Abstract
A large portion of atmospheric aerosol particles consists of secondary material produced by oxidation reactions. The relative importance of secondary organic aerosol (SOA) can increase with improved emission regulations. A relatively simple way to study potential particle formation in the atmosphere is by using oxidation flow reactors (OFRs) which simulate atmospheric ageing. Here we report on the first ambient OFR ageing experiment in Europe, coupled with scanning mobility particle sizer (SMPS), aerosol mass spectrometer (AMS) and proton transfer reaction (PTR)-MS measurements. We found that the simulated ageing did not produce any measurable increases in particle mass or number concentrations during the two months of the campaign due to low concentrations of precursors. Losses in the reactor increased with hydroxyl radical (OH) exposure and with increasing difference between ambient and reactor temperatures, indicating fragmentation and evaporation of semivolatile material.
1. Introduction
Submicron particle mass is often dominated by secondary material formed from atmospheric oxidation of both organic and inorganic precursors [1,2]. Despite major research advances in the fields of atmospheric organic chemistry and secondary organic aerosols (SOA) during the last decade, knowledge and understanding is far from complete [3,4,5]. Knowledge gaps include speciation and the chemical and physical processes governing both formation and removal of SOA mass. Organic precursors (volatile organic compounds, VOCs) are emitted from both anthropogenic and biogenic sources, while inorganic sources are mainly anthropogenic.
In recent years, several research groups have employed oxidation flow reactors (OFRs) to study secondary aerosol particle properties. The most common OFR to date is the potential aerosol mass (PAM) reactor. High oxidant concentrations and a continuous flow enables faster experiments and higher degree of oxidation. OFRs have proved valuable as an alternative or complement to the traditionally used large smog chambers [6,7]. Although mobile smog chambers exist [8], they are not portable in the same way as an OFR. By deploying OFRs at field sites, ideally with complimentary gas phase measurements, the potential aerosol formation of the ambient atmosphere can be studied directly, thereby overriding some of the difficulties in translating simplified laboratory experiments to atmospheric implications. To date, only a handful of such field deployments target ambient air, as opposed to a specific emitter (such as vehicles or biomass burning), have been performed. Since the ambient aerosol is always aged to a certain extent, less pronounced particle production can be expected in environments far away from precursor sources. Most campaigns have seen a net mass loss at the highest OH exposures, consistent with laboratory measurements [7], which is interpreted as a shift from functionalization to fragmentation of SOA precursors. In urban areas, Ortega et al. [9] measured organic mass enhancement factors in Los Angeles of around 1.3 during days and 1.8 during night-time, while George et al. [10] saw significant oxidation but little or no mass enhancement in downtown Toronto. In a pine forest in Colorado, dominated by biogenic sources, the SOA mass enhancement scaled with measured monoterpenes, although OH induced oxidation of unmeasured semi- or intermediate volatility organic compounds was needed to explain the formed mass [11,12]. Similarly to the Los Angeles study, the enhancement was larger at night-time. Using a different reactor, Slowik et al. [13] saw significant losses of organics in a remote Canadian forest influenced by biogenic sources, which they attributed to a volatilization from an increased temperature and OH oxidation. In central Amazonia, Palm et al. [14] again saw maximum enhancement at night-time, while daytime oxidation produced much less SOA, especially during the wet season. Kang et al. [15], measuring in the Yellow Sea, saw net losses of organics but an increase in sulphate aerosol mass. With OFRs now being produced commercially, these types of measurements can be expected to become more frequent.
Here we report, to the best of our knowledge, the first ambient OFR campaign in Europe. Ambient aerosol measurements, alternating through a PAM OFR, were performed during July and August of 2015 at a willow (Salix viminalis) bioenergy plantation in southwestern Sweden. We aimed to study the aerosol formation potential of background air at a rural site, as well as the influence of the fast-growing energy crops. We compare the results to previous literature and discuss correlations between the (lack of) enhancement with temperature, wind and ambient aerosol levels. The sensitivity of ambient OFR measurements, expressed as required precursor concentration and yield as a function of ambient condensation sink, is also estimated.
2. Experimental Methods
2.1. Campaign Site and Set-Up
The measurement site located in Skrehalla (58°17′ N, 12°46′ E) is surrounded mostly by arable land. It is about 80 km north–east of Gothenburg (580 k inhabitants), 30 km to the nearest city (Trollhättan, 49 k inhabitants) and 10 km to three smaller villages (1–3 k inhabitants). A major road from Gothenburg to Stockholm is located 10 km southeast of the site, with around 10 k vehicles per day at the nearest section during summer (accessed on 22 September 2017. Available online: http://vtf.trafikverket.se/). The Salix plantation, about six hectares, was in its third growing season at the time of the campaign and more than two meters tall. Measurements took place during July–August 2015. Meteorological data was retrieved from the SMHI (Swedish Meteorological and Hydrological Institute) station in Hällum, 16 km to the east of the measurement site.
All instruments were placed in an air-conditioned laboratory container. The inlet consisted of quarter inch annealed 316(L) stainless steel tubing and a funnel installed to sample upwards, just above the salix canopy. The flow through the inlet was 6.3 lpm (assumed laminar flow with an approximate Reynolds number of 2100), and tubing length was about 4.5 m which gives a residence time in the inlet tubing of 0.6 s. The instruments were checked remotely, but also serviced once a week. Data from a total of 27 days were analyzed.
2.2. Instrumentation
PAM reactors have been extensively used and characterized during the last years [16,17,18,19]. The version used is a 13 l aluminum cylinder with two UV lamps (peak intensities at 185 and 254 nm) mounted inside to produce ozone and hydroxyl radicals from oxygen and water. The OH exposure was calibrated offline in a laboratory environment, using the decay of 2 ppb SO2. OH exposure during field measurements was calculated using a parameterization from the calibration and measured absolute humidity and ozone. OH exposure during the campaign was mostly in the range 1 × 1011–1 × 1012 molecules cm−3 s. In order to scan OH exposure and potential particle formation, the voltage across the lamps was automatically changed in steps according to a programmed schedule. Flow through the reactor was 5 lpm. Sampling through the reactor during 60 min was alternated by 20 min of ambient sampling. During analysis, the first ten minutes of reactor sampling and first five minutes of ambient sampling were not considered, in order to give flows time to stabilize.
A scanning mobility particle sizer (SMPS, [20]) and a high-resolution time-of-flight aerosol mass spectrometer (AMS from here on, [21]) were used to monitor particle characteristics. The SMPS, consisting of a DMA (TSI 3071) and a CPC (TSI 3775), was used to measure the number size distribution of particles between 11–600 nm in electrical mobility diameter, while the AMS measured chemically resolved mass concentrations in the approximate range 50–1000 nm (vacuum aerodynamic diameter) with less than 100% transmission at the low and high end of the size spectrum. Although ambient number concentrations apparently approached zero at the high end of the SMPS mass spectrum (Figure S1), a small number of large particles (likely measurement artefacts) distorted the volume size distribution. The error in number concentration from this distortion is negligible and relative changes in volume are not affected, but the absolute volume measurements are likely overestimated by on average 20%. Due to the low mass concentration of Aitken mode particles, the AMS lens penetration is not believed to have been an issue. In order to measure dry aerosol particles, a Permapure drier was installed between the OFR and the SMPS and AMS. The AMS ionization efficiency was calibrated three times during the campaign. The airbeam signal and linear interpolations between these calibrations were used to correct the data.
A proton transfer reaction-time of flight-mass spectrometer (PTR-TOF-MS 8000; Ionicon Analytik GmbH, Innsbruck, Austria) was used to monitor ambient VOC concentrations during the experiment. The PTR-TOF-MS uses a soft ionization technique to protonate VOC in ambient air, and reaction products are mass-discriminated through a time-of-flight detection unit with a typical mass resolution of 4000 m/∆m [22,23]. The instrument was operated in a laboratory trailer and ambient air from above the plantation was sampled at a height of 4.7 m a.g.l. through a ca. 30 m long heated PFA inlet (1/2” OD, PFA-T8-062, Swagelok, OH, USA) with a high flow rate (20 lpm) from which the PTR-TOF took a subsample of 200 mL min−1. SO2 was measured using a UV fluorescent monitor (Environnement S.A AF22M). The concentration was, however, at all times close to or below the detection limit of the monitor with a campaign average of 0.1 ppb. This data was not analyzed further.
2.3. Analysis
Particle losses in the reactor were calculated using a three day period with the UV lamps turned off, by taking the ratio of the reactor to ambient measurements. The ambient value was calculated using an average before and after the reactor measurements. The average SMPS volume losses were 6 ± 6% (1σ) and AMS mass losses was 4 ± 4% (1σ). The losses were somewhat size-dependent (Figure S1), but given the low correction and largely similar volume size distribution during the campaign (volume geometric mean diameter 249 ± 38 nm, 1σ), the average values were used to correct the reactor output data. Particle losses in the inlet tubing and reactor bypass were assessed using the Max Planck Particle Loss Calculator [24]. The effect on total volume of this correction was relatively small and stable during the campaign (4.4 ± 0.6%, 1σ). Gas phase losses in the reactor were considered using the model first published in Palm et al. [11] (Figure S2). This model was used to calculate the fate of low-volatile organic compounds (LVOCs) by comparing four competing loss rates, walls of the reactor, fragmentation (assumed after reacting with OH five times), condensation onto particles and exiting the reactor. Wall loss rate, coefficient of eddy diffusion and reaction rate with OH were all similar to those in the original paper (see Figure S2 caption).
A collection efficiency (CE) of 1 for the AMS was assumed after comparison with the SMPS volume concentration. The CE parameterizations of Middlebrock et al. [25] were assessed and resulted in a CE close to 0.5. Changes from the default value where mostly due to acidity effects. Since sulphate relative ionization efficiency (RIE) was not calibrated in this campaign, which can give an inaccurate ammonium ion balance, this parameterization method was deemed uncertain. The data was instead compared to the volume of the SMPS measurements. The volume was converted to mass using an organic density of 1.4 g cm−3 and inorganic density of 1.75 g cm−3. As seen in Figure S3, the AMS and calculated SMPS mass concentrations fall roughly on a 1:1 line, except for an offset of ~0.5 µg m−3 in the SMPS data corresponding to the measurement artefact at large diameters. Figure S3 also shows that there was no difference between OFR and ambient CE (implying that the CE only affects absolute values and not the ratio between reactor and ambient values). The SMPS was regularly checked and serviced but no comparisons to other instruments were made. Although uncommon, CE = 1 have been previously seen [11], and was not investigated further. In order to calculate organonitrates, the nitrate fragmentation pattern in the AMS was analyzed according to the procedure of Farmer et al. [26]. The elemental ratios was calculated using the “improved-ambient” parameterization [27].
The PTR-ToF-MS raw data was analyzed with the PTR-wid software [28] that provides peak detection, a mass scale calibration, and a unified mass list to analyze and convert long-term data sets into mixing ratios. While the analyzing software applies a typical mass dependent transmission function [29,30], the instrument was calibrated against a gas standard mixture (Ionicon Analytik GmbH, Innsbruck, Austria) including e.g., methanol, acetaldehyde, acetone, isoprene, benzene, toluene, o-xylene and α-pinene several times before, during and after the experiment. Thus, for those compounds used in this study the instrument was directly calibrated against those gas standards. The instrument background was measured three times per day automatically for 30 min each, and readings were corrected for this.
3. Results and Discussion
Accounting for reactor losses, OFR processing led to net losses (1−COFR/CAmbient) of 7.5% ± 7.1 (1σ) in SMPS volume concentration and 9.8% ± 10.4 (1σ) in AMS mass concentration. The ambient particle properties were relatively stable during the two months of measurements. The average number concentration was 2100 ± 850 (1σ) and AMS pm1 was 2.8 ± 1.7 (1σ) µg m−3. Particles were dominated by organics (63% ± 13, 1σ) and sulphate (26% ± 10, 1σ). A time series of ambient SMPS number and volume concentrations and AMS fractional composition, covering the entire campaign, is shown in Figure S4. Winds were predominantly from the south and southwest (54% of the time). The number mode was mostly below 100 nm, while the volume geometric mean diameter was 249 ± 38 nm (1σ).
An example of a typical time series is seen in Figure 1, with reactor measurements on light blue background. As expected, the oxidizing environment lead to an increased O:C and decreased H:C ratio, with campaign averages H:C and O:C ratios of 1.47 ± 0.07 (1σ) and 0.68 ± 0.08 (1σ) (OSC = −0.11) for ambient measurements and 1.39 ± 0.08 (1σ) and 0.84 ± 0.11 (1σ) (OSC = 0.30) for reactor measurements. SMPS number and volume concentrations were consistently higher during ambient measurements, and the AMS shows that the losses were composition dependent (note that nitrate and ammonium was increased by a factor of five in the figure). Total losses were dominated by organics while sulphate was not substantially affected by the reactor. This indicates that the losses were due to volatilization by a temperature gradient or heterogeneous fragmentation, or both. Figure 2 shows the absolute difference between reactor and ambient AMS nominal organic mass peaks. Most fragments decrease in a proportional manner to their ambient concentration. The fragment CO2+ (m/z 44), and fragments related to it (m/z 28 and 18), were not lost in the same manner and played a dominant role in the difference between the two measurements. An m/z ratio of 44 is generally related to a higher degree of oxidation, but also to a lower volatility, which again are hard to separate. However, the fact that the reactor output at times had higher concentrations of CO2+ points to the fact that the aerosol was oxidized and that temperature driven evaporation was not the only driving force. Although very low during the campaign, significant losses of nitrate were measured in the reactor. Ammonium nitrate is known to evaporate, but the nitrate present as organonitrates was high for both ambient and reactor measurements with campaign average fractions of 0.80 and 0.96 respectively. Similar to previous OFR field measurements, the ratio of reactor/ambient organics is dependent on OH exposure [9,11]. Figure 3 shows that the organic losses increase with OH exposure. It is however hard to quantify the relative contribution from temperature and fragmentation effects, since the temperature in the OFR can be expected to increase with higher lamp intensities, and loss of the most volatile compounds most likely affects the elemental ratios in a similar manner in both cases. However, in a lab environment, the temperature increase in the reactor was measured to be below 2 °C at an OH exposure of 7 × 1011 molecules cm−3 s and a room temperature of 22 °C. This moderate increase in reactor temperature would not evaporate much of the particle mass, but the reactor has a longer residence time than the bypass sampling line which increases the time the aerosol spends in the air conditioned room before being measured. Although there were times when enhancement in the reactor occurred (reactor/ambient ratio >1), inspection of the time series showed that most, if not all, of these points are due to ambient particle mass concentration changes and the fact that losses/enhancements are calculated by taking the average of ambient concentrations before and after the reactor measurements. The lack of a maximum reactor/ambient ratio in Figure 3 indicates that there were very few occasions when there was a net production of SOA in the reactor.
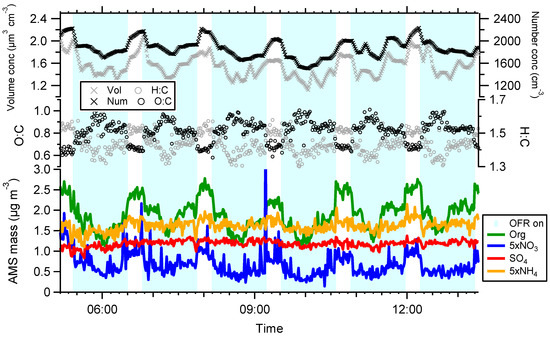
Figure 1.
Typical time series from the campaign showing scanning mobility particle sizer (SMPS) volume and number concentrations, O:C and H:C elemental ratios and chemically resolved mass concentration (NO3 and NH4 are offset a factor 5 for clarity). Reactor measurements, highlighted in light blue gives lower volume and number concentrations, but higher O:C. Organics and nitrate is lost while sulphate is not affected.
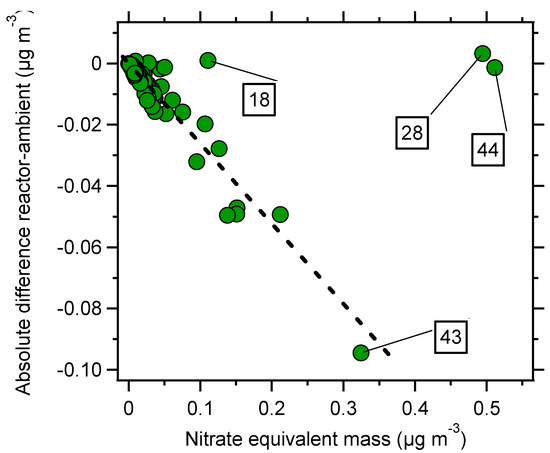
Figure 2.
Aerosol mass spectrometer (AMS) mass peak difference between reactor and ambient measurements as a function of nitrate equivalent mass (signal strength). Losses due to diffusion or impaction would result in a linear decrease (given uniform particle composition). The dashed line shows the correlation between loss and signal excluding the fragments m/z 44, 28 and 18 (all belonging to the ion CO2+) which shows a deviation consistent with evaporation and/or oxidation.
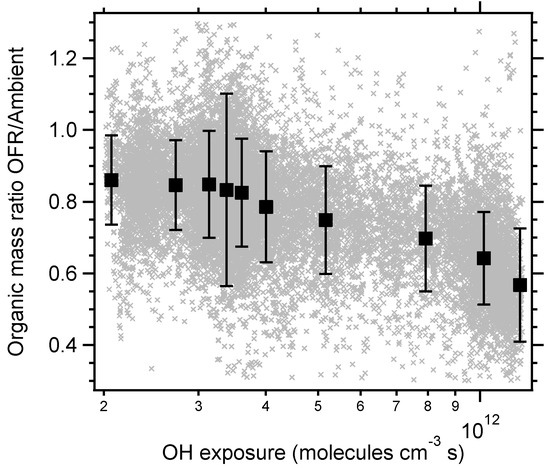
Figure 3.
The ratio between reactor and ambient organic mass (with a 4% loss correction) as a function of OH exposure during the whole campaign. The 10% OH exposure quantiles with error bars corresponding to 1σ are shown for clarity. Some data included in the quantiles are outside the axis ranges.
If the losses were due to an increased temperature in the reactor, a correlation with ambient temperature should be seen, since more semi-volatile material would have been condensed at low ambient temperature. Figure 4 shows that there was a significant (p < 0.01) temperature effect up to ~18 °C, but above that value there was still a net loss in the reactor, likely due to heterogeneous oxidation. The temperature effect also gave rise to a diurnal pattern with maximum loss during night when temperatures were lower. This is in contrast to previous OFR field campaigns in which maximum enhancement was seen during the night when ambient OH exposure and boundary layer height generally is lower. Although the particle mass concentrations during this campaign were low, which leads to lower yields in the OFR, and there was likely an effect of the temperature difference, the main reason behind the lack of net production in the OFR seen in this campaign is believed to be low production of SOA in the OFR due to low ambient VOC concentrations, as measured by the PTR-MS. The total monoterpene concentration was on average ~140 ppt (0.79 µg m−3) with a diurnal pattern with minimum during daytime. The sum of benzene, toluene and xylenes were even lower at 96 ppt (0.36 µg m−3), showing the same diurnal pattern. Further indication that little SOA formation took place in the reactor is the lack of nucleated particles. The rapid oxidation in OFRs normally produces substantial amounts of small particles, given that enough precursors are available. In this campaign, ~10% net loss in particle number was observed in the reactor (no correction applied).
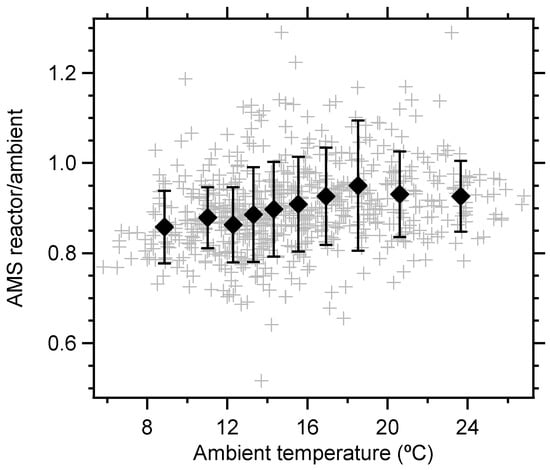
Figure 4.
Ratio between reactor and ambient total AMS mass concentrations (hourly values, loss corrected) as a function of temperature. 10% temperature quantiles show a significant trend below 18 °C.
The sensitivity of OFR measurements can be estimated using assumptions on particle mass yield and SOA partitioning. For example, if the minimum detectable SOA mass is 0.5 µg m−3 and the precursor particle mass yield is assumed to be 10%, we need to oxidize a precursor concentration of 5 µg m−3. Incorporating the effective condensation sink effect from the model of Palm et al. [11] increases the precursor concentration needed. In Figure 5, we calculated the SOA precursor concentration needed to see a 0.2 µg m−3 absolute increase, which is possible to measure with a variety of instruments, or a 5% mass based increase, which would cancel out the reactor losses in this campaign, as a function of effective condensation sink at four different yields. The approximate mass concentration is shown on the top axis (parameterized from the condensation sink of this campaign). The campaign average mass and the sum of monoterpenes and aromatics are also plotted. The α-pinene SOA mass yields from Ahlberg et al. (2017) is added as a reference. This yield was ~10% at the average campaign particle mass concentration. This value is an underestimation due to the condensation sink effect [31], but is far from the 50% yield needed to see a 0.2 µg m−3 increase. The average yield of an ambient precursor mixture is unknown, but it is not likely to be 50%. In this campaign, the concentration of monoterpenes and aromatics likely would have to be almost an order of magnitude higher to produce any visible mass in the reactor. However, many other precursors exist, some which would not be visible in the PTR-MS [11].

Figure 5.
The precursor concentration needed to measure either a 0.2 µg m−3 or a 5% increase in particle mass at different effective condensation sinks and particle mass yields (boxes). The top axis shows the corresponding mass concentration at each condensation sink parameterized from the campaign data. The blue star shows the campaign average particle mass concentration and the concentration of common precursors (monoterpenes, benzene, toluene and xylenes), with ±1σ in grey. For comparison, the secondary organic aerosols (SOA) mass yield of α-pinene from Ahlberg et al. (2017) as a function of organic mass is added (green axes and line).
To assess the potential losses of VOCs in the inlet, the penetration efficiency was calculated [32]. The penetration depends on the diffusion coefficient, flow rate, length of the tubing and wall accommodation coefficients. Assuming a diffusion coefficient of 7 × 10−2 cm2 s−1 [11] and that anything hitting the walls will stay there, the penetration would be only 3%. If the wall accommodation coefficient is 0.1 the penetration of the same molecule is 90%. Figure S5 shows the calculated penetration for diffusion coefficients between 1–10 × 10−2 cm2 s−1 [33,34] and mass accommodation coefficients of 0.01–1. It is clear that the uncertainty in wall accommodation had a much larger effect on penetration than the uncertainty of the diffusion coefficient. Although inlet losses may have been a factor for some species, there are compensating effects of wall loss, diffusion and particle accommodation. For example, a low volatility molecule with high wall accommodation generally also means that the diffusion is slower and the likelihood that the molecule is already bound to a particle is higher. On the other hand, a more volatile SOA precursor molecule is less likely to stick to the walls, and more likely to desorb if it does. The accommodation coefficient on Teflon is orders of magnitude lower than 0.1 [35,36], but to our knowledge, no literature data on wall accommodation coefficients on stainless steel exist. Pagonis et al. [37] investigated the effect of partitioning between gas and Teflon tubing for several VOCs, and found that significant delays due to the retention times could occur. The time-scales for equilibrium partitioning in the inlet is unknown, but the duration of the measurements, together with the PTR-MS measurements, favors the conclusion that the lack of reactor SOA production was not due to VOC losses.
4. Conclusions
We present the first ambient oxidation flow reactor measurements in Europe. The measurements took place during summer at a rural site surrounded by arable land, with 30 km to the nearest city. We saw virtually no increases in particle mass or number concentrations during the two months of the campaign. PTR-MS measurements showed that precursor concentrations were too low to form a significant amount of secondary material. A net particle mass loss of ~10%, which was correlated to ambient temperature and OH exposure, was seen. We attribute this to both evaporation of organic material in the reactor due to differences between ambient and reactor temperatures, and to fragmentation reactions. These effects are hard to separate, but it is clear that the change in elemental ratios was not only driven by evaporation, since the AMS mass fragment CO2+ was often increased in the reactor output. This could be due to both gas and particle phase reactions. Reactor processing led to an increase in carbon oxidation state, which is commonly linked to water uptake.
The conditions at the site are representative of many parts of northern Europe. The results are due to the relatively clean air of the site, but also because of the fast atmospheric conversion of trace gases. Net losses due to temperature driven evaporation can be limited by keeping the reactor at ambient temperatures, as has been done on other campaigns (e.g., [9,11]). However, this is not always possible at field sites and would likely not have affected the conclusions of this campaign. The sensitivity of the reactor measurements at different precursor concentrations and effective condensation sinks was calculated. It was shown that, with an ambient pm1 concentration of ~3 µg m−3, an order of magnitude higher precursor concentrations would have been needed to form significant amounts of SOA in the reactor. Sites closer to precursor emissions and with relatively high ambient aerosol mass concentrations will give much stronger responses in an OFR. These measurements show that further processing of relatively clean ambient aerosols can result in lower mass concentrations.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4433/10/7/408/s1, Figure S1: Average size spectrum and reactor losses. Figure S2: Modeled fractional fate of LVOCs. Figure S3: AMS vs. SMPS mass concentrations. Figure S4: SMPS concentrations and chemical composition. Figure S5: Calculated penetration through the inlet.
Author Contributions
Conceptualization, B.S., T.H. and E.A.; methodology, B.S., T.H., A.K., G.F., and E.A.; software, B.S.; validation, E.A., T.H. and A.E.; formal analysis, E.A., T.H.; investigation, E.A., S.A., T.H., T.K., G.F., A.K. and B.S. data curation, E.A. and T.H.; writing—original draft preparation, E.A.; writing—review and editing, E.A., S.A., A.E., T.H., T.K., W.H.B., G.F., P.R., A.K. and B.S.; supervision, B.S. and A.K.; project administration, E.A. and B.S.; funding acquisition, B.S.
Funding
This work was supported by The Swedish Research Council Formas [grant number 2011-1101-19993-37] and the strategic research area MERGE at Lund University.
Acknowledgments
We thank L.C. and A.J., for providing the research indrastructure in Skrehalla.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Allan, J.D.; Coe, H.; Ulbrich, I.; Alfarra, M.R.; Takami, A.; Middlebrook, A.M.; Sun, Y.L.; et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef]
- Pandis, S.N.; Skyllakou, K.; Florou, K.; Kostenidou, E.; Kaltsonoudis, C.; Hasa, E.; Presto, A.A. Urban particulate matter pollution: A tale of five cities. Faraday Discuss. 2016, 189, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Glasius, M.; Goldstein, A.H. Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry. Environ. Sci. Technol. 2016, 50, 2754–2764. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, M.; Cappa, C.D.; Fan, J.; Goldstein, A.H.; Guenther, A.B.; Jimenez, J.L.; Kuang, C.; Laskin, A.; Martin, S.T.; Ng, N.L.; et al. Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Rev. Geophys. 2017, 55, 509–559. [Google Scholar] [CrossRef]
- Jimenez, J.L. Concluding remarks: Faraday Discussion on chemistry in the urban atmosphere. Faraday Discuss. 2016, 189, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Bruns, E.A.; El Haddad, I.; Keller, A.; Klein, F.; Kumar, N.K.; Pieber, S.M.; Corbin, J.C.; Slowik, J.G.; Brune, W.H.; Baltensperger, U.; et al. Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition. Atmos. Meas. Tech. 2015, 8, 2315–2332. [Google Scholar] [CrossRef]
- Lambe, A.T.; Chhabra, P.S.; Onasch, T.B.; Brune, W.H.; Hunter, J.F.; Kroll, J.H.; Cummings, M.J.; Brogan, J.F.; Parmar, Y.; Worsnop, D.R.; et al. Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield. Atmos. Chem. Phys. 2015, 15, 3063–3075. [Google Scholar] [CrossRef]
- Platt, S.M.; Haddad, I.E.; Zardini, A.A.; Clairotte, M.; Astorga, C.; Wolf, R.; Slowik, J.G. Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber. Atmos. Chem. Phys. 2013, 13, 9141–9158. [Google Scholar] [CrossRef]
- Ortega, A.M.; Hayes, P.L.; Peng, Z.; Palm, B.B.; Hu, W.; Day, D.A.; Li, R.; Cubison, M.J.; Brune, W.H.; Graus, M.; et al. Real-time measurements of secondary organic aerosol formation and aging from ambient air in an oxidation flow reactor in the Los Angeles area. Atmos. Chem. Phys. 2016, 16, 7411–7433. [Google Scholar] [CrossRef]
- George, I.J.; Slowik, J.; Abbatt, J.P.D. Chemical aging of ambient organic aerosol from heterogeneous reaction with hydroxyl radicals. Geophys. Res. Lett. 2008, 35. [Google Scholar] [CrossRef]
- Palm, B.B.; Campuzano-Jost, P.; Ortega, A.M.; Day, D.A.; Kaser, L.; Jud, W.; Karl, T.; Hansel, A.; Hunter, J.F.; Cross, E.S.; et al. In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor. Atmos. Chem. Phys. 2016, 16, 2943–2970. [Google Scholar] [CrossRef]
- Palm, B.B.; Campuzano-Jost, P.; Day, D.A.; Ortega, A.M.; Fry, J.L.; Brown, S.S.; Zarzana, K.J.; Dube, W.; Wagner, N.L.; Draper, D.C.; et al. Secondary organic aerosol formation from in situ OH, O-3, and NO3 oxidation of ambient forest air in an oxidation flow reactor. Atmos. Chem. Phys. 2017, 17, 5331–5354. [Google Scholar] [CrossRef]
- Slowik, J.G.; Wong, J.P.S.; Abbatt, J.P.D. Real-time, controlled OH-initiated oxidation of biogenic secondary organic aerosol. Atmos. Chem. Phys. 2012, 12, 9775–9790. [Google Scholar] [CrossRef]
- Palm, B.B.; Sá, S.S.D.; Day, D.A.; Campuzano-Jost, P.; Hu, W.; Seco, R.; Sjostedt, S.J.; Park, J.H.; Guenther, A.B.; Kim, S.; et al. Secondary organic aerosol formation from ambient air in an oxidation flow reactor in central Amazonia. Atmos. Chem. Phys. Discuss. 2018, 18, 467–493. [Google Scholar] [CrossRef]
- Kang, E.; Lee, M.; Brune, W.H. Taehyung Lee Photochemical aging of organic and inorganic ambient aerosol from the Potential Aerosol Mass (PAM) reactor experiment in East Asia. Atmos. Chem. Phys. Discuss. 2017. [Google Scholar] [CrossRef]
- Kang, E.; Root, M.J.; Toohey, D.W.; Brune, W.H. Introducing the concept of Potential Aerosol Mass (PAM). Atmos. Chem. Phys. 2007, 7, 5727–5744. [Google Scholar] [CrossRef]
- Lambe, A.T.; Ahern, A.T.; Williams, L.R.; Slowik, J.G.; Wong, J.P.S.; Abbatt, J.P.D.; Brune, W.H.; Ng, N.L.; Wright, J.P.; Croasdale, D.R.; et al. Characterization of aerosol photooxidation flow reactors: Heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements. Atmos. Meas. Tech. 2011, 4, 445–461. [Google Scholar] [CrossRef]
- Li, R.; Palm, B.B.; Ortega, A.M.; Hlywiak, J.; Hu, W.; Peng, Z.; Day, D.A.; Knote, C.; Brune, W.H.; De Gouw, J.A.; et al. Modeling the Radical Chemistry in an Oxidation Flow Reactor: Radical Formation and Recycling, Sensitivities, and the OH Exposure Estimation Equation. J. Phys. Chem. A 2015, 119, 4418–4432. [Google Scholar] [CrossRef]
- Peng, Z.; Day, D.A.; Stark, H.; Li, R.; Lee-Taylor, J.; Palm, B.B.; Brune, W.H.; Jimenez, J.L. HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling. Atmos. Meas. Tech. 2015, 8, 4863–4890. [Google Scholar] [CrossRef]
- Wiedensohler, A.; Birmili, W.; Nowak, A.; Sonntag, A.; Weinhold, K.; Merkel, M.; Wehner, B.; Tuch, T.; Pfeifer, S.; Fiebig, M.; et al. Mobility particle size spectrometers: Harmonization of technical standards and data structure to facilitate high quality long-term observations of atmospheric particle number size distributions. Atmos. Meas. Tech. 2012, 5, 657–685. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S.; et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Graus, M.; Müller, M.; Hansel, A. High Resolution PTR-TOF: Quantification and Formula Confirmation of VOC in Real Time. J. Am. Soc. Mass Spectrom. 2010, 21, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Lindinger, W.; Hansel, A.; Jordan, A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) - Medical applications, food control and environmental research. Int. J. Mass Spectrom. 1998, 173, 191–241. [Google Scholar] [CrossRef]
- Von der Weiden, S.L.; Drewnick, F.; Borrmann, S. Particle Loss Calculator—A new software tool for the assessment of the performance of aerosol inlet systems. Atmos. Meas. Tech. 2009, 2, 479–494. [Google Scholar] [CrossRef]
- Middlebrook, A.M.; Bahreini, R.; Jimenez, J.L.; Canagaratna, M.R. Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data. Aerosol Sci. Technol. 2012, 46, 258–271. [Google Scholar] [CrossRef]
- Farmer, D.K.; Matsunaga, A.; Docherty, K.S.; Surratt, J.D.; Seinfeld, J.H.; Ziemann, P.J.; Jimenez, J.L. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. Proc. Natl. Acad. Sci. USA 2010, 107, 6670–6675. [Google Scholar] [CrossRef] [PubMed]
- Canagaratna, M.R.; Jimenez, J.L.; Kroll, J.H.; Chen, Q.; Kessler, S.H.; Massoli, P.; Hildebrandt Ruiz, L.; Fortner, E.; Williams, L.R.; Wilson, K.R.; et al. Worsnop: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: Characterization, improved calibration, and implications. Atmos. Chem. Phys. 2015, 15, 253–272. [Google Scholar] [CrossRef]
- Holzinger, R. PTRwid: A new widget tool for processing PTR-TOF-MS data. Atmos. Meas. Tech. 2015, 8, 3903–3922. [Google Scholar] [CrossRef]
- Holzinger, R.; Kasper-Giebl, A.; Staudinger, M.; Schauer, G.; Röckmann, T. Analysis of the chemical composition of organic aerosol at the Mt. Sonnblick observatory using a novel high mass resolution thermal-desorption proton-transfer-reaction mass-spectrometer (hr-TD-PTR-MS). Atmos. Chem. Phys. 2010, 10, 10111–10128. [Google Scholar] [CrossRef]
- Cappellin, L.; Biasioli, F.; Schuhfried, E.; Soukoulis, C.; Märk, T.D.; Gasperi, F. Extending the dynamic range of proton transfer reaction time-of-flight mass spectrometers by a novel dead time correction. Rapid Commun. Mass Spectrom. 2011, 25, 179–183. [Google Scholar] [CrossRef]
- Ahlberg, E.; Eriksson, A.; Brune, W.H.; Roldin, P.; Svenningsson, B. Effect of salt seed particle surface area, composition and phase on secondary organic aerosol mass yields in oxidation flow reactors. Atmos. Chem. Phys. 2019, 19, 2701–2712. [Google Scholar] [CrossRef]
- Hinds, W.C. Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Lugg, G.A. Diffusion Coefficients of Some Organic and Other Vapors in Air. Anal. Chem. 1968, 40, 1072–1077. [Google Scholar] [CrossRef]
- Tang, M.J.; Shiraiwa, M.; Pöschl, U.; Cox, R.A.; Kalberer, M. Compilation and evaluation of gas phase diffusion coefficients of reactive trace gases in the atmosphere: Volume 2. Diffusivities of organic compounds, pressure-normalised mean free paths, and average Knudsen numbers for gas uptake calculations. Atmos. Chem. Phys. 2015, 15, 5585–5598. [Google Scholar] [CrossRef]
- Mcmurry, P.H.; Grosjean, D. Gas and Aerosol Wall Losses in Teflon Film Smog Chambers. Environ. Sci. Technol. 1985, 19, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, A.; Ziemann, P.J. Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements. Aerosol Sci. Technol. 2010, 44, 881–892. [Google Scholar] [CrossRef]
- Pagonis, D.; Krechmer, J.E.; de Gouw, J.; Jimenez, J.L.; Ziemann, P.J. Effects of Gas-Wall Partitioning in Teflon Tubing and Instrumentation on Time-Resolved Measurements of Gas-Phase Organic Compounds. Atmos. Meas. Tech. Discuss. 2017, 10, 4687–4696. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).