Testing Iron Stable Isotope Ratios as a Signature of Biomass Burning


Abstract
:1. Introduction
2. Experiments
2.1. Sampling
2.2. Acid Digestion and Leaching Experiments
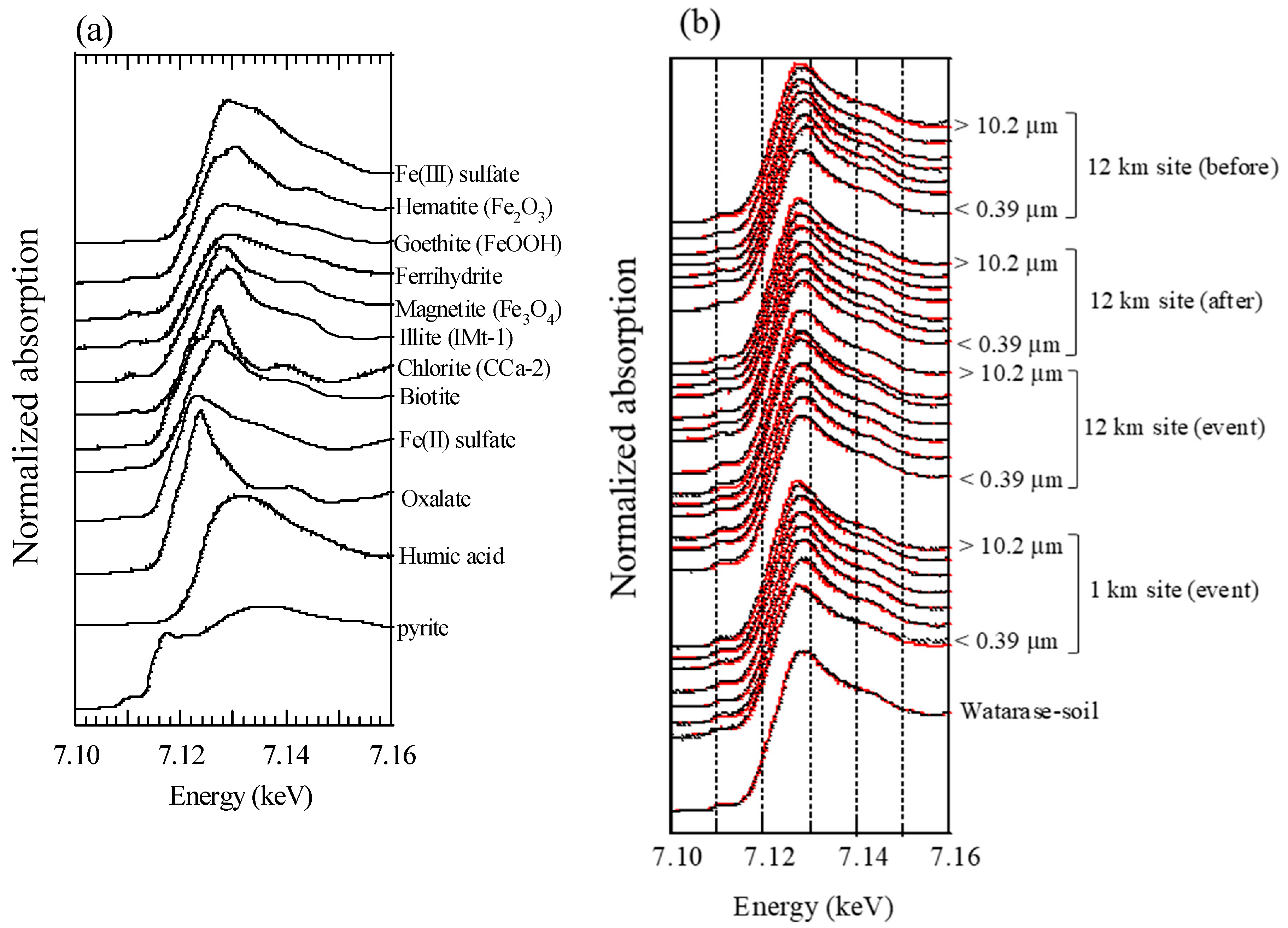
2.3. X-Ray Absorption Fine Structure (XAFS) Analysis
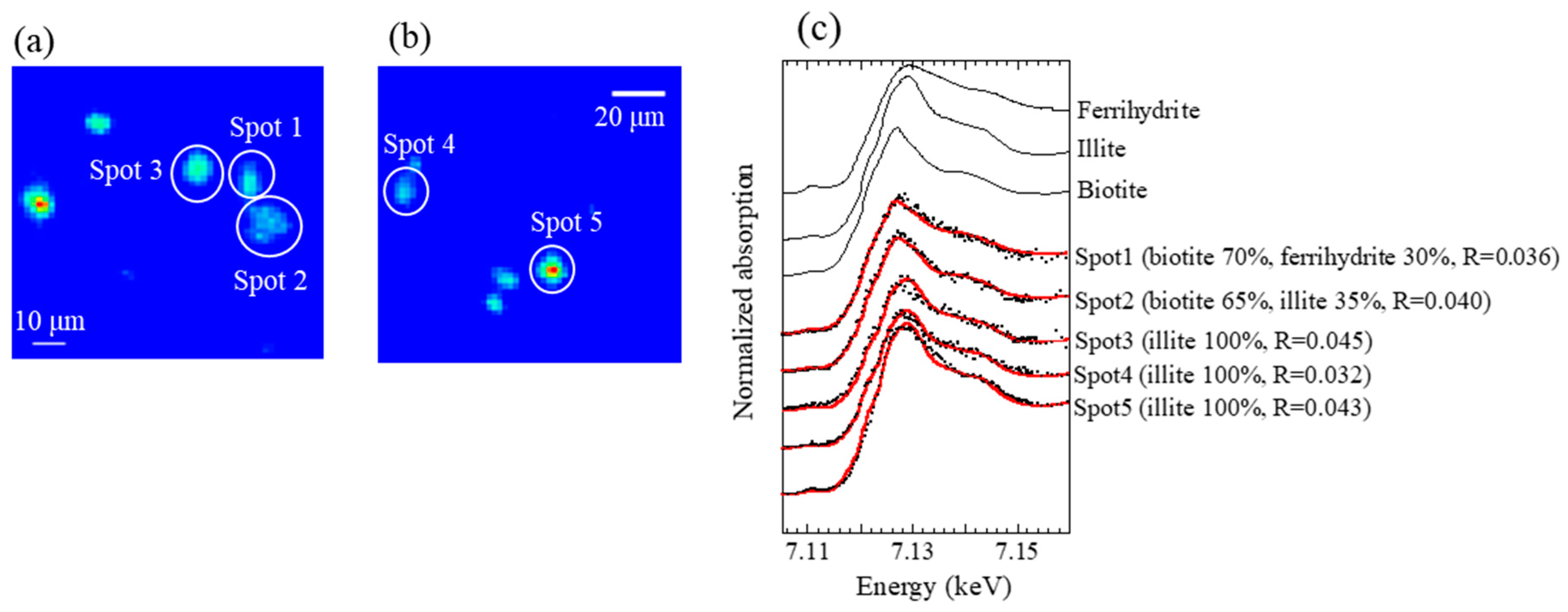
2.4. Scanning Transmission X-Ray Microscope (STXM) Analysis
2.5. Iron Isotope Analysis
3. Results
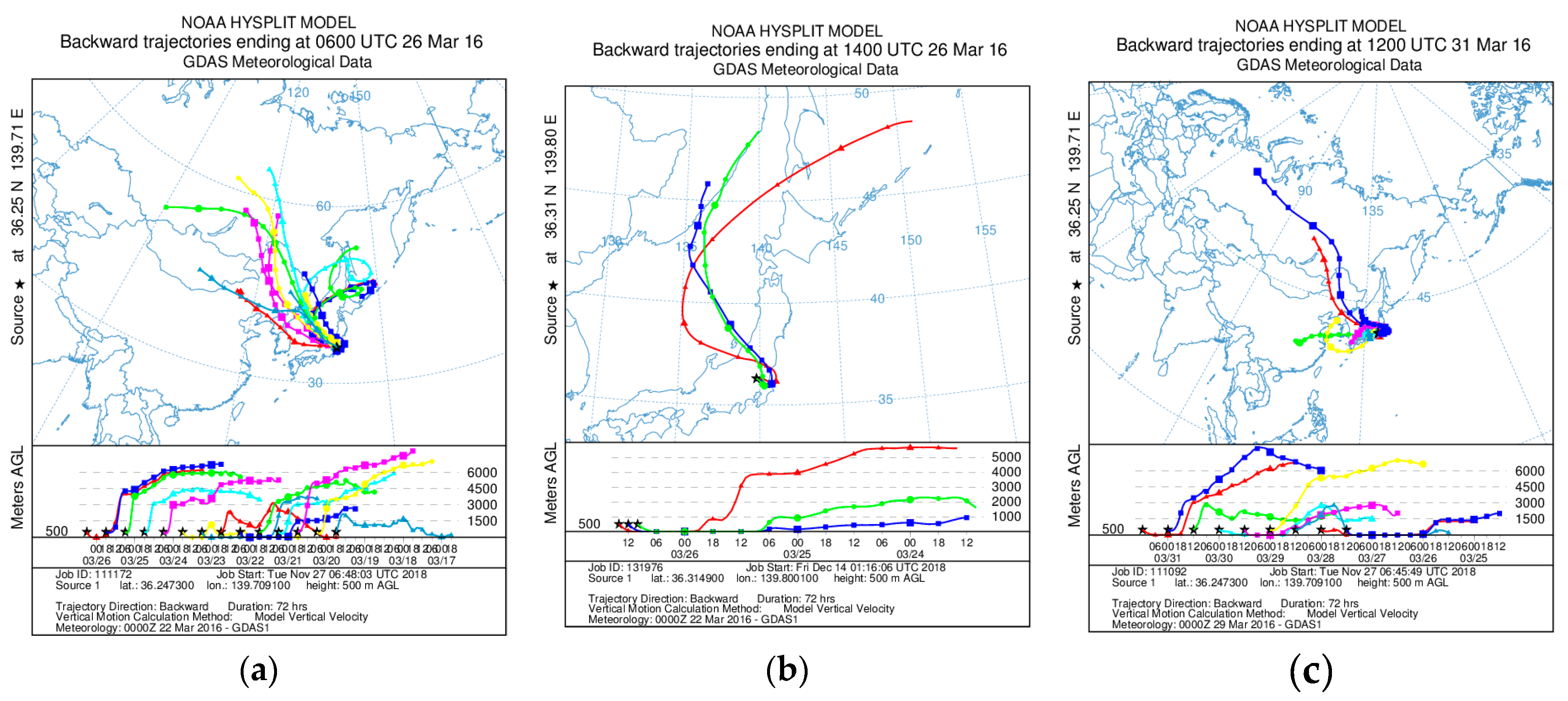
3.1. Characteristics of Aerosols
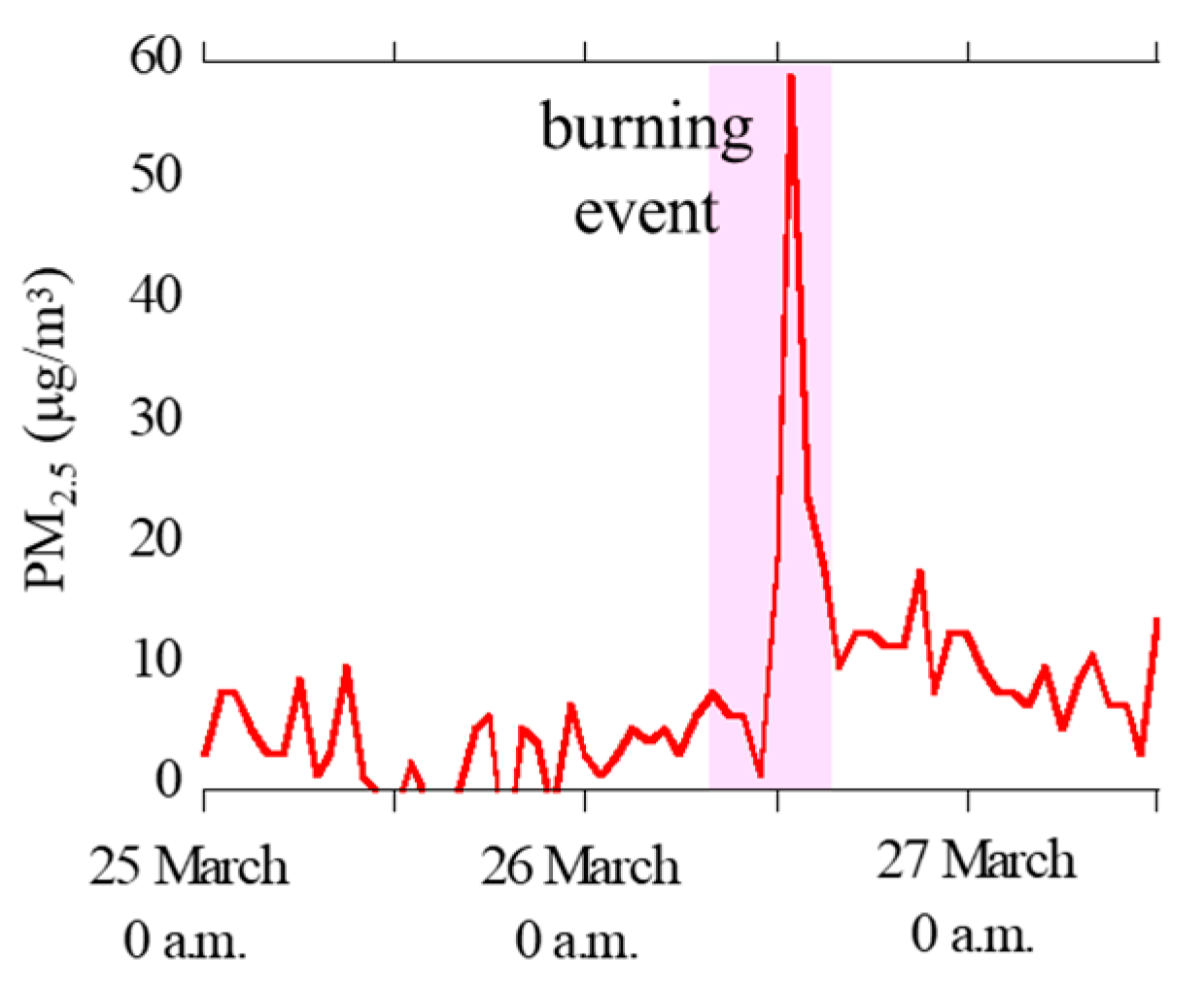
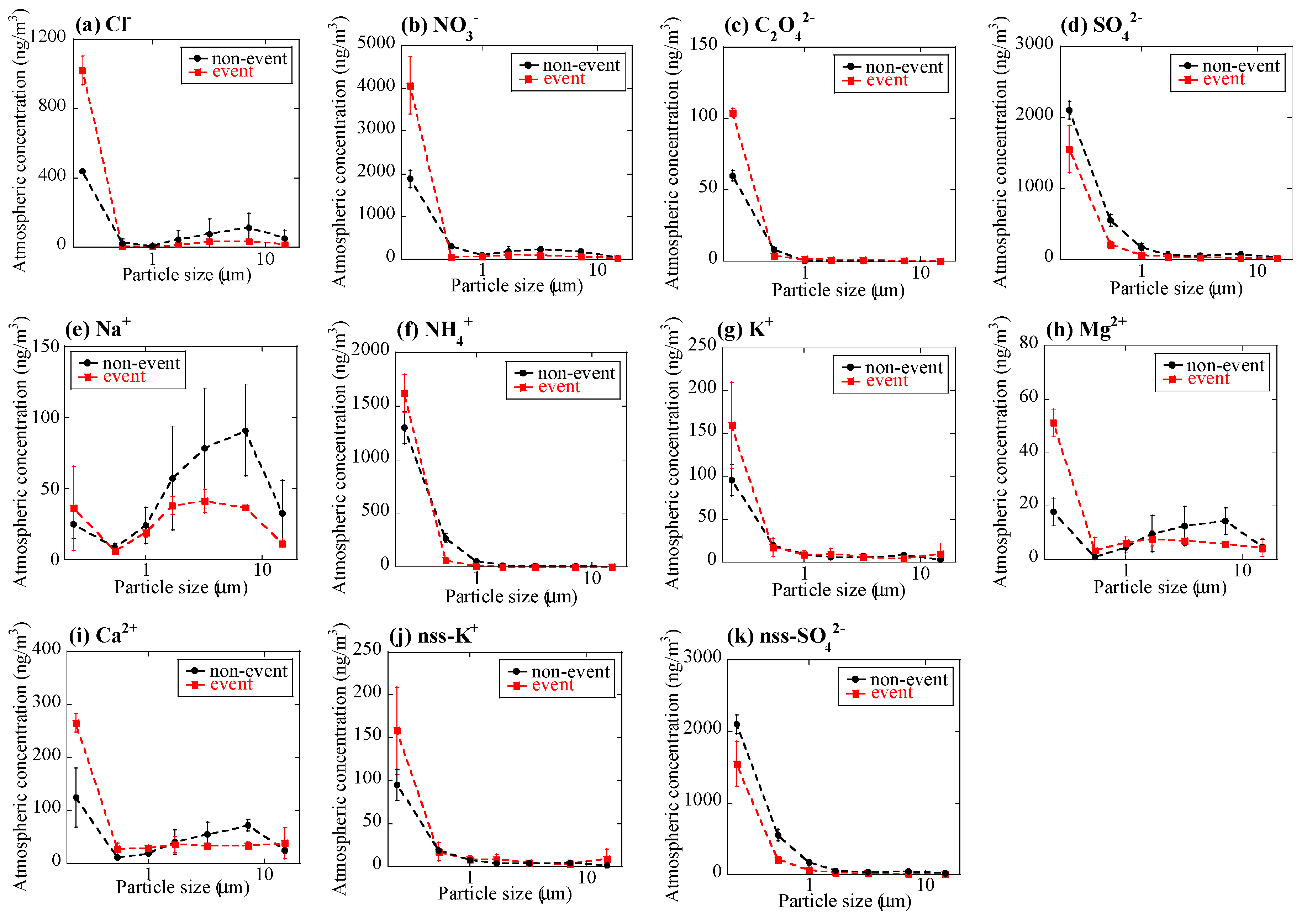
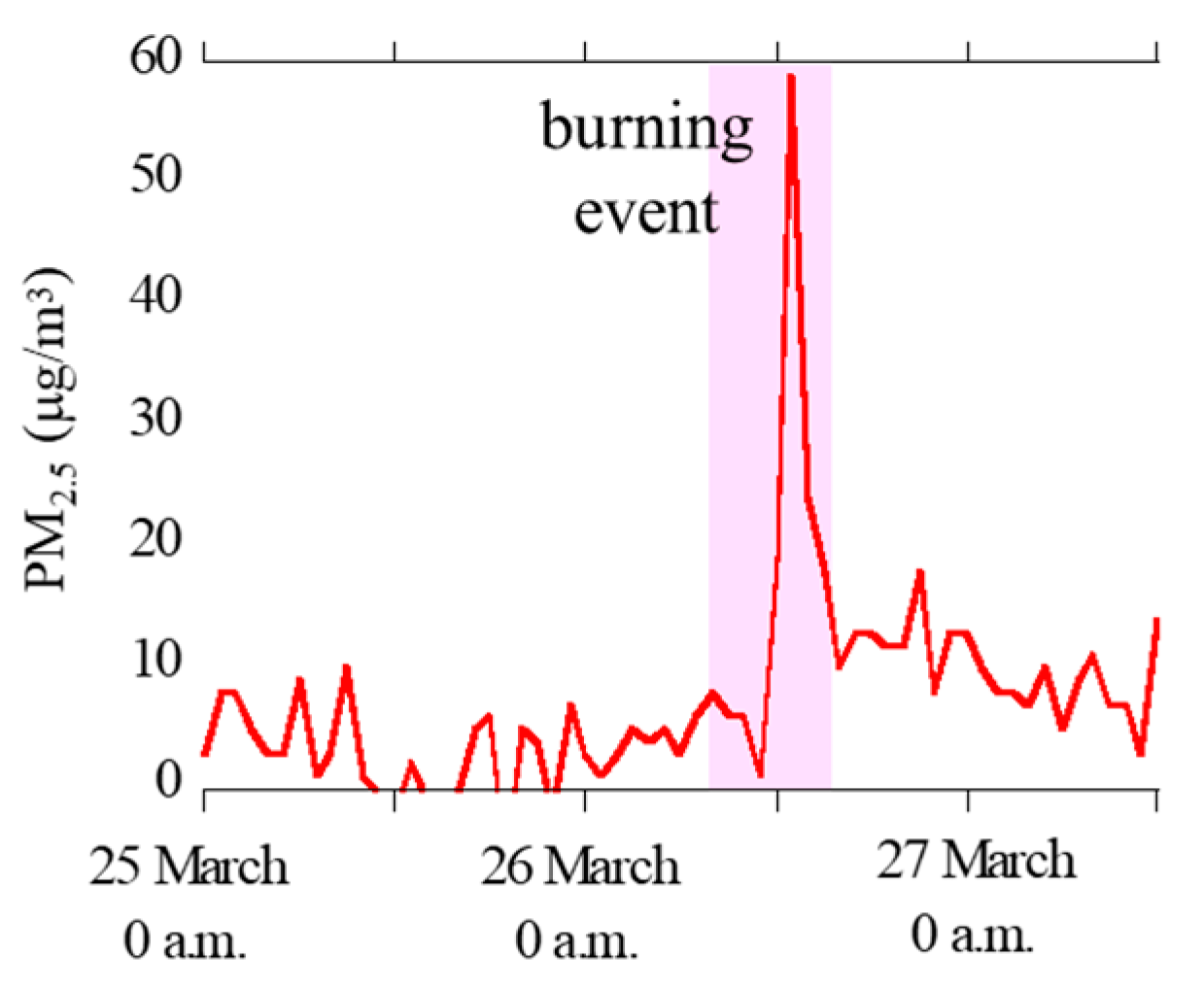
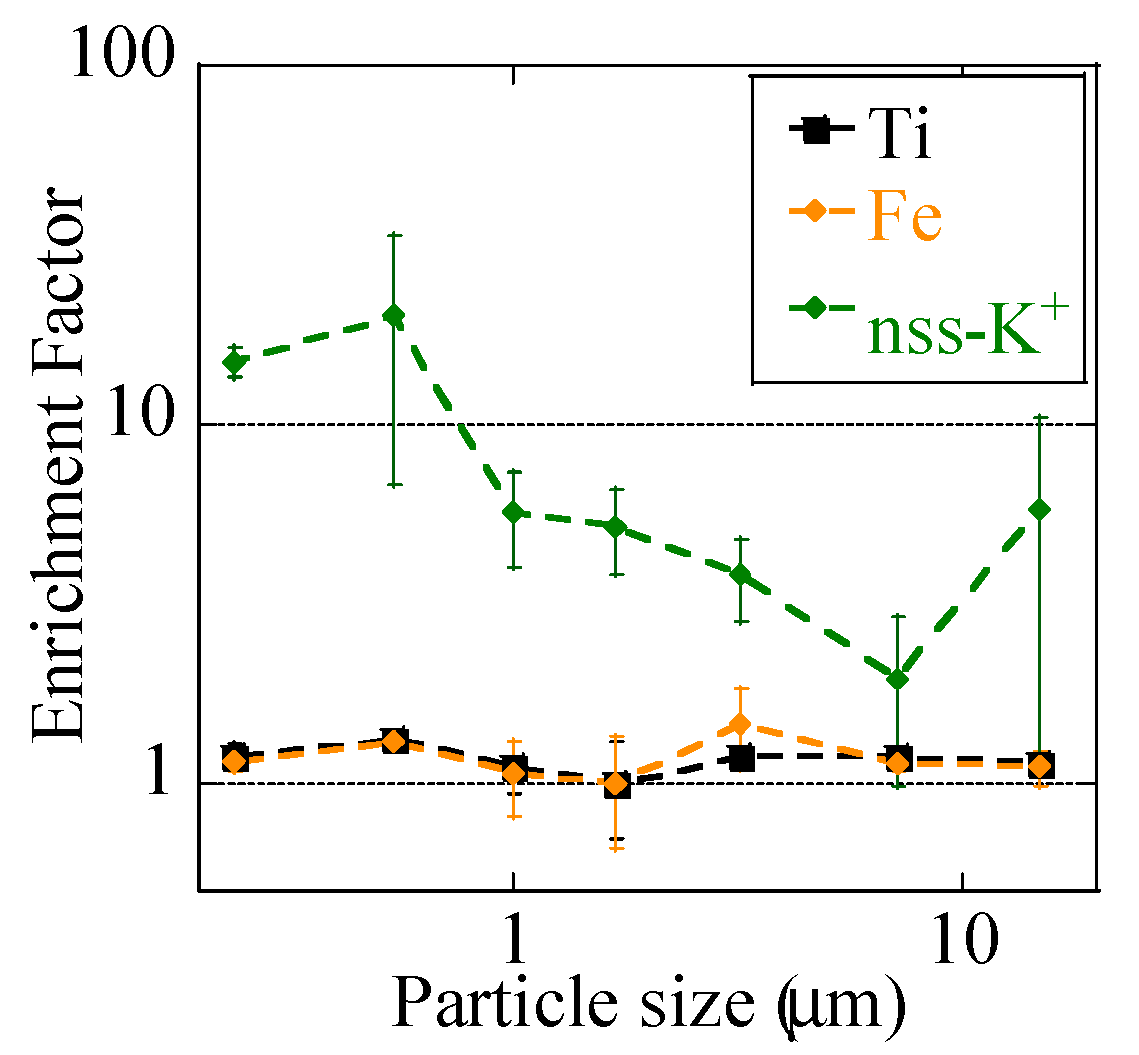
3.1.1. Concentrations of PM2.5, Major Ions, Fe, and Other Trace Elements
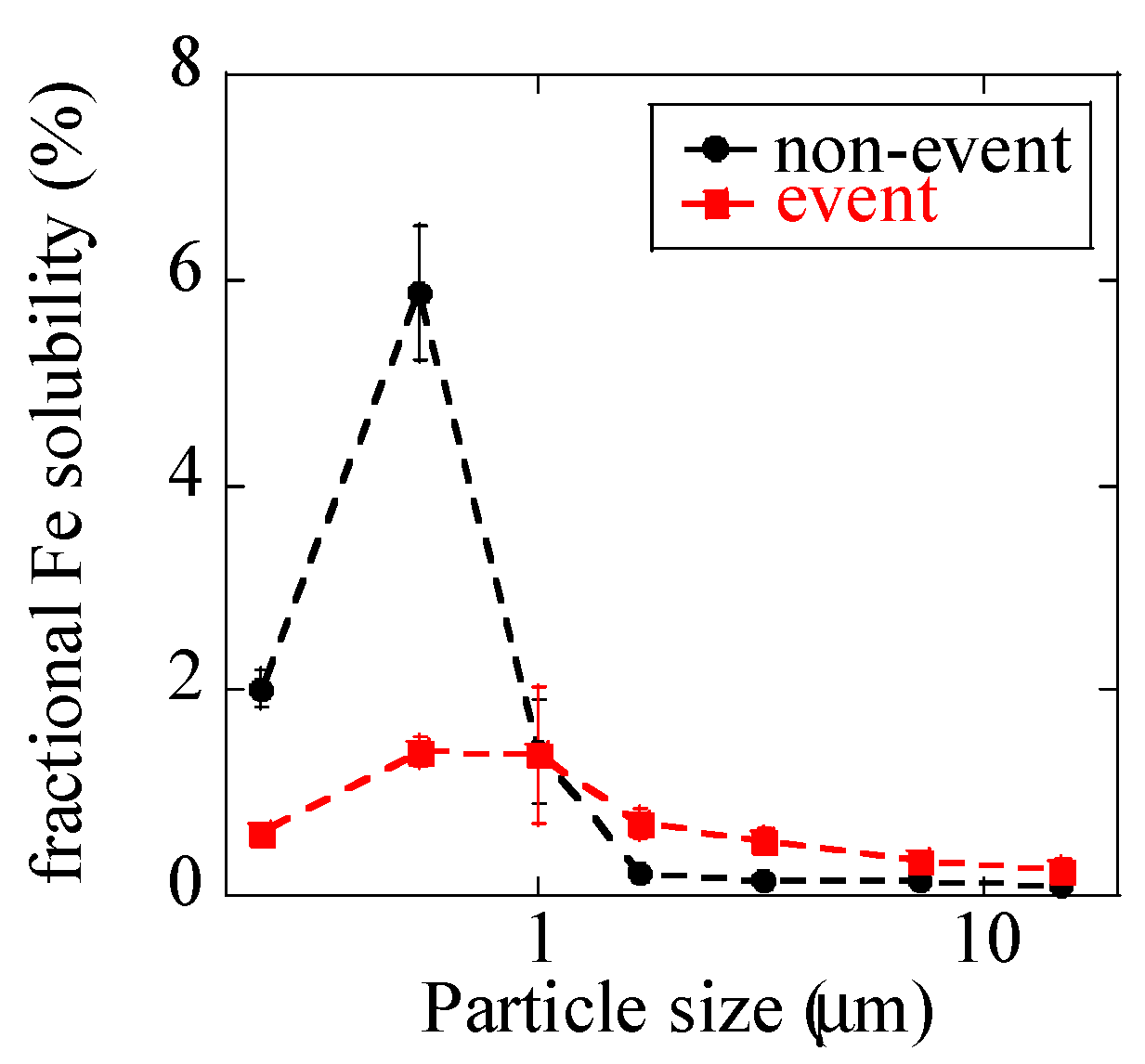
3.1.2. Fractional Fe Solubility
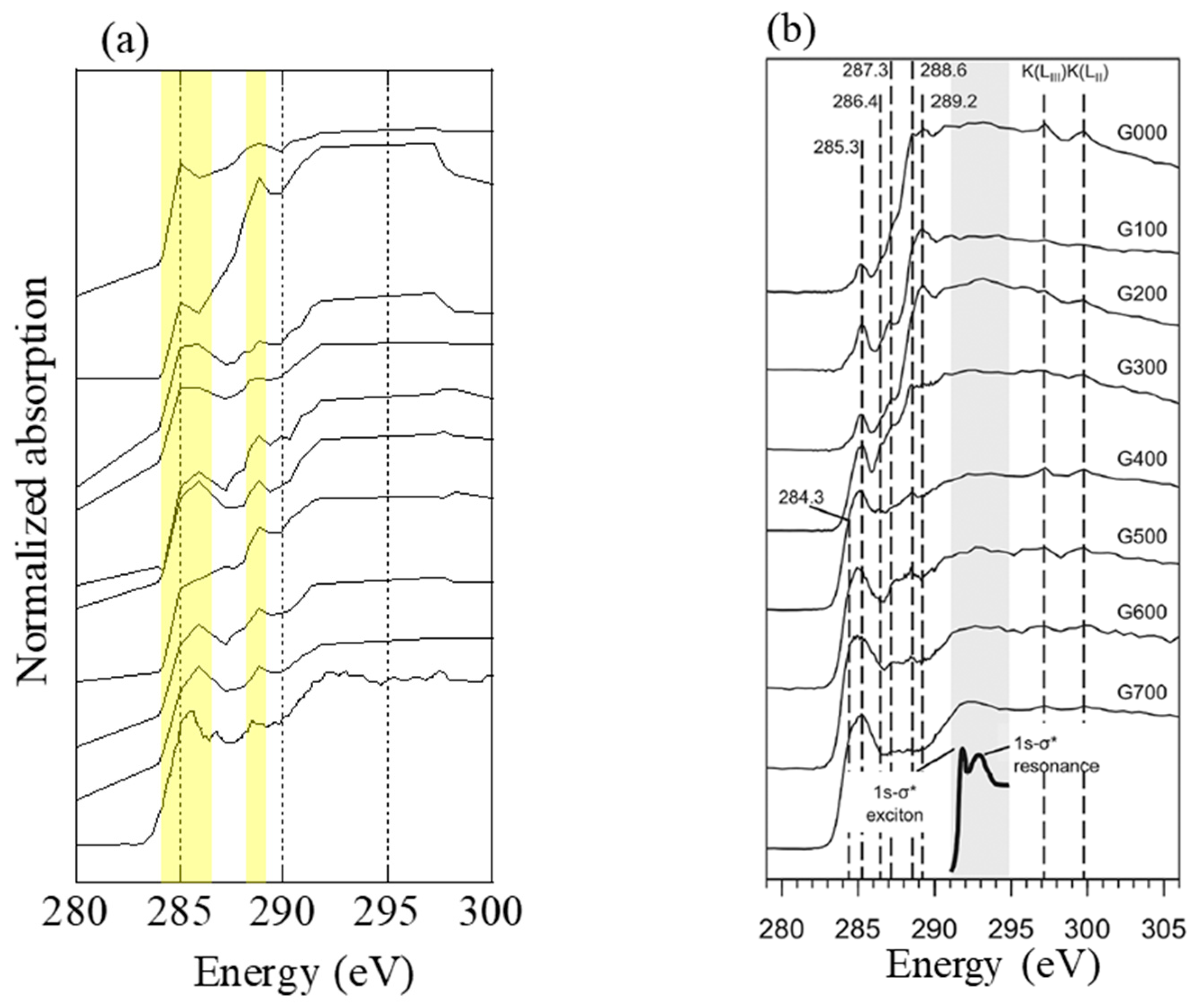
3.1.3. Estimation of Combustion Temperature from C K-edge XANES
3.2. Iron Isotope Ratios
3.2.1. Soil, Reed, and Residual Ash
3.2.2. Bulk Aerosol
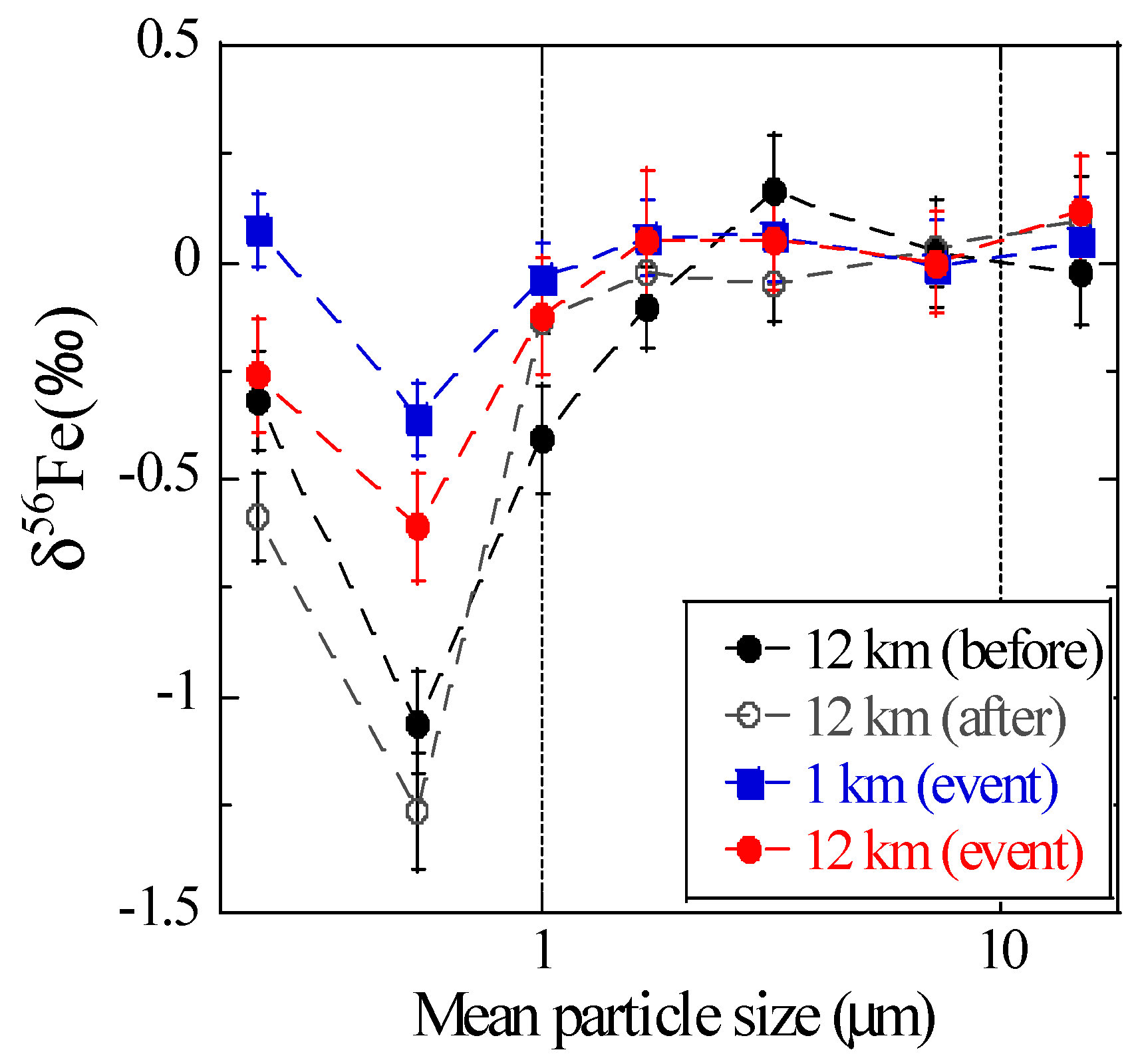
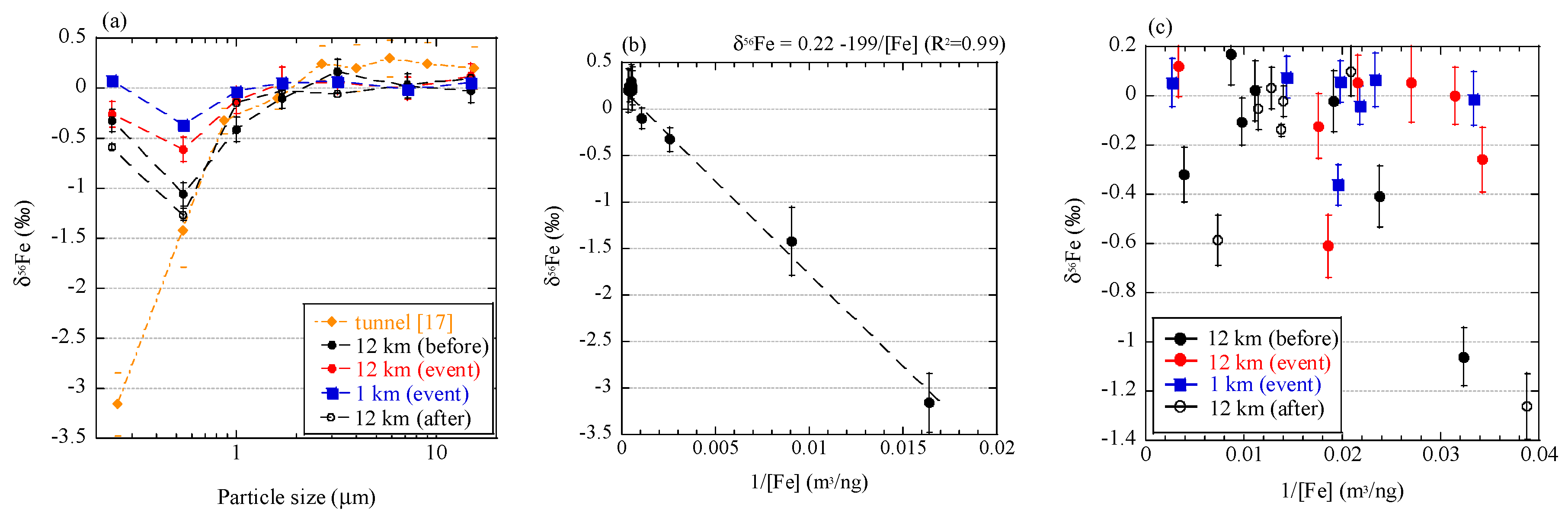
3.2.3. Soluble Fe Fraction of Aerosols
4. Discussion
4.1. Atmospheric Concentration and Fractional Solubility of Fe Emitted by Biomass Burning
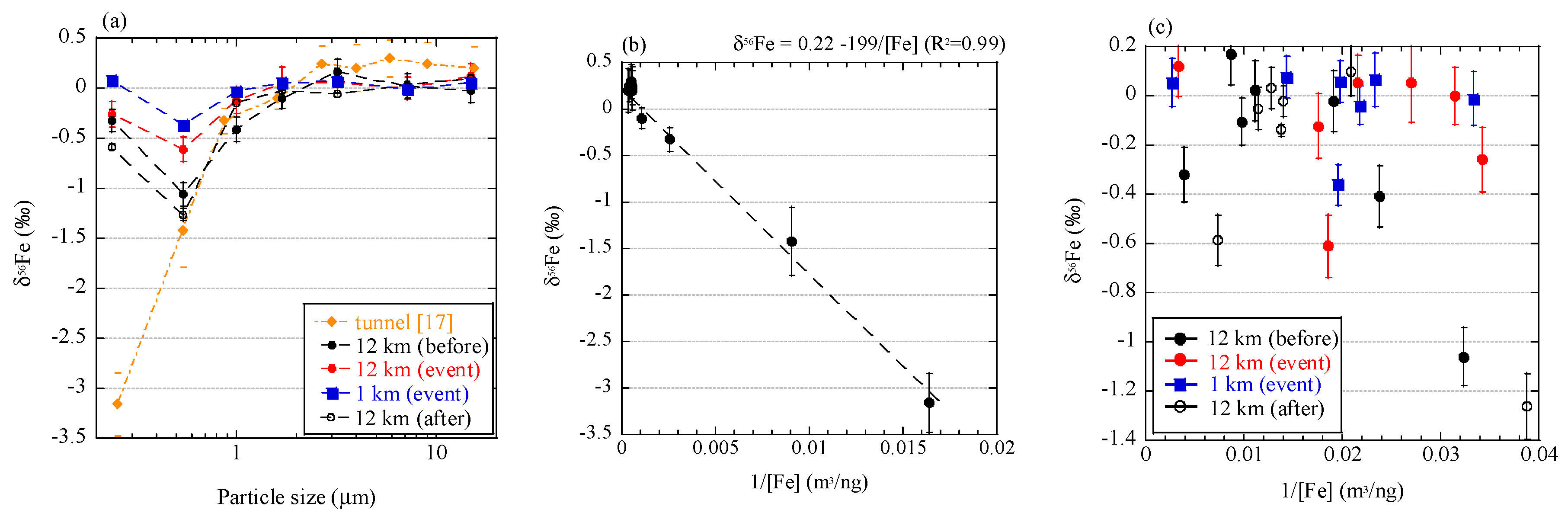
4.2. Can Low δ56Fe Values Be Used as a Tracer of Biomass Burning?
4.3. Fe Isotope Fractionation During Combustion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
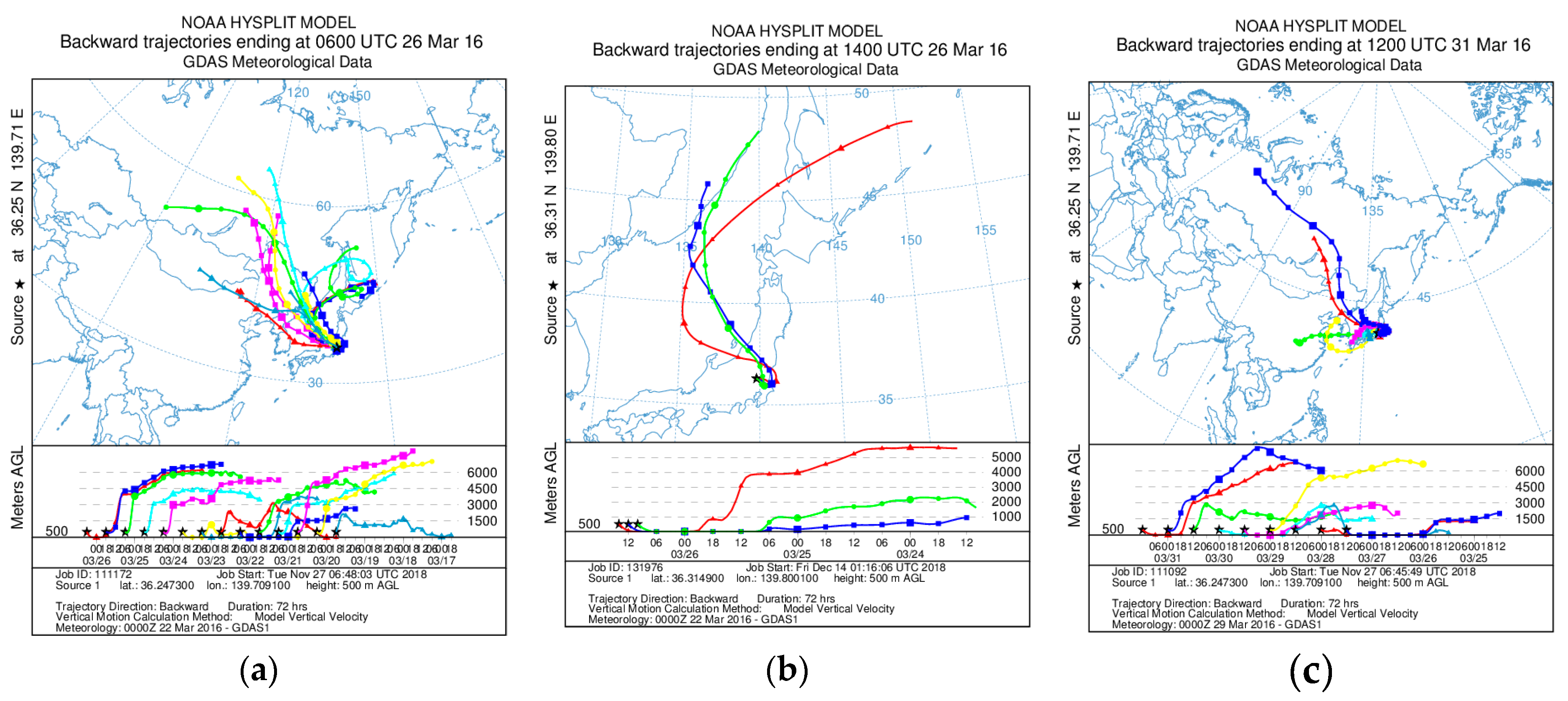
Appendix B
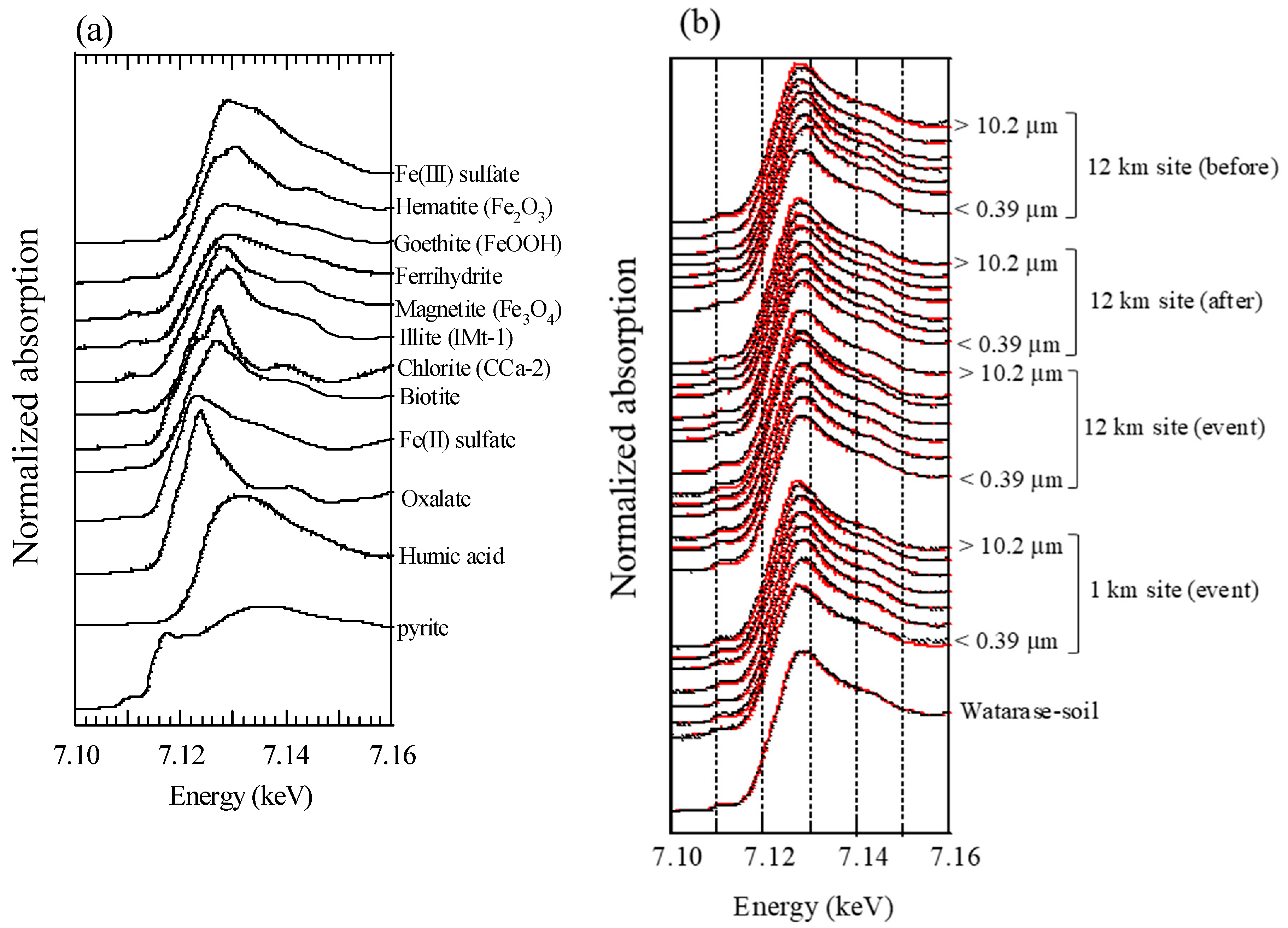
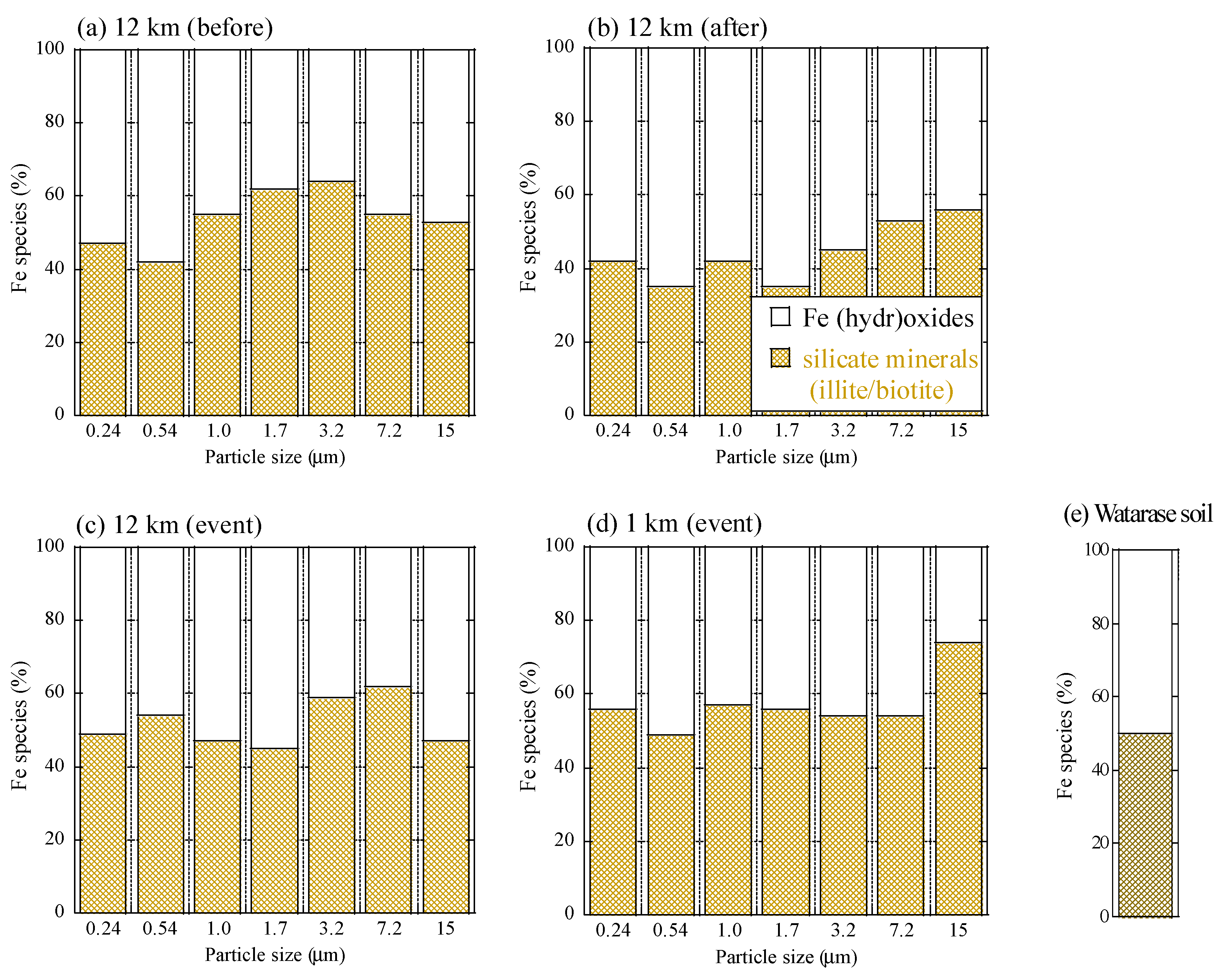


Appendix C

References
- Martin, J.H.; Fitzwater, S.E. Iron deficiency limits phytoplankton growth in the north-east Pacific subarctic. Nature 1988, 331, 341–343. [Google Scholar] [CrossRef]
- Martin, J.H.; Coale, K.H.; Johnson, K.S.; Fitzwater, S.E.; Gordon, R.M.; Tanner, S.J.; Hunter, C.N.; Elrod, V.A.; Nowicki, J.L.; Coley, T.L.; et al. Testing the iron hypothesis in ecosystems of the equatorial Pacific Ocean. Nature 1994, 371, 123–129. [Google Scholar] [CrossRef]
- Boyd, P.W.; Jickells, T.; Law, C.S.; Blain, S.; Boyle, E.A.; Buesseler, K.O.; Coale, K.H.; Cullen, J.J.; de Baar, H.J.W.; Follows, M.; et al. Mesoscale iron enrichment experiments 1993–2005: Synthesis and future directions. Science 2007, 315, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.M.; Mills, M.M.; Arrigo, K.R.; Berman-Frank, I.; Bopp, L.; Boyd, P.W.; Galbraith, E.D.; Geider, R.J.; Guieu, C.; Jaccard, S.L.; et al. Processes and patterns of oceanic nutrient limitation. Nat. Geosci. 2013, 6, 701–710. [Google Scholar] [CrossRef]
- Conway, T.M.; John, S.G. Quantification of dissolved iron sources to the North Atlantic Ocean. Nature 2014, 511, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Sedwick, P.N.; Sholkovitz, E.R.; Church, T.M. Impact of anthropogenic combustion emissions on the fractional solubility of aerosol iron: Evidence from the Sargasso Sea. Geochem. Geophys. Geosyst. 2007, 8, 1–41. [Google Scholar] [CrossRef]
- Takahashi, Y.; Higashi, M.; Furukawa, T.; Mitsunobu, S. Change of iron species and iron solubility in Asian dust during the long-range transport from western China to Japan. Atmos. Chem. Phys. 2011, 11, 11237–11252. [Google Scholar] [CrossRef]
- Ito, A. Atmospheric processing of combustion aerosols as a source of bioavailable iron. Environ. Sci. Technol. Lett. 2015, 2, 70–75. [Google Scholar] [CrossRef]
- Schroth, A.W.; Crusius, J.; Sholkovitz, E.R.; Bostick, B.C. Iron solubility driven by speciation in dust sources to the ocean. Nat. Geosci. 2009, 2, 337–340. [Google Scholar] [CrossRef]
- Myriokefalitakis, S.; Ito, A.; Kanakidou, M.; Nenes, A.; Krol, M.C.; Mahowald, N.M.; Scanza, R.A.; Hamilton, D.S.; Johnson, M.S.; Meskhidze, N.; et al. Reviews and syntheses: The GESAMP atmospheric iron deposition model intercomparison study. Biogeosciences 2018, 15, 6659–6684. [Google Scholar] [CrossRef]
- Sanderson, P.; Delgado-Saborit, J.M.; Harrison, R.M. A review of chemical and physical characterisation of atmospheric metallic nanoparticles. Atmos. Environ. 2014, 94, 353–365. [Google Scholar] [CrossRef]
- Kopcewicz, B.; Kopcewicz, M.; Pietruczuk, A. The Mössbauer study of atmospheric iron-containing aerosol in the coarse and PM2.5 fractions measured in rural site. Chemosphere 2015, 131, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Moteki, N.; Adachi, K.; Ohata, S.; Yoshida, A.; Harigaya, T.; Koike, M.; Kondo, Y. Anthropogenic iron oxide aerosols enhance atmospheric heating. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, M.; Yoshikawa, A.; Muraki, H.; Arao, K.; Uno, I. Transport of mineral and anthropogenic aerosols during a Kosa event over East Asia. J. Geophys. Res. 2002, 107, 4059. [Google Scholar] [CrossRef]
- Maher, B.A.; Ahmed, I.A.M.; Karloukovski, V.; MacLaren, D.A.; Foulds, P.G.; Allsop, D.; Mann, D.M.A.; Torres-Jardón, R.; Calderon-Garciduenas, L. Magnetite pollution nanoparticles in the human brain. Proc. Natl. Acad. Sci. USA 2016, 113, 10797–10801. [Google Scholar] [CrossRef] [PubMed]
- Labatut, M.; Lacan, F.; Pradoux, C.; Chemeleff, J.; Radic, A.; Murray, J.W.; Poitrasson, F.; Johansen, A.M.; Thil, F. Iron sources and dissolved-particulate interactions in the seawater of the Western Equatorial Pacific, iron isotope perspectives. Glob. Biogeochem. Cycles 2014, 28, 1044–1065. [Google Scholar] [CrossRef]
- Kurisu, M.; Sakata, K.; Miyamoto, C.; Takaku, Y.; Iizuka, T.; Takahashi, Y. Variation of iron isotope ratios in anthropogenic materials emitted through combustion processes. Chem. Lett. 2016, 45, 970–972. [Google Scholar] [CrossRef]
- Kurisu, M.; Takahashi, Y.; Iizuka, T.; Uematsu, M. Very low isotope ratio of iron in fine aerosols related to its contribution to the surface ocean. J. Geophys. Res. Atmos. 2016, 121, 11119–11136. [Google Scholar] [CrossRef]
- Kurisu, M.; Adachi, K.; Sakata, K.; Takahashi, Y. Stable isotope ratios of combustion iron produced by evaporation in a steel plant. ACS Earth Space Chem. under review. [CrossRef]
- Beard, B.L.; Johnson, C.M.; Skulan, J.L.; Nealson, K.H.; Cox, L.; Sun, H. Application of Fe isotopes to tracing the geochemical and biological cycling of Fe. Chem. Geol. 2003, 195, 87–117. [Google Scholar] [CrossRef]
- Beard, B.L.; Johnson, C.M.; Von Damm, K.L.; Poulson, R.L. Iron isotope constraints on Fe cycling and mass balance in oxygenated Earth oceans. Geology 2003, 31, 629–632. [Google Scholar] [CrossRef]
- Guieu, C.; Bonnet, S.; Wagener, T.; Loÿe-Pilot, M.D. Biomass burning as a source of dissolved iron to the open ocean? Geophys. Res. Lett. 2005, 32, 1–5. [Google Scholar] [CrossRef]
- Paris, R.; Desboeufs, K.V.; Formenti, P.; Nava, S.; Chou, C. Chemical characterisation of iron in dust and biomass burning aerosols during AMMA-SOP0/DABEX: Implication for iron solubility. Atmos. Chem. Phys. 2010, 10, 4273–4282. [Google Scholar] [CrossRef]
- Winton, V.H.L.; Edwards, R.; Bowie, A.R.; Keywood, M.; Williams, A.G.; Chambers, S.D.; Selleck, P.W.; Desservettaz, M.; Mallet, M.D.; Paton-Walsh, C. Dry season aerosol iron solubility in tropical northern Australia. Atmos. Chem. Phys. 2016, 16, 12829–12848. [Google Scholar] [CrossRef]
- Dauphas, N.; Janney, P.E.; Mendybaev, R.A.; Wadhwa, M.; Richter, F.M.; Davis, A.M.; Van Zuilen, M.; Hines, R.; Foley, C.N. Hromatographic separation and multicollection-ICPMS analysis of iron. Investigating mass-dependent and -independent isotope effects. Anal. Chem. 2004, 76, 5855–5863. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Dauphas, N.; Teng, F. Non-traditional fractionation of non-traditional isotopes: Evaporation, chemical diffusion and Soret diffusion. Chem. Geol. 2009, 258, 92–103. [Google Scholar] [CrossRef]
- Guelke, M.; Von Blanckenburg, F. Fractionation of stable iron isotopes in higher plants. Environ. Sci. Technol. 2007, 41, 1896–1901. [Google Scholar] [CrossRef] [PubMed]
- Mead, C.; Herckes, P.; Majestic, B.J.; Anbar, A.D. Source apportionment of aerosol iron in the marine environment using iron isotope analysis. Geophys. Res. Lett. 2013, 40, 5722–5727. [Google Scholar] [CrossRef]
- Streets, D.G.; Yarber, K.F.; Woo, J.-H.; Carmichael, G.R. Biomass burning in Asia: Annual and seasonal estimates and atmospheric emissions. Glob. Biogeochem. Cycles 2003, 17, 1–20. [Google Scholar] [CrossRef]
- Chen, J.; Li, C.; Ristovski, Z.; Milic, A.; Gu, Y.; Islam, M.S.; Wang, S.; Hao, J.; Zhang, H.; He, C.; Guo, H.; et al. A review of biomass burning: Emissions and impacts on air quality, health and climate in China. Sci. Total Environ. 2017, 579, 1000–1034. [Google Scholar] [CrossRef] [PubMed]
- Japan Meteorological Agency. Available online: www.jma.go.jp/jp/yoho/ (accessed on 11 October 2017).
- Sakata, K.; Kurisu, M.; Tanimoto, H.; Sakaguchi, A.; Uematsu, M.; Miyamoto, C.; Takahashi, Y. Custom-made PTFE filters for ultra-clean size-fractionated aerosol sampling for trace metals. Mar. Chem. 2018, 206, 100–108. [Google Scholar] [CrossRef]
- Keene, W.C.; Pszenny, A.A.P.; Galloway, J.N.; Hawley, M.E. Sea-salt corrections and interpretation of constituent ratios in marine precipitation. J. Geophys. Res. 1986, 91, 6647. [Google Scholar] [CrossRef]
- Luo, C.; Mahowald, N.; Bond, T.; Chuang, P.Y.; Artaxo, P.; Siefert, R.; Chen, Y.; Schauer, J. Combustion iron distribution and deposition. Glob. Biogeochem. Cycles 2008, 22, 1–17. [Google Scholar] [CrossRef]
- Hsu, S.C.; Wong, G.T.F.; Gong, G.C.; Shiah, F.K.; Huang, Y.T.; Kao, S.J.; Tsai, F.; Candice Lung, S.C.; Lin, F.J.; Lin, I.I.; et al. Sources, solubility, and dry deposition of aerosol trace elements over the East China Sea. Mar. Chem. 2010, 120, 116–127. [Google Scholar] [CrossRef]
- Morton, P.L.; Landing, W.M.; Hsu, S.C.; Milne, A.; Aguilar-Islas, A.M.; Baker, A.R.; Bowie, A.R.; Buck, C.S.; Gao, Y.; Gichuki, S.; et al. Methods for the sampling and analysis of marine aerosols: Results from the 2008 GEOTRACES aerosol intercalibration experiment. Limnol. Oceanogr. Methods 2013, 11, 62–78. [Google Scholar] [CrossRef]
- Takeichi, Y.; Inami, N.; Suga, H.; Takahashi, Y.; Ono, K. Compact scanning transmission X-ray microscope at the photon factory. AIP Conf. Proc. 2016, 1696. [Google Scholar] [CrossRef]
- Keiluweit, M.; Nico, P.S.; Johnson, M.G. Dynamic molecular structure of plant biomass-derived black carbon (biochar). Environ. Sci. Technol. 2010, 44, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Cheng, C.H.; Huang, Y.H.; Chen, C.-T.; Lai, C.M.; Menyailo, O.V.; Fan, L.J.; Yang, Y.W. Converting leguminous green manure into biochar: Changes in chemical composition and C and N mineralization. Geoderma 2014, 232–234, 581–588. [Google Scholar] [CrossRef]
- Albarède, F.; Telouk, P.; Blichert-Toft, J.; Boyet, M.; Agranier, A.; Nelson, B. Precise and accurate isotopic measurements using multiple-collector ICPMS. Geochim. Cosmochim. Acta 2004, 68, 2725–2744. [Google Scholar] [CrossRef]
- Weyer, S.; Anbar, A.D.; Brey, G.P.; Münker, C.; Mezger, K.; Woodland, A.B. Iron isotope fractionation during planetary differentiation. Earth Planet. Sci. Lett. 2005, 240, 251–264. [Google Scholar] [CrossRef]
- Atmospheric Environmental Information System. Available online: http://atmospheric-monitoring.jp/pref/tochigi/index.html (accessed on 1 February 2019).
- Andreae, M.O.; Browell, E.V.; Garstang, G.L.; Harriss, R.C.; Hill, G.F.; Jacobs, D.J.; Pereira, M.C.; Sachse, G.W.; Setzer, A.W.; Silva Dias, P.L.; et al. Biomass-burning emissions and associated haze layers over Amazonia. J. Geophys. Res. 1988, 93, 1509–1527. [Google Scholar] [CrossRef]
- Deng, C.; Zhuang, G.; Huang, K.; Li, J.; Zhang, R.; Wang, Q.; Liu, T.; Sun, Y.; Guo, Z.; Fu, J.S.; et al. Chemical characterization of aerosols at the summit of Mountain Tai in Central East China. Atmos. Chem. Phys. 2011, 11, 7319–7332. [Google Scholar] [CrossRef]
- Lamarque, J.F.; Bond, T.C.; Eyring, V.; Granier, C.; Heil, A.; Klimont, Z.; Lee, D.; Liousse, C.; Mieville, A.; Owen, B.; et al. Historical (1850-2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: Methodology and application. Atmos. Chem. Phys. 2010, 10, 7017–7039. [Google Scholar] [CrossRef]
- Echalar, F.; Gaudichet, A.; Cachier, H.; Artaxo, P. Aerosol emissions by tropical forest and savanna biomass burning: characteristic trace elements and fluxes. Geophys. Res. Lett. 1995, 22, 3039–3042. [Google Scholar] [CrossRef]
- Nriagu, J.O.; Pacnya, J.M. Quantative assessment of worldwide contamination of air, water and soils by trace metals. Nature 1988, 333, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Artaxo, P.; Storms, H.; Bruynseels, F.; Van Grieken, R.; Maenhaut, W. Composition and sources of aerosols from the Amazon Basin. J. Geophys. Res. 1988, 93, 1605–1615. [Google Scholar] [CrossRef]
- Yamada, E.; Funoki, S.; Abe, Y.; Umemura, S.; Yamaguchi, D.; Fuse, Y. Size distribution and characteristics of chemical components in ambient particulate matter. Anal. Sci. 2005, 21, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Cuiping, L.; Chuangzhi, W.; Yanyongjie; Haitao, H. Chemical elemental characteristics of biomass fuels in China. Biomass Bioenergy 2004, 27, 119–130. [Google Scholar] [CrossRef]
- Barker, A.V.; Pilbeam, D.J. Handbook of Plant Nutrition; CRC press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Taylor, S.R. Abundance of chemical elements in the continental crust: A new table. Geochim. Cosmochim. Acta 1964, 28, 1273–1285. [Google Scholar] [CrossRef]
- Andreae, M.O. Soot carbon and excess fire potassium: Long-range transport of combustion-derived aerosols. Science (80-.) 1983, 220, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Mahowald, N.M.; Hamilton, D.S.; Mackey, K.R.M.; Moore, J.K.; Baker, A.R.; Scanza, R.A.; Zhang, Y. Aerosol trace metal leaching and impacts on marine microorganisms. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Andreae, M.O.; Artaxo, P.; Fischer, H.; Freitas, S.R.; Gregoire, J.-M.; Hansel, A.; Hoor, P.; Kormann, R.; Krejci, R.; Lange, L.; et al. Transport of biomass burning smoke to the upper troposphere by deep convection in the equatorial region. Geophys. Res. Lett. 2001, 28, 951–954. [Google Scholar] [CrossRef]
- Artaxo, P.; Martins, J.V.; Yamasoe, M.A.; Procópio, A.S.; Pauliquevis, T.M.; Andreae, M.O.; Guyon, P.; Gatti, L.V.; Leal, A.M.C. Physical and chemical properties of aerosols in the wet and dry seasons in Rondônia, Amazonia. J. Geophys. Res. D Atmos. 2002, 107, 1–14. [Google Scholar] [CrossRef]
- Siefert, R.L.; Webb, S.M.; Hoffmann, M.R. Determination of photochemically available iron in ambient aerosols. J. Geophys. Res. 1996, 101, 14441–14449. [Google Scholar] [CrossRef]
- Miller, A.; Ahlstrand, G.; Kittelson, D.; Zachariah, M. The fate of metal (Fe) during diesel combustion: Morphology, chemistry, and formation pathways of nanoparticles. Combust. Flame 2007, 149, 129–143. [Google Scholar] [CrossRef]
- Kubaschewski, O.; Alcock, C.B. Metallurgical Thermochemistry, 3rd ed.; Pergamon: Oxford, UK, 1979. [Google Scholar]
- Symonds, R.B.; Reed, M.H.; Rose, W.I. Origin, speciation, and fluxes of trace-element gases at Augustine volcano, Alaska: Insights into magma degassing and fumarolic processes. Geochim. Cosmochim. Acta 1992, 56, 633–657. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. Noaa’s hysplit atmospheric transport and dispersion modeling system. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Takahashi, Y.; Furukawa, T.; Kanai, Y.; Uematsu, M.; Zheng, G.; Marcus, M.A. Seasonal changes in Fe species and soluble Fe concentration in the atmosphere in the Northwest Pacific region based on the analysis of aerosols collected in Tsukuba, Japan. Atmos. Chem. Phys. 2013, 13, 7695–7710. [Google Scholar] [CrossRef]
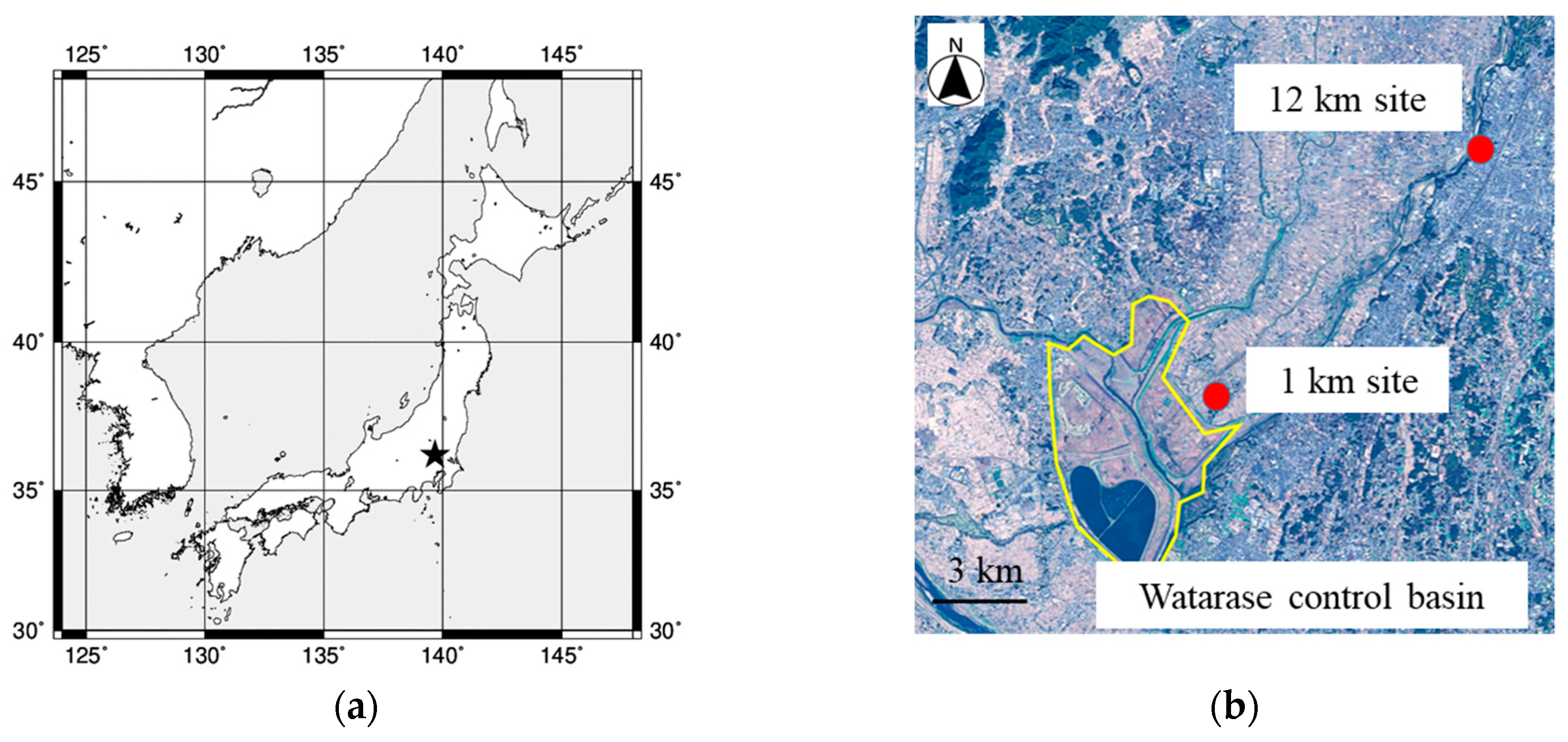





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Start | End | Total Flow Volume (m3) |
|---|---|---|---|
| 12 km (before) | 19 March 2016, 12:22 p.m. | 26 March 2016, 6:47 a.m. | 5265.3 |
| 12 km (event) | 26 March 2016, 7:07 a.m. | 26 March 2016, 6:15 p.m. | 385.6 |
| 1 km (event) | 26 March 2016, 7:59 a.m. | 26 March 2016, 4:55 p.m. | 300.4 |
| 12 km (after) | 26 March 2016, 6:34 a.m. | 31 March 2016 1:42 p.m. | 3996.8 |
| Element | Watarase Soil (wt %) | Reed (wt %, Dry Biomass Basis) | Various Plants [50,51] (wt %, Dry Biomass Basis) |
|---|---|---|---|
| K | 0.64 | 0.26 | 0.1–7 |
| Al | 4.2 | 0.016 | 0.001–0.5 |
| Ti | 0.20 | 0.00083 | 0.001–0.02 |
| Fe | 2.2 | 0.013 | 0.002–0.5 |
| Zn | 0.04 | 0.00097 | 0.001–0.02 |
| Pb | 0.002 | 0.00025 | 0.00001–0.006 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurisu, M.; Takahashi, Y. Testing Iron Stable Isotope Ratios as a Signature of Biomass Burning. Atmosphere 2019, 10, 76. https://doi.org/10.3390/atmos10020076
Kurisu M, Takahashi Y. Testing Iron Stable Isotope Ratios as a Signature of Biomass Burning. Atmosphere. 2019; 10(2):76. https://doi.org/10.3390/atmos10020076
Chicago/Turabian StyleKurisu, Minako, and Yoshio Takahashi. 2019. "Testing Iron Stable Isotope Ratios as a Signature of Biomass Burning" Atmosphere 10, no. 2: 76. https://doi.org/10.3390/atmos10020076
APA StyleKurisu, M., & Takahashi, Y. (2019). Testing Iron Stable Isotope Ratios as a Signature of Biomass Burning. Atmosphere, 10(2), 76. https://doi.org/10.3390/atmos10020076