Aqueous-Phase Brown Carbon Formation from Aromatic Precursors under Sunlight Conditions




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Laboratory Experiments
2.3. Kinetic Analysis
2.4. Determination of Absorption Properties
3. Results and Discussion
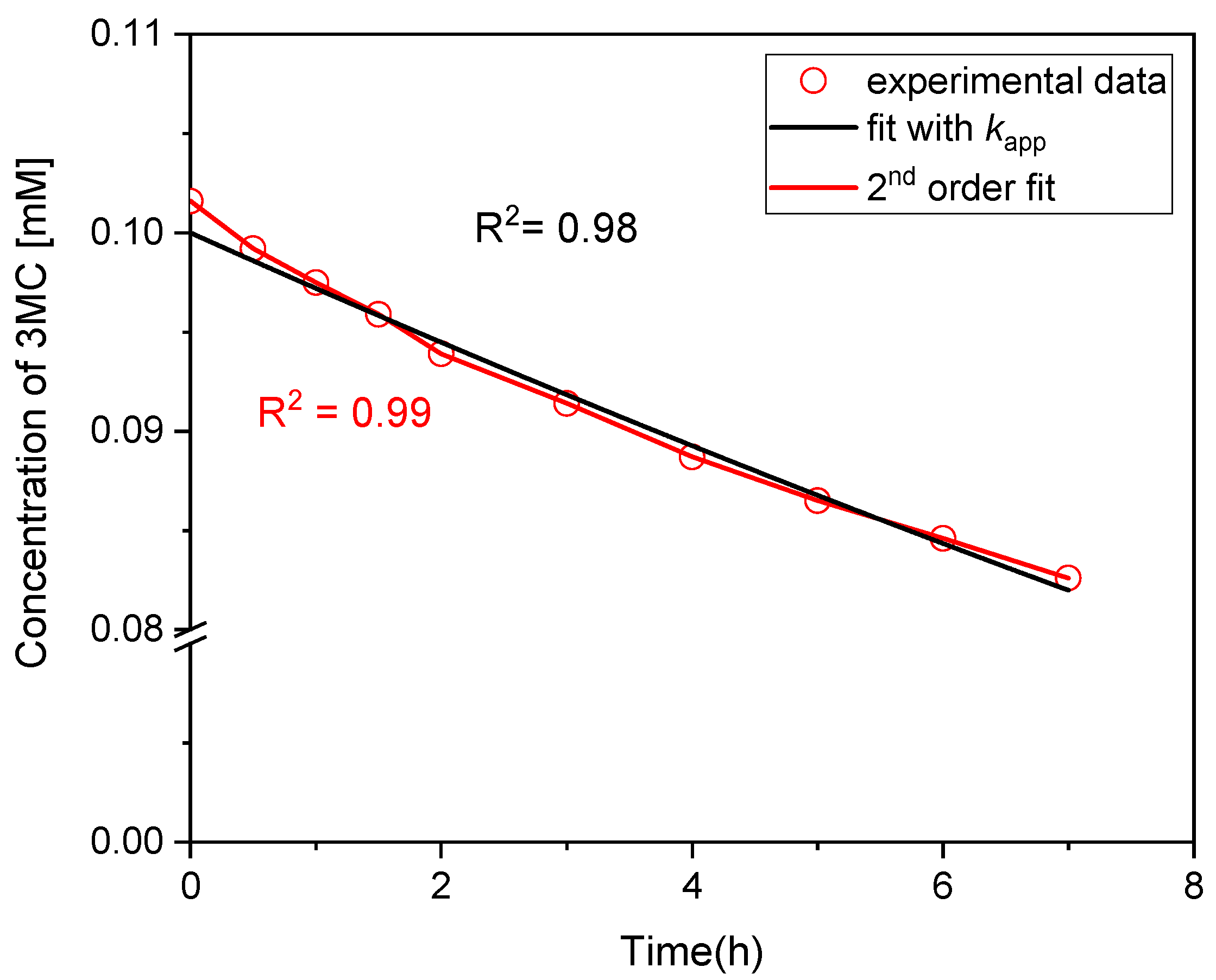
3.1. Kinetic Analysis of 3MC Photolysis in Mildly Acidic NaNO2 Solution
3.1.1. Kinetic Analysis at Low NaNO2 Concentrations
3.1.2. Kinetic Analysis at High NaNO2 Concentrations
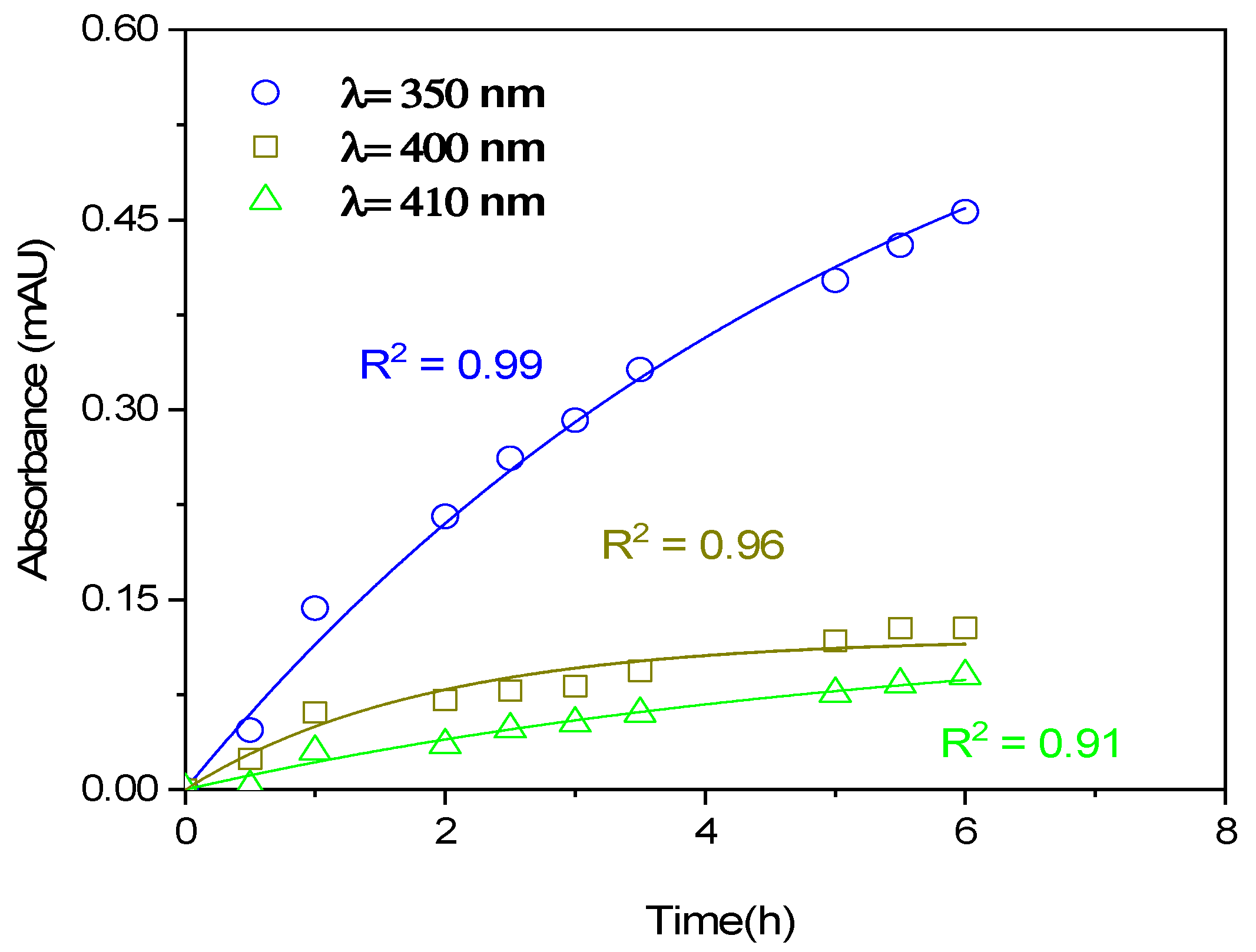
3.2. Absorption Spectra and BrC Formation
3.2.1. Low NaNO2 Concentrations
3.2.2. High NaNO2 Concentrations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laskin, A.; Laskin, J.; Nizkorodov, S.A. Chemistry of Atmospheric Brown Carbon. Chem. Rev. 2015, 115, 4335–4382. [Google Scholar] [CrossRef]
- Teich, M.; van Pinxteren, D.; Wang, M.; Kecorius, S.; Wang, Z.B.; Muller, T.; Mocnik, G.; Herrmann, H. Contributions of nitrated aromatic compounds to the light absorption of water-soluble and particulate brown carbon in different atmospheric environments in Germany and China. Atmos. Chem. Phys. 2017, 17, 1653–1672. [Google Scholar] [CrossRef]
- Xie, M.J.; Chen, X.; Hays, M.D.; Holder, A.L. Composition and light absorption of N-containing aromatic compounds in organic aerosols from laboratory biomass burning. Atmos. Chem. Phys. 2019, 19, 2899–2915. [Google Scholar] [CrossRef]
- Bond, T.C. Spectral dependence of visible light absorption by carbonaceous particles emitted from coal combustion. Geophys. Res. Lett. 2001, 28, 4075–4078. [Google Scholar] [CrossRef]
- Feng, Y.; Ramanathan, V.; Kotamarthi, V.R. Brown carbon: A significant atmospheric absorber of solar radiation? Atmos. Chem. Phys. 2013, 13, 8607–8621. [Google Scholar] [CrossRef]
- Andreae, M.O.; Gelencser, A. Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols. Atmos. Chem. Phys. 2006, 6, 3131–3148. [Google Scholar] [CrossRef]
- Saleh, R.; Robinson, E.S.; Tkacik, D.S.; Ahern, A.T.; Liu, S.; Aiken, A.C.; Sullivan, R.C.; Presto, A.A.; Dubey, M.K.; Yokelson, R.J. Brownness of organics in aerosols from biomass burning linked to their black carbon content. Nat. Geosci. 2014, 7, 647–650. [Google Scholar] [CrossRef]
- Pflieger, M.; Kroflič, A. Acute toxicity of emerging atmospheric pollutants from wood lignin due to biomass burning. J. Hazard. Mater. 2017, 338, 132–139. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Gu, R.; Wang, H.; Yao, L.; Wen, L.; Zhu, F.; Wang, W.; Xue, L.; Yang, L. Observations of fine particulate nitrated phenols in four sites in northern China: concentrations, source apportionment, and secondary formation. Atmos. Chem. Phys. 2018, 18, 4349–4359. [Google Scholar] [CrossRef]
- Saleh, R.; Hennigan, C.J.; McMeeking, G.R.; Chuang, W.K.; Robinson, E.S.; Coe, H.; Donahue, N.M.; Robinson, A.L. Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions. Atmos. Chem. Phys. 2013, 13, 7683–7693. [Google Scholar] [CrossRef]
- Kroflič, A.; Grilc, M.; Grgić, I. Unraveling Pathways of Guaiacol Nitration in Atmospheric Waters: Nitrite, A Source of Reactive Nitronium Ion in the Atmosphere. Environ. Sci. Technol. 2015, 49, 9150–9158. [Google Scholar] [CrossRef]
- McNeill, V.F. Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols. Environ. Sci. Technol. 2015, 49, 1237–1244. [Google Scholar] [CrossRef]
- Harrison, M.A.J.; Barra, S.; Borghesi, D.; Vione, D.; Arsene, C.; Olariu, R.L. Nitrated phenols in the atmosphere: a review. Atmos. Environ. 2005, 39, 231–248. [Google Scholar] [CrossRef]
- Hoffmann, E.H.; Tilgner, A.; Wolke, R.; Boge, O.; Walter, A.; Herrmann, H. Oxidation of substituted aromatic hydrocarbons in the tropospheric aqueous phase: Kinetic mechanism development and modelling. Phys. Chem. Chem. Phys. 2018, 20, 10960–10977. [Google Scholar] [CrossRef]
- Claeys, M.; Vermeylen, R.; Yasmeen, F.; Gomez-Gonzalez, Y.; Chi, X.G.; Maenhaut, W.; Meszaros, T.; Salma, I. Chemical characterisation of humic-like substances from urban, rural and tropical biomass burning environments using liquid chromatography with UV/vis photodiode array detection and electrospray ionisation mass spectrometry. Environ. Chem. 2012, 9, 273–284. [Google Scholar] [CrossRef]
- Kitanovski, Z.; Grgić, I.; Vermeylen, R.; Claeys, M.; Maenhaut, W. Liquid chromatography tandem mass spectrometry method for characterization of monoaromatic nitro-compounds in atmospheric particulate matter. J. Chromatogr. A 2012, 1268, 35–43. [Google Scholar] [CrossRef]
- Kahnt, A.; Behrouzi, S.; Vermeylen, R.; Shalamzari, M.S.; Vercauteren, J.; Roekens, E.; Claeys, M.; Maenhaut, W. One-year study of nitro-organic compounds and their relation to wood burning in PM10 aerosol from a rural site in Belgium. Atmos. Environ. 2013, 81, 561–568. [Google Scholar] [CrossRef]
- Chow, K.S.; Huang, X.H.H.; Yu, J.Z. Quantification of nitroaromatic compounds in atmospheric fine particulate matter in Hong Kong over 3 years: Field measurement evidence for secondary formation derived from biomass burning emissions. Environ. Chem. 2016, 13, 665–673. [Google Scholar] [CrossRef]
- Wang, X.F.; Gu, R.R.; Wang, L.W.; Xu, W.X.; Zhang, Y.T.; Chen, B.; Li, W.J.; Xue, L.K.; Chen, J.M.; Wang, W.X. Emissions of fine particulate nitrated phenols from the burning of five common types of biomass. Environ. Pollut. 2017, 230, 405–412. [Google Scholar] [CrossRef]
- Pereira, K.L.; Hamilton, J.F.; Rickard, A.R.; Bloss, W.J.; Alam, M.S.; Camredon, M.; Ward, M.W.; Wyche, K.P.; Munoz, A.; Vera, T.; et al. Insights into the Formation and Evolution of Individual Compounds in the Particulate Phase during Aromatic Photo-Oxidation. Environ. Sci. Technol. 2015, 49, 13168–13178. [Google Scholar] [CrossRef]
- Yuan, B.; Liggio, J.; Wentzell, J.; Li, S.-M.; Stark, H.; Roberts, J.M.; Gilman, J.; Lerner, B.; Warneke, C.; Li, R. Secondary formation of nitrated phenols: Insights from observations during the Uintah Basin Winter Ozone Study (UBWOS) 2014. Atmos. Chem. Phys. 2016, 16, 2139–2153. [Google Scholar] [CrossRef]
- Frka, S.; Šala, M.; Kroflič, A.; Huš, M.; Čusak, A.; Grgić, I. Quantum Chemical Calculations Resolved Identification of Methylnitrocatechols in Atmospheric Aerosols. Environ. Sci. Technol. 2016, 50, 5526–5535. [Google Scholar] [CrossRef] [PubMed]
- Kroflič, A.; Huš, M.; Grilc, M.; Grgić, I. Underappreciated and Complex Role of Nitrous Acid in Aromatic Nitration under Mild Environmental Conditions: The Case of Activated Methoxyphenols. Environ. Sci. Technol. 2018, 52, 13756–13765. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.B.; Tan, Y.; Perri, M.J.; Seitzinger, S.P.; Turpin, B.J. Aqueous chemistry and its role in secondary organic aerosol (SOA) formation. Atmos. Chem. Phys. 2010, 10, 10521–10539. [Google Scholar] [CrossRef]
- Ervens, B.; Turpin, B.J.; Weber, R.J. Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): A review of laboratory, field and model studies. Atmos. Chem. Phys. 2011, 11, 11069–11102. [Google Scholar] [CrossRef]
- Iinuma, Y.; Boge, O.; Grafe, R.; Herrmann, H. Methyl-Nitrocatechols: Atmospheric Tracer Compounds for Biomass Burning Secondary Organic Aerosols. Environ. Sci. Technol. 2010, 44, 8453–8459. [Google Scholar] [CrossRef]
- Vidović, K.; Jurković, D.L.; Šala, M.; Kroflič, A.; Grgić, I. Nighttime Aqueous-Phase Formation of Nitrocatechols in the Atmospheric Condensed Phase. Environ. Sci. Technol. 2018, 52, 9722–9730. [Google Scholar] [CrossRef]
- Vidović, K.; Kroflič, A.; Jovanovič, P.; Šala, M.; Grgić, I. Electrochemistry as a Tool for Studies of Complex Reaction Mechanisms: The Case of the Atmospheric Aqueous-Phase Aging of Catechols. Environ. Sci. Technol. 2019, 53, 11195–11203. [Google Scholar] [CrossRef]
- Albinet, A.; Minero, C.; Vione, D. UVA irradiation induces direct phototransformation of 2, 4-dinitrophenol in surface water samples. Chemosphere 2010, 80, 759–763. [Google Scholar] [CrossRef]
- Zhao, S.F.; Ma, H.J.; Wang, M.; Cao, C.Q.; Xiong, J.; Xu, Y.S.; Yao, S.D. Study on the mechanism of photo-degradation of p-nitrophenol exposed to 254 nm UV light. J. Hazard. Mater. 2010, 180, 86–90. [Google Scholar] [CrossRef]
- Slade, J.H.; Knopf, D.A. Heterogeneous OH oxidation of biomass burning organic aerosol surrogate compounds: Assessment of volatilisation products and the role of OH concentration on the reactive uptake kinetics. Phys. Chem. Chem. Phys. 2013, 15, 5898–5915. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Lee, A.K.Y.; Huang, L.; Li, X.; Yang, F.; Abbatt, J.P.D. Photochemical processing of aqueous atmospheric brown carbon. Atmos. Chem. Phys. 2015, 15, 6087–6100. [Google Scholar] [CrossRef]
- Hems, R.F.; Abbatt, J.P.D. Aqueous Phase Photo-oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption. ACS Earth Space Chem. 2018, 2, 225–234. [Google Scholar] [CrossRef]
- Palumbo, A.; Napolitano, A.; d’Ischia, M. Nitrocatechols versus nitrocatecholamines as novel competitive inhibitors of neuronal nitric oxide synthase: Lack of the aminoethyl side chain determines loss of tetrahydrobiopterin-antagonizing properties. Bioorg. Med. Chem. Lett. 2002, 12, 13–16. [Google Scholar] [CrossRef]
- Kitanovski, Z.; Grgić, I.; Yasmeen, F.; Claeys, M.; Čusak, A. Development of a liquid chromatographic method based on ultraviolet-visible and electrospray ionization mass spectrometric detection for the identification of nitrocatechols and related tracers in biomass burning atmospheric organic aerosol. Rapid Commun. Mass Spectrom. 2012, 26, 793–804. [Google Scholar] [CrossRef]
- Kitanovski, Z.; Čusak, A.; Grgić, I.; Claeys, M. Chemical characterization of the main products formed through aqueous-phase photonitration of guaiacol. Atmos. Meas. Tech. 2014, 7, 2457–2470. [Google Scholar] [CrossRef]
- Vione, D.; Maurino, V.; Minero, C.; Pelizzetti, E. Phenol photonitration upon UV irradiation of nitrite in aqueous solution I: Effects of oxygen and 2-propanol. Chemosphere 2001, 45, 893–902. [Google Scholar] [CrossRef]
- He, L.; Schaefer, T.; Otto, T.; Kroflič, A.; Herrmann, H. Kinetic and Theoretical Study of the Atmospheric Aqueous-Phase Reactions of OH Radicals with Methoxyphenolic Compounds. J. Phys. Chem. A 2019, 123, 7828–7838. [Google Scholar] [CrossRef]
- Kroflič, A.; Schaefer, T.; Huš, M.; Phuoc Le, H.; Otto, T.; Herrmann, H. OH radicals reactivity towards phenol-related pollutants in water: temperature dependence of the rate constants and novel insights into the [OH–phenol]˙ adduct formation. Phys. Chem. Chem. Phys. 2020, 22, 1324–1332. [Google Scholar] [CrossRef]
- Nakayama, T.; Sato, K.; Matsumi, Y.; Imamura, T.; Yamazaki, A.; Uchiyama, A. Wavelength and NOx dependent complex refractive index of SOAs generated from the photooxidation of toluene. Atmos. Chem. Phys. 2013, 13, 531–545. [Google Scholar] [CrossRef]
- Li, Y.J.; Huang, D.D.; Cheung, H.Y.; Lee, A.K.Y.; Chan, C.K. Aqueous-phase photochemical oxidation and direct photolysis of vanillin—A model compound of methoxy phenols from biomass burning. Atmos. Chem. Phys. 2014, 14, 2871–2885. [Google Scholar] [CrossRef]
- Bluvshtein, N.; Lin, P.; Flores, J.M.; Segev, L.; Mazar, Y.; Tas, E.; Snider, G.; Weagle, C.; Brown, S.S.; Laskin, A.; et al. Broadband optical properties of biomass-burning aerosol and identification of brown carbon chromophores. J. Geophys. Res. Atmos. 2017, 122, 5441–5456. [Google Scholar] [CrossRef]
- Liu, J.M.; Lin, P.; Laskin, A.; Laskin, J.; Kathmann, S.M.; Wise, M.; Caylor, R.; Imholt, F.; Selimovic, V.; Shilling, J.E. Optical properties and aging of light-absorbing secondary organic aerosol. Atmos. Chem. Phys. 2016, 16, 12815–12827. [Google Scholar] [CrossRef]
- Lin, P.; Liu, J.M.; Shilling, J.E.; Kathmann, S.M.; Laskin, J.; Laskin, A. Molecular characterization of brown carbon (BrC) chromophores in secondary organic aerosol generated from photo-oxidation of toluene. Phys. Chem. Chem. Phys. 2015, 17, 23312–23325. [Google Scholar] [CrossRef]
- Jiang, H.H.; Frie, A.L.; Lavi, A.; Chen, J.Y.; Zhang, H.F.; Bahreini, R.; Lin, Y.H. Brown Carbon Formation from Nighttime Chemistry of Unsaturated Heterocyclic Volatile Organic Compounds. Environ. Sci. Technol. Lett. 2019, 6, 184–190. [Google Scholar] [CrossRef]
- Hoffer, A.; Gelencser, A.; Guyon, P.; Kiss, G.; Schmid, O.; Frank, G.P.; Artaxo, P.; Andreae, M.O. Optical properties of humic-like substances (HULIS) in biomass-burning aerosols. Atmos. Chem. Phys. 2006, 6, 3563–3570. [Google Scholar] [CrossRef]
- Lack, D.A.; Richardson, M.S.; Law, D.; Langridge, J.M.; Cappa, C.D.; McLaughlin, R.J.; Murphy, D.M. Aircraft Instrument for Comprehensive Characterization of Aerosol Optical Properties, Part 2: Black and Brown Carbon Absorption and Absorption Enhancement Measured with Photo Acoustic Spectroscopy. Aerosol Sci. Technol. 2012, 46, 555–568. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | 3MC [mM] | NaNO2 [mM] | 3M5NC/3M4NC | pH | MNC Product Yield after 8 h |
|---|---|---|---|---|---|
| 1 | 0.1 | 0.1 | 5 | 3.9 | 0.17 |
| 2 | 0.1 | 0.5 | 6 | 4.5 | 0.45 |
| 3 | 0.1 | 1.0 | 96 | 4.6 | 0.40 |
| 4 | 0.1 | 2.0 | 98 | 4.9 | 0.36 |
| 5 | 0.1 | 5.0 | 63 | 5 | 0.27 |
| Experiment | NaNO2 [mM] | Sunlight | kapp (s−1) | k2nd (M−1s−1) | k2nd(global) (M−1·s−1) |
|---|---|---|---|---|---|
| 1 | 0.1 | YES | 7.88 × 10−6 | 0.168 | − |
| 2 | 0.5 | YES | 4.47 × 10−5 | 0.089 | 0.075 |
| 3 | 1.0 | YES | 7.30 × 10−5 | 0.073 | |
| 4 | 2.0 | YES | 1.30 × 10−4 | 0.065 | |
| 5 | 5.0 | YES | 2.40 × 10−4 | 0.048 | |
| 6 | 1.0 | NO | 4.26 × 10−5 | 0.043 | 0.032 |
| 7 | 2.0 | NO | 5.03 × 10−5 | 0.025 |
| Experiment | NaNO2 (mM) | Sunlight | MACBrC(350nm) (m2/g) | MACBrC(400nm) (m2/g) | MACBrC(400nm) (m2/g) |
|---|---|---|---|---|---|
| 1 | 0.1 | YES | 1.80 | 0.87 | 0.43 |
| 2 | 0.5 | YES | 3.10 | 1.26 | 0.94 |
| 3 | 1 | YES | 3.44 | 1.34 | 1.02 |
| 4 | 2 | YES | 3.77 | 1.54 | 1.17 |
| 5 | 5 | YES | 4.09 | 1.74 | 1.42 |
| 6 | 1 | NO | 5.41 | 1.73 | 1.75 |
| 7 | 2 | NO | 5.63 | 1.90 | 1.38 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidović, K.; Kroflič, A.; Šala, M.; Grgić, I. Aqueous-Phase Brown Carbon Formation from Aromatic Precursors under Sunlight Conditions. Atmosphere 2020, 11, 131. https://doi.org/10.3390/atmos11020131
Vidović K, Kroflič A, Šala M, Grgić I. Aqueous-Phase Brown Carbon Formation from Aromatic Precursors under Sunlight Conditions. Atmosphere. 2020; 11(2):131. https://doi.org/10.3390/atmos11020131
Chicago/Turabian StyleVidović, Kristijan, Ana Kroflič, Martin Šala, and Irena Grgić. 2020. "Aqueous-Phase Brown Carbon Formation from Aromatic Precursors under Sunlight Conditions" Atmosphere 11, no. 2: 131. https://doi.org/10.3390/atmos11020131
APA StyleVidović, K., Kroflič, A., Šala, M., & Grgić, I. (2020). Aqueous-Phase Brown Carbon Formation from Aromatic Precursors under Sunlight Conditions. Atmosphere, 11(2), 131. https://doi.org/10.3390/atmos11020131