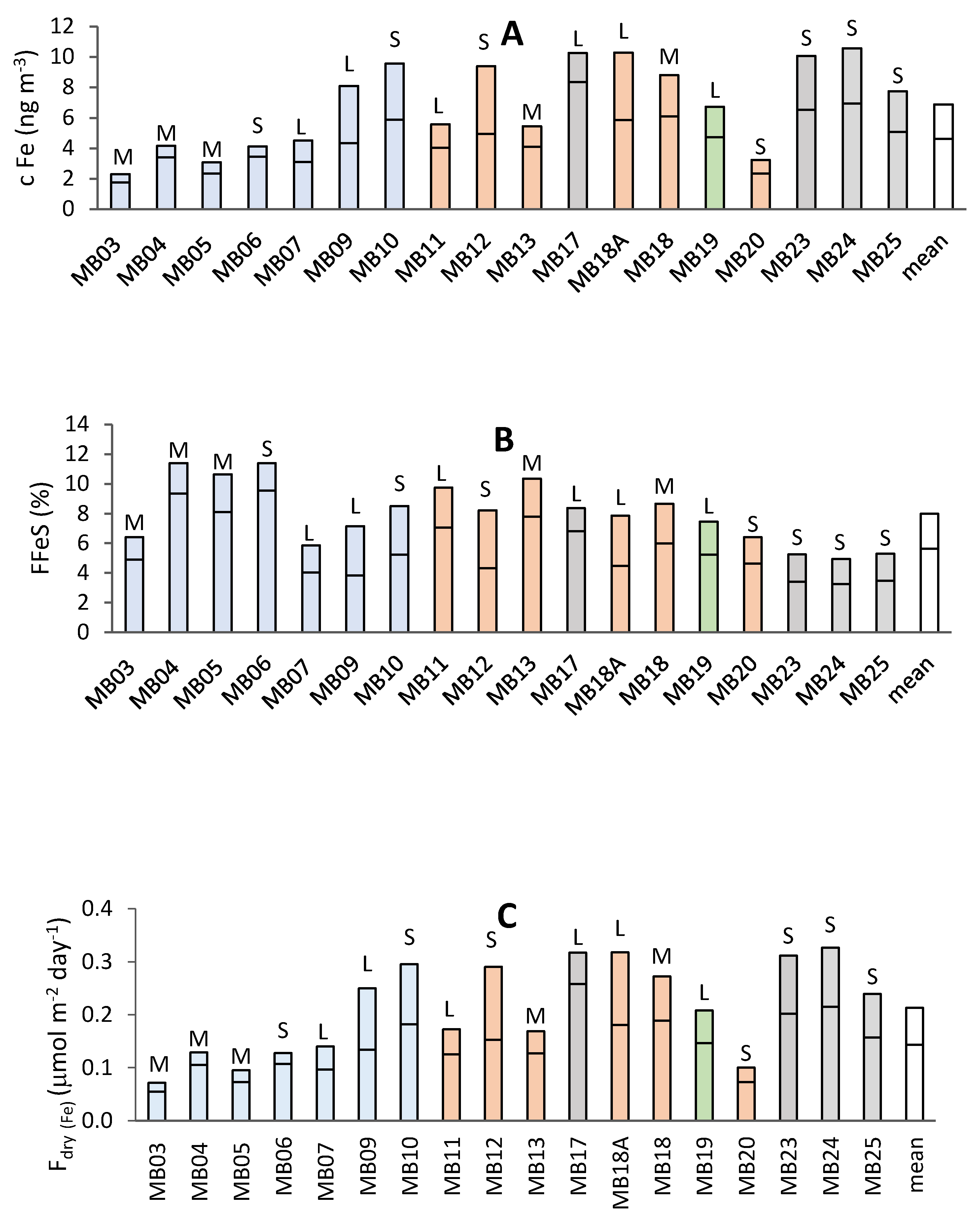
3.1. Iron in Aerosols
3.1.1. Iron Provenance
3.1.2. Iron Solubility
3.1.3. Drivers of Iron Solubility
Non-Crustal Emissions vs. Iron Solubility
Combustion Products vs. Iron Solubility
Aging Processes vs. Iron Solubility
3.1.4. Estimation of Fe Deposition Fluxes
3.2. Iron in Rain Water
3.3. Atmospheric Deposition of Coral Toxins and Other Bioactive Metals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Analytical Methods Check
Appendix A.1. Iron Blanks


Appendix A.2. Digestion Procedure Recovery
Appendix A.3. Precision of the Leaching Protocol and Digestion
References
- Jickells, T.; Moore, C.M. The Importance of Atmospheric Deposition for Ocean Productivity. Annu. Rev. Ecol. Evol. Syst. 2015, 46, 481–501. [Google Scholar] [CrossRef]
- Jickells, T.D. Global Iron Connections Between Desert Dust, Ocean Biogeochemistry, and Climate. Science 2005, 308, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Perron, M.M.G. Assessment of leaching protocols to determine the solubility of trace metals in aerosols. Talanta 2020, 208, 120377. [Google Scholar] [CrossRef] [PubMed]
- Morel, F.M.M.; Price, N. The Biogeochemical Cycles of Trace Metals in the Oceans. Science 2003, 300, 944–947. [Google Scholar] [CrossRef]
- Henderson, G. GEOTRACES—An international study of the global marine biogeochemical cycles of trace elements and their isotopes. Chemie Der Erde Geochem. 2007, 67, 85–131. [Google Scholar]
- Lohan, M.C.; Tagliabue, A. Oceanic Micronutrients: Trace Metals that are Essential for Marine Life. Elements 2018, 14, 385–390. [Google Scholar] [CrossRef]
- Lønborg, C. The Great Barrier Reef: A source of CO 2 to the atmosphere. Mar. Chem. 2019, 210, 24–33. [Google Scholar] [CrossRef]
- Morrison, T.; Hughes, T. Climate change and the Great Barrier Reef. Policy Information Brief 1; National Climate Change Adaptation Research Facility: Gold Coast, Australia, 2016. [Google Scholar]
- Brodie, J. Dispersal of suspended sediments and nutrients in the Great Barrier Reef lagoon during river-discharge events: Conclusions from satellite remote sensing and concurrent flood-plume sampling. Mar. Freshw. Res. 2010, 61, 651–664. [Google Scholar] [CrossRef]
- Brand, L.E. Minimum iron requirements of marine phytoplankton and the implications for the biogeochemical control of new production. Limnol. Oceanogr. 1991, 36, 1756–1771. [Google Scholar] [CrossRef]
- Kustka, A.B. Iron requirements for dinitrogen- and ammonium-supported growth in cultures of Trichodesmium (IMS 101): Comparison with nitrogen fixation rates and iron: Carbon ratios of field populations. Limnol. Oceanogr. 2003, 48, 1869–1884. [Google Scholar] [CrossRef]
- Cropp, R.A. The likelihood of observing dust-stimulated phytoplankton growth in waters proximal to the Australian continent. J. Mar. Syst. 2013, 117–118, 43–52. [Google Scholar] [CrossRef]
- Shaw, E.C.; Gabric, A.J.; McTainsh, G.H. Impacts of aeolian dust deposition on phytoplankton dynamics in Queensland coastal waters. Mar. Freshw. Res. 2008, 59, 951–962. [Google Scholar] [CrossRef]
- Shaw, E.C.; Gabric, A.J.; McTainsh, G.H. Response to comment on Impacts of aeolian dust deposition on phytoplankton dynamics in Queensland coastal waters. Mar. Freshw. Res. 2010, 61, 504–506. [Google Scholar] [CrossRef]
- Mackie, D.S. Biogeochemistry of iron in Australian dust: From eolian uplift to marine uptake. Geochem. Geophys. Geosyst. 2008, 9, Q03–Q08. [Google Scholar] [CrossRef]
- Boyd, P.W.; Mackie, D.S.; Hunter, K.A. Aerosol iron deposition to the surface ocean - Modes of iron supply and biological responses. Mar. Chem. 2010, 120, 128–143. [Google Scholar] [CrossRef]
- Falkowski, P.G. Evolution of the nitrogen cycle and its influence on the biological sequestration of CO2 in the ocean. Nature 1997, 387, 272–275. [Google Scholar] [CrossRef]
- Berman-Frank, I. Iron availability, cellular iron quotas, and nitrogen fixation in Trichodesmium. Limnol. Oceanogr. 2001, 46, 1249–1260. [Google Scholar] [CrossRef]
- Mills, M.M. Iron and phosphorus co-limit nitrogen fixation in the eastern tropical North Atlantic. Nature 2004, 429, 292–294. [Google Scholar] [CrossRef]
- LaRoche, J.; Breitbarth, E. Importance of the diazotrophs as a source of new nitrogen in the ocean. J. Sea Res. 2005, 53, 67–91. [Google Scholar] [CrossRef]
- Kroon, F.J. Sources, presence and potential effects of contaminants of emerging concern in the marine environments of the Great Barrier Reef and Torres Strait, Australia. Sci. Total Environ. 2020, 719, 135140. [Google Scholar] [CrossRef]
- Taylor, M.P. Atmospherically deposited trace metals from bulk mineral concentrate port operations. Sci. Total Environ. 2015, 515–516, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Reichelt-Brushett, A.; Michalek-Wagner, K. Effects of copper on the fertilization success of the soft coral Lobophytum compactum. Aquat. Toxicol. 2005, 74, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Reichelt-Brushett, A.J.; Harrison, P.L. The effect of copper, zinc and cadmium on fertilization success of gametes from scleractinian reef corals. Mar. Pollut. Bull. 1999, 38, 182–187. [Google Scholar] [CrossRef]
- Deschaseaux, E. High zinc exposure leads to reduced dimethylsulfoniopropionate (DMSP) levels in both the host and endosymbionts of the reef-building coral Acropora aspera. Mar. Pollut. Bull. 2018, 126, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Mahowald, N.M. Observed 20th century desert dust variability: Impact on climate and biogeochemistry. Atmos. Chem. Phys. 2010, 10, 10875–10893. [Google Scholar] [CrossRef]
- Strzelec, M. Atmospheric Trace Metal Deposition from Natural and Anthropogenic Sources in Western Australia. Submited to Atmosphere in March 2020.
- Winton, V.H.L. Dry season aerosol iron solubility in tropical northern Australia. Atmos. Chem. Phys. Discuss. 2016, 16, 12829–12848. [Google Scholar] [CrossRef]
- Winton, V.H.L. Fractional iron solubility of atmospheric iron inputs to the Southern Ocean. Mar. Chem. 2015, 177, 20–32. [Google Scholar] [CrossRef]
- Journet, E. Mineralogy as a critical factor of dust iron solubility. Geophys. Res. Lett. 2008, 35, 12829–12848. [Google Scholar] [CrossRef]
- Ito, A. Pyrogenic iron: The missing link to high iron solubility in aerosols. Sci. Adv. 2019, 5, eaau7671. [Google Scholar] [CrossRef]
- Scanza, R.A. Atmospheric processing of iron in mineral and combustion aerosols: Development of an intermediate-complexity mechanism suitable for Earth system models. Atmos. Chem. Phys. 2018, 18, 14175–14196. [Google Scholar] [CrossRef]
- Desboeufs, K.V. Dissolution and solubility of trace metals from natural and anthropogenic aerosol particulate matter. Chemosphere 2005, 58, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Schroth, A.W. Iron solubility driven by speciation in dust sources to the ocean. Nat. Geosci. 2009, 2, 337–340. [Google Scholar] [CrossRef]
- Huang, X.-F. Water-soluble organic carbon and oxalate in aerosols at a coastal urban site in China: Size distribution characteristics, sources, and formation mechanisms. J. Geophys. Res. Atmos. 2006, 111, D22212. [Google Scholar] [CrossRef]
- Matsuki, A. Morphological and chemical modification of mineral dust: Observational insight into the heterogeneous uptake of acidic gases. Geophys. Res. Lett. 2005, 32, L22806. [Google Scholar] [CrossRef]
- Zuo, Y.; Zhan, J. Effects of oxalate on Fe-catalyzed photooxidation of dissolved sulfur dioxide in atmospheric water. Atmos. Environ. 2005, 39, 27–37. [Google Scholar] [CrossRef]
- Cwiertny, D.M. Characterization and acid-mobilization study of iron-containing mineral dust source materials. J. Geophys. Res. Atmos. 2008, 113, D05202. [Google Scholar] [CrossRef]
- Fu, H. Photoreductive dissolution of Fe-containing mineral dust particles in acidic media. J. Geophys. Res. Atmos. 2010, 115, D11304. [Google Scholar] [CrossRef]
- Dupart, Y. Mineral dust photochemistry induces nucleation events in the presence of SO2. Proc. Natl. Acad. Sci. USA 2012, 109, 20842–20847. [Google Scholar] [CrossRef]
- Shi, Z. Formation of iron nanoparticles and increase in iron reactivity in mineral dust during simulated cloud processing. Environ. Sci. Technol. 2009, 43, 6592–6596. [Google Scholar] [CrossRef]
- Mackie, D.S. Simulating the cloud processing of iron in Australian dust: pH and dust concentration. Geophys. Res. Lett. 2005, 32, L06809. [Google Scholar] [CrossRef]
- Spokes, L.J.; Jickells, T.D. Factors controlling the solubility of aerosol trace metals in the atmosphere and on mixing into seawater. Aquat. Geochem. 1995, 1, 355–374. [Google Scholar] [CrossRef]
- Winton, V.H.L. Multiple sources of soluble atmospheric iron to Antarctic waters. Glob. Biogeochem. Cycles 2016, 30, 421–437. [Google Scholar] [CrossRef]
- Sedwick, P.N.; Sholkovitz, E.R.; Church, T.M. Impact of anthropogenic combustion emissions on the fractional solubility of aerosol iron: Evidence from the Sargasso Sea. Geochem. Geophys. Geosyst. 2007, 8, Q10Q06. [Google Scholar] [CrossRef]
- Fu, H.B. Fractional iron solubility of aerosol particles enhanced by biomass burning and ship emission in Shanghai, East China. Sci. Total Environ. 2014, 481, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Shafizadeh, F. Production of levoglucosan and glucose from pyrolysis of cellulosic materials. J. Appl. Polym. Sci. 1979, 23, 3525–3539. [Google Scholar] [CrossRef]
- Simoneit, B.R.T. Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles. Atmos. Environ. 1999, 33, 173–182. [Google Scholar] [CrossRef]
- Rodriguez, E.S. Analysis of levoglucosan and its isomers in atmospheric samples by ion chromatography with electrospray lithium cationisation—Triple quadrupole tandem mass spectrometry. J. Chromatogr. A 2019, 1610, 460557. [Google Scholar] [CrossRef]
- Yu, X. Recyclable silver nanoplate-decorated copper membranes for solid-phase extraction coupled with surface-enhanced Raman scattering detection. Anal. Methods 2018, 10, 1353–1361. [Google Scholar] [CrossRef]
- Liang, L. Biomass burning impacts on ambient aerosol at a background site in East China: Insights from a yearlong study. Atmos. Res. 2020, 231, 104660. [Google Scholar] [CrossRef]
- Catelani, T.; Pratesi, G.; Zoppi, M. Raman Characterization of Ambient Airborne Soot and Associated Mineral Phases. Aerosol Sci. Technol. 2014, 48, 13–21. [Google Scholar] [CrossRef]
- Desboeufs, K.; Cautenet, G. Transport and mixing zone of desert dust and sulphate over Tropical Africa and the Atlantic Ocean region. Atmos. Chem. Phys. 2005, 5, 5615–5644. [Google Scholar] [CrossRef]
- Siefert, R.L.; Johansen, A.M.; Hoffmann, M.R. Chemical characterization of ambient aerosol collected during the southwest monsoon and intermonsoon seasons over the Arabian Sea: Labile-Fe(II) and other trace metals. J. Geophys. Res. Atmos. 1999, 104, 3511–3526. [Google Scholar] [CrossRef]
- Takahashi, Y. Seasonal changes in Fe species and soluble Fe concentration in the atmosphere in the Northwest Pacific region based on the analysis of aerosols collected in Tsukuba, Japan. Atmos. Chem. Phys. 2013, 13, 7695–7710. [Google Scholar] [CrossRef]
- Rudnick, R.; Gao, S. Composition of the Continental Crust. Treatise Geochem 3:1-64. Treatise Geochem. 2003, 3, 1–64. [Google Scholar]
- Mackie, D.S. Soil abrasion and eolian dust production: Implications for iron partitioning and solubility. Geochem. Geophys. Geosyst. 2006, 7, Q12Q03. [Google Scholar] [CrossRef]
- Vassilev, S.V. An overview of the composition and application of biomass ash. Part 1. Phase–mineral and chemical composition and classification. Fuel 2013, 105, 40–76. [Google Scholar] [CrossRef]
- Vassilev, S.V.; Vassileva, C.G.; Baxter, D. Trace element concentrations and associations in some biomass ashes. Fuel 2014, 129, 292–313. [Google Scholar] [CrossRef]
- Butler, A. Acquisition and Utilization of Transition Metal Ions by Marine Organisms. Science 1998, 281, 207–209. [Google Scholar] [CrossRef]
- Morel, F.M.M.; Milligan, A.J.; Saito, M.A. 6.05—Marine Bioinorganic Chemistry: The Role of Trace Metals in the Oceanic Cycles of Major Nutrients. In Treatise on Geochemistry; Holland, H.D., Turekian, K.K., Eds.; Pergamon: Oxford, UK, 2003. [Google Scholar]
- Fishwick, M.P. Impact of surface ocean conditions and aerosol provenance on the dissolution of aerosol manganese, cobalt, nickel and lead in seawater. Mar. Chem. 2018, 198, 28–43. [Google Scholar] [CrossRef]
- ARC Centre for Excellence for Climate System Science. Available online: https://www.climatescience.org.au/content/1130-reef-rainforest-campaign (accessed on 13 April 2020).
- AIRBOX, Reef to Reefforest. Available online: https://airbox.earthsci.unimelb.edu.au/reef-to-rainforest/ (accessed on 13 April 2020).
- AIRBOX A Mobile Air Chemistry Laboratory. Available online: https://airbox.earthsci.unimelb.edu.au/ (accessed on 13 April 2020).
- Duce, R.A. The atmospheric input of trace species to the world ocean. Glob. Biogeochem. Cycles 1991, 5, 193–259. [Google Scholar] [CrossRef]
- Baker, A.R. Dry and wet deposition of nutrients from the tropical Atlantic atmosphere: Links to primary productivity and nitrogen fixation. Deep Sea Res. Part I Oceanogr. Res. Pap. 2007, 54, 1704–1720. [Google Scholar] [CrossRef]
- Perron, M.M.G. Origin, transport and deposition of aerosol iron to Australian coastal waters. Atmos. Environ. 2020, 228, 117432. [Google Scholar] [CrossRef]
- Morton, P.L. Methods for the sampling and analysis of marine aerosols: Results from the 2008 GEOTRACES aerosol intercalibration experiment. Limnol. Oceanogr. Methods 2013, 11, 62–78. [Google Scholar] [CrossRef]
- Cutter, G. Sampling and Sample-Handling Protocols for GEOTRACES Cruises. Version 3. Available online: https://epic.awi.de/id/eprint/34484/ (accessed on 13 April 2020).
- Angel, B. Spatial variability of cadmium, copper, manganese, nickel and zinc in the Port Curtis Estuary, Queensland, Australia. Mar. Freshw. Res. 2010, 61, 170–183. [Google Scholar] [CrossRef]
- Stein, A.F. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-time Environmental Applications and Display System: Ready. Environ. Model. Softw. 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Bowie, A.R. Modern sampling and analytical methods for the determination of trace elements in marine particulate material using magnetic sector inductively coupled plasma–mass spectrometry. Anal. Chim. Acta 2010, 676, 15–27. [Google Scholar] [CrossRef]
- Keene, W.C. Sea-salt corrections and interpretation of constituent ratios in marine precipitation. J. Geophys. Res. Atmos. 1986, 91, 6647–6658. [Google Scholar] [CrossRef]
- Thermo Scientific Model 5012 MAAP Multi Angle Absorption Photometer. Available online: https://www.aires.com.br/wp-content/uploads/2018/01/5012-MAAP.pdf (accessed on 13 April 2020).
- Aerodyne Research, Aerosol Mass Spectrometer. Available online: http://www.aerodyne.com/products/aerosol-mass-spectrometer (accessed on 13 April 2020).
- Alfarra, M.R. Identification of the Mass Spectral Signature of Organic Aerosols from Wood Burning Emissions. Environ. Sci. Technol. 2007, 41, 5770–5777. [Google Scholar] [CrossRef]
- Goldstein, J.I. Scanning Electron Microscopy and X-Ray Microanalysis, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Wedepohl, H.K. The composition of the continental crust. Geochim. Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Buck, C.S. Trace element concentrations, elemental ratios, and enrichment factors observed in aerosol samples collected during the US GEOTRACES eastern Pacific Ocean transect (GP16). Chem. Geol. 2019, 511, 212–224. [Google Scholar] [CrossRef]
- Evans, J.D. Straightforward Statistics for the Behavioral Sciences; Thomson Brooks/Cole Publishing Co.: Belmont, CA, USA, 1996. [Google Scholar]
- Goldich, S.S. A Study in Rock-Weathering. J. Geol. 1938, 46, 17–58. [Google Scholar] [CrossRef]
- Mahowald, N.M. Atmospheric iron deposition: Global distribution, variability, and human perturbations. Annu. Rev. Mar. Sci. 2009, 1, 245–278. [Google Scholar] [CrossRef] [PubMed]
- Mahowald, N.M. Aerosol trace metal leaching and impacts on marine microorganisms. Nat. Commun. 2018, 9, 2614. [Google Scholar] [CrossRef]
- Srinivas, B.; Sarin, M.M.; Kumar, A. Impact of anthropogenic sources on aerosol iron solubility over the Bay of Bengal and the Arabian Sea. Biogeochemistry 2012, 110, 257–268. [Google Scholar] [CrossRef]
- Schneider, L.; Allen, K.; Haberle, S. Mercury Pollution from Decades Past May Have Been re-Released by Tasmania’s Bushfires. The Conversations 2019. Available online: https://theconversation.com/mercury-pollution-from-decades-past-may-have-been-re-released-by-tasmanias-bushfires-114603 (accessed on 13 April 2020).
- Schneider, L. How significant is atmospheric metal contamination from mining activity adjacent to the Tasmanian Wilderness World Heritage Area? A spatial analysis of metal concentrations using air trajectories models. Sci. Total Environ. 2019, 656, 250–260. [Google Scholar] [CrossRef]
- Available online: https://asmc.com.au/ (accessed on 13 April 2020).
- Chen, Z. Characterization of aerosols over the Great Barrier Reef: The influence of transported continental sources. Sci. Total Environ. 2019, 690, 426–437. [Google Scholar] [CrossRef]
- Pósfai, M. Individual aerosol particles from biomass burning in southern Africa: 1. Compositions and size distributions of carbonaceous particles. J. Geophys. Res. 2003, 109, 1–9. [Google Scholar] [CrossRef]
- Pósfai, M. Atmospheric tar balls: Particles from biomass and biofuel burning. J. Geophys. Res. Atmos. 2004, 109, D06213. [Google Scholar] [CrossRef]
- Cullis, C.F.; Hirschler, M.M. Atmospheric sulphur: Natural and man-made sources. Atmos. Environ. 1980, 14, 1263–1278. [Google Scholar] [CrossRef]
- Galbally, I.; Roy, C.R. The fate of nitrogen compounds in the atmosphere. In Gaseous Loss of Nitrogen from Plant-Soil Systems; Springer: Dordrecht, The Netherlands, 1983. [Google Scholar]
- Viemeister, P.E. Lightning and the origin of nitrates found in precipitation. J. Meteorol. 1960, 17, 681–683. [Google Scholar] [CrossRef][Green Version]
- Butterbach-Bahl, K. Nitrous oxide emissions from soils: How well do we understand the processes and their controls? Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 368, 20130122. [Google Scholar] [CrossRef] [PubMed]
- Jickells, T. The cycling of organic nitrogen through the atmosphere. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 368, 20130115. [Google Scholar] [CrossRef] [PubMed]
- Sholkovitz, E.R. Fractional solubility of aerosol iron: Synthesis of a global-scale data set. Geochim. Cosmochim. Acta 2012, 89, 173–189. [Google Scholar] [CrossRef]
- Theodosi, C.; Markaki, Z.; Mihalopoulos, N. Iron speciation, solubility and temporal variability in wet and dry deposition in the Eastern Mediterranean. Mar. Chem. 2010, 120, 100–107. [Google Scholar] [CrossRef]
- Séguret, M.J.M. Iron solubility in crustal and anthropogenic aerosols: The Eastern Mediterranean as a case study. Mar. Chem. 2011, 126, 229–238. [Google Scholar] [CrossRef]
- Heimburger, A.; Losno, R.; Triquet, S. Solubility of iron and other trace elements in rainwater collected on the Kerguelen Islands (South Indian Ocean). Biogeosciences 2013, 10, 6617–6628. [Google Scholar] [CrossRef]
- Shotyk, W. A peat bog record of natural, pre-anthropogenic enrichments of trace elements in atmospheric aerosols since 12 370 14C yr BP, and their variation with Holocene climate change. Earth Planet. Sci. Lett. 2002, 199, 21–37. [Google Scholar] [CrossRef]
- Boutron, C.F. Changes in cadmium concentrations in Antarctic ice and snow during the past 155,000 years. Earth Planet. Sci. Lett. 1993, 117, 431–441. [Google Scholar] [CrossRef]
- Haynes, D.; Johnson, J. Organochlorine, Heavy Metal and Polyaromatic Hydrocarbon Pollutant Concentrations in the Great Barrier Reef (Australia) Environment: A Review. Mar. Pollut. Bull. 2000, 41, 267–278. [Google Scholar] [CrossRef]
- Fabricius, K.E. Changes in water clarity in response to river discharges on the Great Barrier Reef continental shelf: 2002–2013, Estuarine. Coast. Shelf Sci. 2016, 173, A1–A15. [Google Scholar] [CrossRef]
- Mitchelmore, C.; Verde, A.; Weis, V. Uptake and partitioning of copper and cadmium in the coral Pocillopora damicornis. Aquat. Toxicol. 2007, 85, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Denton, G.R.W.; Burdon-Jones, C. Trace metals in corals from the Great Barrier Reef. Mar. Pollut. Bull. 1986, 17, 209–213. [Google Scholar] [CrossRef]
- Esslemont, G. Heavy metals in seawater, marine sediments and corals from the Townsville section, Great Barrier Reef Marine Park, Queensland. Mar. Chem. 2000, 71, 215–231. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All | Marine | Sea Breeze | Terrestrial | Land Breeze | |
|---|---|---|---|---|---|
| df (n − 2) | 16 | 5 | 4 | ||
| (p = 0.05) | 0.468 | 0.755 | 0.811 | ||
| Cd | 0.574 | 0.582 | 0.964 | 0.711 | 0.603 |
| Co | 0.546 | 0.571 | 0.0894 | 0.720 | 0.648 |
| Cu | 0.575 | 0.651 | 0.555 | −0.315 | 0.321 |
| Mn | 0.576 | 0.582 | 0.122 | 0.878 | 0.112 |
| Mo | 0.624 | 0.629 | 0.608 | 0.702 | 0.005 |
| Pb | 0.667 | 0.667 | 0.604 | 0.540 | 0.833 |
| V | 0.618 | 0.651 | 0.892 | 0.833 | 0.711 |
| Zn | 0.677 | 0.675 | 0.786 | 0.579 | 0.167 |
| Element | Riverine (Dissolved) * | Dry Deposition (Labile) | Wet Deposition (Soluble) |
|---|---|---|---|
| trace metal deposition (μmol m−2 y−1) | |||
| Cd | 0.19 ± 0.17 | 0.04 ± 0.06 | 1.26 ± 0.54 |
| Cu | 22.0 ± 4.9 | 0.6 ± 0.4 | 8.0 ± 0.7 |
| Zn | 5.5 ± 2.5 | 7.9 ± 1.4 | 40.1 ± 5.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strzelec, M.; Proemse, B.C.; Gault-Ringold, M.; Boyd, P.W.; Perron, M.M.G.; Schofield, R.; Ryan, R.G.; Ristovski, Z.D.; Alroe, J.; Humphries, R.S.; et al. Atmospheric Trace Metal Deposition near the Great Barrier Reef, Australia. Atmosphere 2020, 11, 390. https://doi.org/10.3390/atmos11040390
Strzelec M, Proemse BC, Gault-Ringold M, Boyd PW, Perron MMG, Schofield R, Ryan RG, Ristovski ZD, Alroe J, Humphries RS, et al. Atmospheric Trace Metal Deposition near the Great Barrier Reef, Australia. Atmosphere. 2020; 11(4):390. https://doi.org/10.3390/atmos11040390
Chicago/Turabian StyleStrzelec, Michal, Bernadette C. Proemse, Melanie Gault-Ringold, Philip W. Boyd, Morgane M. G. Perron, Robyn Schofield, Robert G. Ryan, Zoran D. Ristovski, Joel Alroe, Ruhi S. Humphries, and et al. 2020. "Atmospheric Trace Metal Deposition near the Great Barrier Reef, Australia" Atmosphere 11, no. 4: 390. https://doi.org/10.3390/atmos11040390
APA StyleStrzelec, M., Proemse, B. C., Gault-Ringold, M., Boyd, P. W., Perron, M. M. G., Schofield, R., Ryan, R. G., Ristovski, Z. D., Alroe, J., Humphries, R. S., Keywood, M. D., Ward, J., & Bowie, A. R. (2020). Atmospheric Trace Metal Deposition near the Great Barrier Reef, Australia. Atmosphere, 11(4), 390. https://doi.org/10.3390/atmos11040390