Assembly of the Boechera retrofracta Genome and Evolutionary Analysis of Apomixis-Associated Genes



Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Information
2.2. Sequencing Strategy
2.3. Raw Data Filtration and Pre-Processing
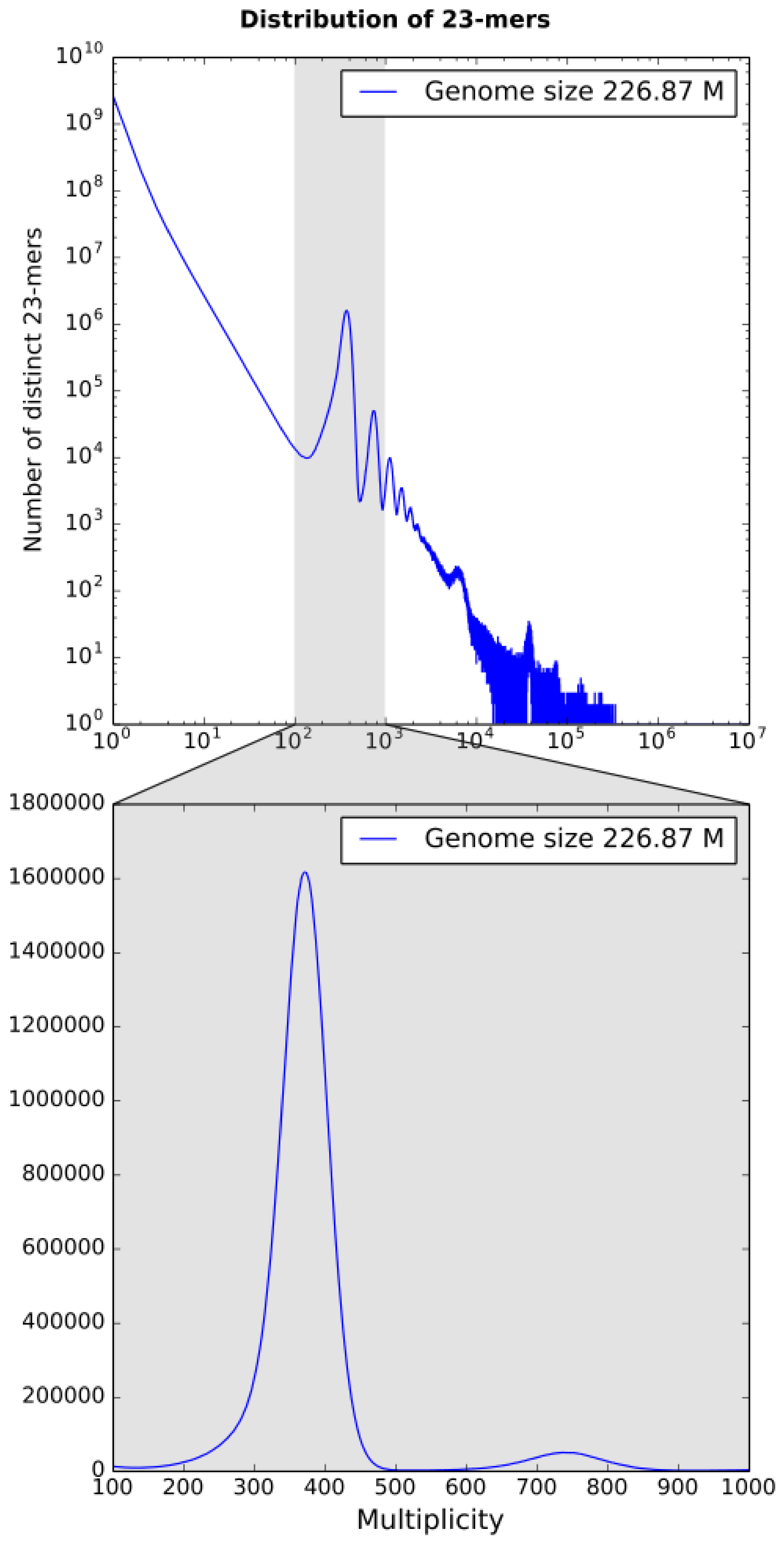
2.4. Genome Size Estimation
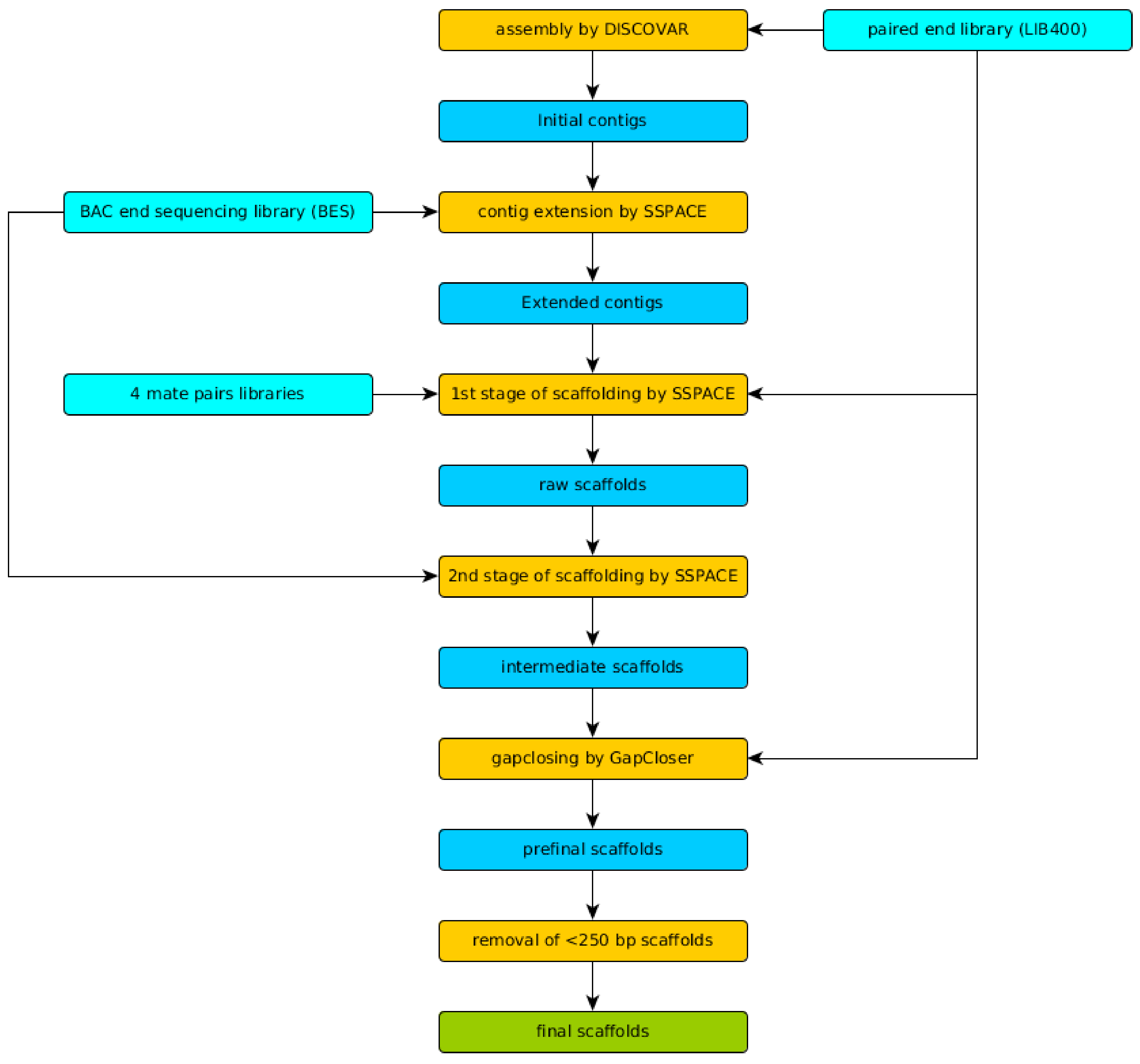
2.5. Genome Assembly and Quality Evaluation
2.6. Repeats Analysis
2.7. Variants Calling and Genotyping
2.8. Prediction of Protein-Coding Genes and Non-Coding RNA
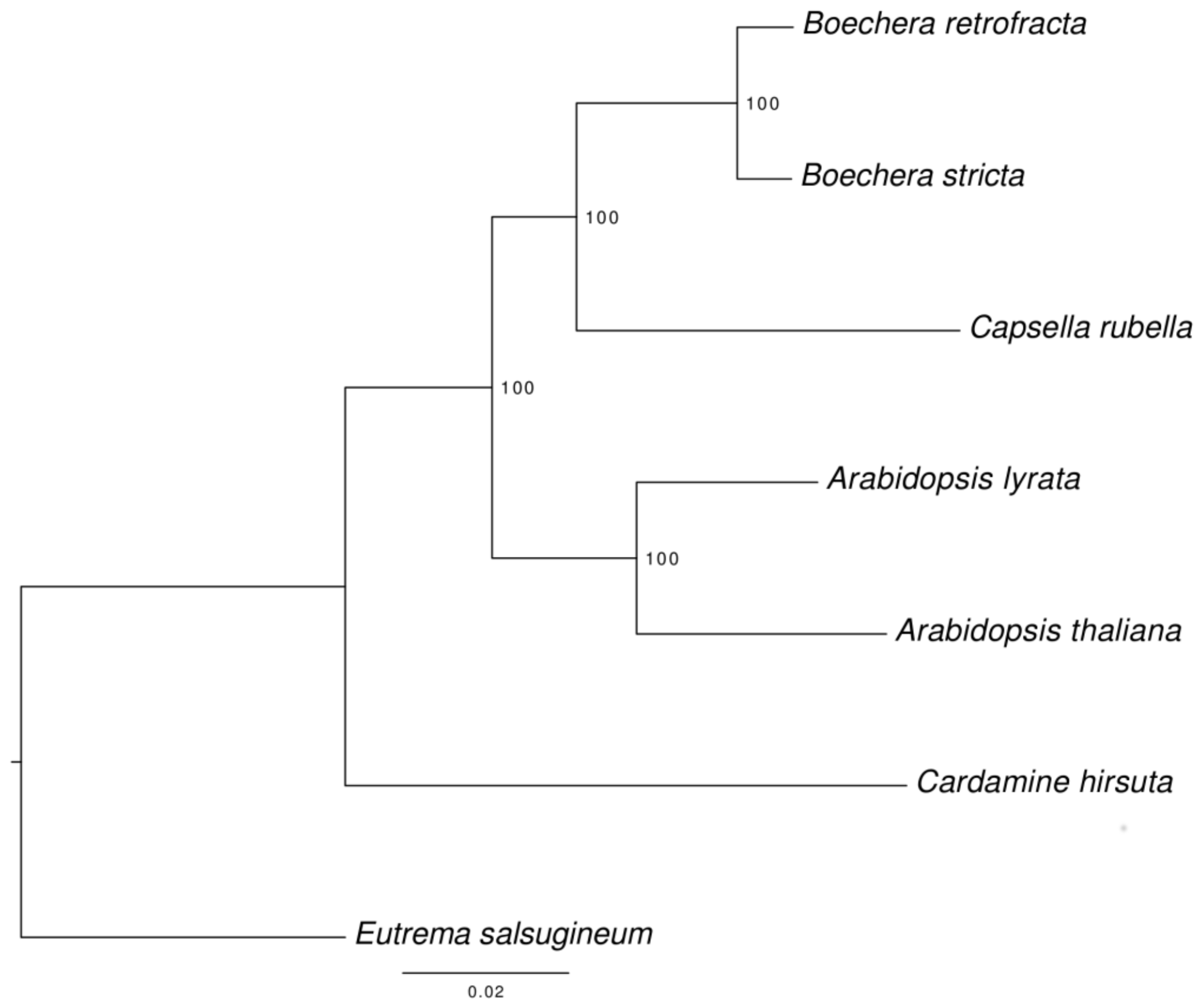
2.9. Phylogenetic Analysis
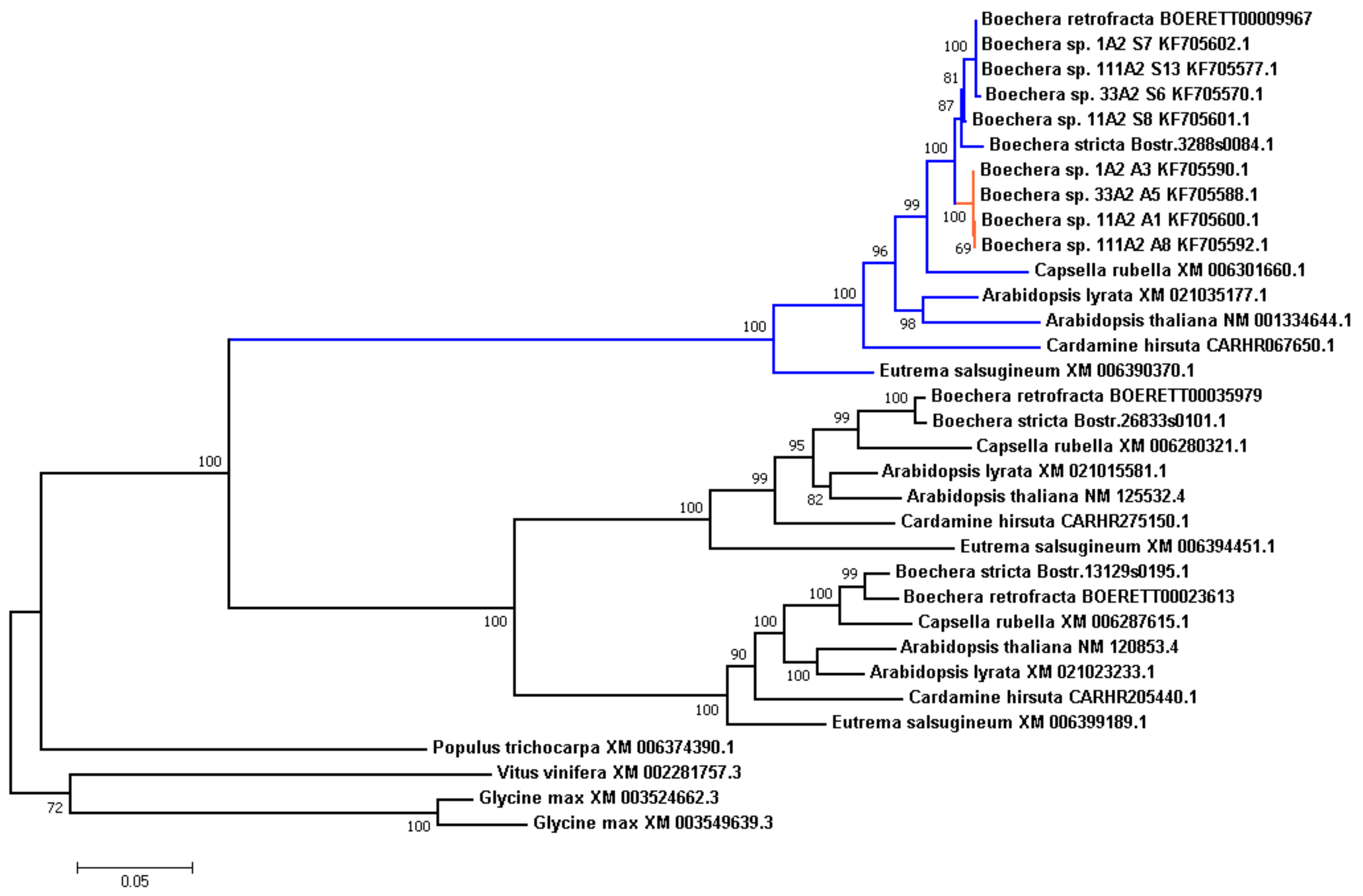
2.10. APOLLO Evolution Analysis
2.11. Whole-Genome Comparison
3. Results
3.1. k-mer Based Statistics
3.2. Genome Assembly and Evaluation
3.3. Repeats Annotation
3.4. Variant Calling and Genotyping
3.5. Prediction of Protein-Coding Genes and Non-Coding RNAs
3.6. Species Tree Reconstruction
3.7. Analysis of Evolution of the APOLLO Locus
3.8. Whole-Genome Comparison
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Library Type | Platform | Read Length | Mean Insert Size (bp) | Number of Reads Pairs |
|---|---|---|---|---|---|
| LIB400 | paired ends | Illumina | 250 | 402 | 189788627 |
| LIB4000R | mate pairs | Roche | - | 4014 | 3259085 |
| LIB5000 | mate pairs | Illumina | 150 | 4877 | 19083787 |
| LIB7000 | mate pairs | Illumina | 150 | 6882 | 34066282 |
| LIB24000R | mate pairs | Roche | - | 24,332 | 672098 |
| BES | BAC end sequencing | Sanger | - | 147,708 | 17775 |

Appendix B
Appendix C

References
- Sharbel, T.F.; Mitchell-Olds, T. Recurrent polyploid origins and chloroplast phylogeography in the Arabis holboellii complex (Brassicaceae). Heredity 2001, 87, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Schranz, M.E.; Dobes, C.; Koch, M.A.; Mitchell-Olds, T. Sexual reproduction, hybridization, apomixis and polyploidization in the genus Boechera (Brassicaceae). Am. J. Bot. 2005, 92, 1797–1810. [Google Scholar] [CrossRef] [PubMed]
- Naumova, T.N. Apomixis and amphimixis in flowering plants. Cytol. Genet. 2008, 3, 51–63. [Google Scholar] [CrossRef]
- Aliyu, O.M.; Schranz, M.E.; Sharbel, T.F. Quantitative variation for apomictic reproduction in the genus Boechera (Brassicaceae). Am. J. Bot. 2010, 97, 1719–1731. [Google Scholar] [CrossRef] [PubMed]
- Koltunow, A.M.; Grossniklaus, U. Apomixis: A developmental perspective. Annu. Rev. Plant Biol. 2003, 54, 547–574. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Leal, D.; Vielle-Calzada, J.P. Regulation of apomixis: Learning from sexual experience. Curr. Opin. Plant Biol. 2012, 15, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Brukhin, V. Molecular and genetic regulation of apomixis. Russ. J. Genet. 2017, 53, 943–964. [Google Scholar] [CrossRef]
- Windham, M.D.; Al-Shehbaz, I.A. New and noteworthy species of Boechera (Brassicaceae) II: Apomictic hybrids. Harv. Pap. Bot. 2007, 11, 257–274. [Google Scholar] [CrossRef]
- Windham, M.D.; Al-Shehbaz, I.A. New and noteworthy species of Boechera (Brassicaceae) III: Additional sexual diploids and apomictic hybrids. Harv. Pap. Bot. 2007, 12, 235–257. [Google Scholar] [CrossRef]
- Lovell, J.T.; Williamson, R.J.; Wright, S.I.; McKay, J.K.; Sharbel, T.F. Mutation Accumulation in an Asexual Relative of Arabidopsis. PLoS Genet. 2017, 13, e1006550. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, C.A.; Song, B.H.; Lee, C.R.; Mitchell-Olds, T. Boechera, a model system for ecological genomics. Mol. Ecol. 2011, 20, 4843–4857. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.B.; Alexander, P.J.; Allphin, L.; Al-Shehbaz, I.A.; Rushworth, C.; Bailey, C.D.; Windham, M.D. Does hybridization drive the transition to asexuality in diploid Boechera? Evolution 2011, 66, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Kantama, L.; Sharbel, T.F.; Schranz, M.E.; Mitchell-Olds, T.; de Vries, S.; de Jong, H. Diploid apomicts of the Boechera holboellii complex display large-scale chromosome substitutions and aberrant chromosomes. Proc. Natl. Acad. Sci. USA 2007, 104, 14026–14031. [Google Scholar] [CrossRef] [PubMed]
- Mandáková, T.; Schranz, M.E.; Sharbel, T.F.; de Jong, H.; Lysak, M.A. Karyotype evolution in apomictic Boechera and the origin of the aberrant chromosomes. Plant J. 2015, 82, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Windham, M.D.; Al-Shehbaz, I.A. New and noteworthy species of Boechera I: Sexual diploids. Harv. Pap. Bot. 2006, 11, 61–88. [Google Scholar] [CrossRef]
- Lee, C.R.; Wang, B.; Mojica, J.P.; Mandáková, T.; Prasad, K.V.S.K.; Goicoechea, J.L.; Perera, N.; Hellsten, U.; Hundley, H.N.; Johnson, J.; Grimwood, J.; et al. Young inversion with multiple linked QTLs under selection in a hybrid zone. Nat. Ecol. Evol. 2017, 1, 0119. [Google Scholar] [CrossRef] [PubMed]
- Why Sequence Boechera holboellii? Available online: http://jgi.doe.gov/why-sequence-boechera-holboellii/ (accessed on 1 November 2017).
- Weisenfeld, N.I.; Yin, S.; Sharpe, T.; Lau, B.; Hegarty, R.; Holmes, L.; Sogoloff, B.; Tabbaa, D.; Williams, L.; Russ, C.; Nusbaum, C.; et al. Comprehensive variation discovery in single human genomes. Nat. Genet. 2014, 46, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Starostina, E.; Tamazian, G.; Dobrynin, P.; O’Brien, S.; Komissarov, A. Cookiecutter: A tool for kmer-based read filtering and extraction. bioRxiv 2015. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Leggett, R.M.; Clavijo, B.J.; Clissold, L.; Clark, M.D.; Caccamo, M. NextClip: An analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 2013, 30, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Kliver, S. CreClip. Available online: https://github.com/mahajrod/CreClip (accessed on 1 July 2016).
- Kliver, S.; Tamazian, G.; O’Brien, S.J.; Brukhin, V.; Komissarov, A. KrATER (K-mer Analysis Tool Easy to Run). Available online: https://github.com/mahajrod/KrATER (accessed on 1 November 2017).
- Anderson, J.T.; Willis, J.H.; Mitchell-Olds, T. Evolutionary genetics of plant adaptation. Trends Genet. 2011, 27, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2010, 27, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; Tang, J. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R. RepeatModeler Open, version 1.0; Institute for Systems Biology: Seattle, WA, USA, 2008. [Google Scholar]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open, version 4.0; Institute for Systems Biology: Seattle, WA, USA, 2013. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Gertz, E.M.; Schäffer, A.A.; Agarwala, R. WindowMasker: Window-based masker for sequenced genomes. Bioinformatics 2005, 22, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43. [Google Scholar] [CrossRef]
- Slater, G.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32 (Suppl. 1), D138–D141. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Victorian Bioinformatics Consortium. Barrnap. Available online: http://www.vicbioinformatics.com/software.barrnap.shtml (accessed on 1 November 2017).
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 November 2017).
- Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 2014, 1079, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Paten, B.; Earl, D.; Nguyen, N.; Diekhans, M.; Zerbino, D.; Haussler, D. Cactus: Algorithms for genome multiple sequence alignment. Genome Res. 2011, 21, 1512–1528. [Google Scholar] [CrossRef] [PubMed]
- Cheong, W.H.; Tan, Y.C.; Yap, S.J.; Ng, K.P. ClicO FS: An interactive web-based service of Circos. Bioinformatics 2015, 31, 3685–3687. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.E.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Corral, J.M.; Vogel, H.; Aliyu, O.M.; Hensel, G.; Thiel, T.; Kumlehn, J.; Sharbel, T.F. A conserved apomixis-specific polymorphism is correlated with exclusive exonuclease expression in premeiotic ovules of apomictic Boechera species. Plant Physiol. 2013, 163, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Phytozome v12.1 Database. Available online: https://phytozome.jgi.doe.gov/pz/portal.html (accessed on 30 January 2018).




| Parameter | Contigs | Extended Contigs | Raw Scaffolds | Intermediate Scaffolfs | Gap Closed Scaffolds | Final Scaffolds |
|---|---|---|---|---|---|---|
| Longest contig | 791,985 | 792,340 | 8,101,256 | 9,045,706 | 9,049,080 | 9,049,080 |
| Ns | 28,100 | 28,100 | 11,890,519 | 16,366,994 | 12,409,189 | 12,409,189 |
| Total length | 225,649 216 | 226,402,628 | 236,469,041 | 240,945,496 | 241,014,839 | 222,253,471 |
| Scaffold Length Cutoff | Contigs | Extended Contigs | Raw Scaffolds | Intermediate Scaffolfs | Gap Closed Scaffolds | Final Scaffolds |
|---|---|---|---|---|---|---|
| all | 85,286 | 84,648 | 1,256,534 | 1,898,006 | 1,898,985 | 2,297,899 |
| ≥100 | 85,286 | 84,648 | 1,256,534 | 1,898,006 | 1,898,985 | 2,297,899 |
| ≥250 | 101,388 | 100,393 | 1,442,421 | 2,296,484 | 2,297,899 | 2,297,899 |
| ≥500 | 115,732 | 115,486 | 1,538,795 | 2,678,857 | 2,680,941 | 2,680,941 |
| ≥1000 | 122,300 | 121,678 | 1,704,064 | 2,678,857 | 2,680,941 | 2,680,941 |
| Class | Number of Elements | Total Length (bp) | Fraction of Assembly (%) |
|---|---|---|---|
| SINEs | 577 | 125,298 | 0.06 |
| LINEs | 7075 | 4,351,241 | 1.96 |
| LTR elements | 51,040 | 40,608,195 | 18.27 |
| DNA elements | 31,638 | 12,868,684 | 5.79 |
| Unclassified | 82,693 | 24,363,135 | 10.96 |
| Total interspersed repeats | - | 82,316,553 | 37.04 |
| Small RNA | 5461 | 1,599,354 | 0.72 |
| Satellites | 1541 | 573,026 | 0.26 |
| Simple repeats | 2044 | 363,642 | 0.16 |
| Low complexity | 56 | 7456 | 0 |
| Tool | Number of Repeats | Total Length (Mbp) |
|---|---|---|
| RepeatMasker | 173,023 | 82.31 |
| TRF | 100,593 | 17.41 |
| Windowmasker | 1,104,650 | 64.20 |
| tRNA Type | Number |
|---|---|
| tRNAs decoding standard 20 AA | 1126 |
| Selenocysteine tRNAs (TCA) | 0 |
| Possible suppressor tRNAs (CTA,TTA) | 3 |
| tRNAs with undetermined isotypes | 5 |
| Resolution of Brassicaceae Phylogeny Using Nuclear Genes Uncovers Nested Radiations and Supports Convergent Morphological Evolution Predicted pseudogenes | 32 |
| Total tRNAs | 1166 |
| rRNA | Complete (≥80% of Expected Length) | Partial (<80% of Expected Length) |
|---|---|---|
| 5.8S | 178 | 53 |
| 5S | 601 | 104 |
| 28S | 0 | 1782 |
| 18S | 1 | 1458 |
| 12S | 0 | 173 |
| 16S | 0 | 607 |
| Boechera retrofracta | Boechera stricta v.1.2 | Arabidopsis lyrata v2.1 | Arabidopsis thaliana TAIR10 | |
|---|---|---|---|---|
| Total length | 227 M | 184 M | 207 Mb | 135 Mb |
| Chromosomes | n = 7 | n = 7 | n = 8 | n = 5 |
| Protein-coding loci | 27,048 | 27,416 | 31,073 | 27,416 |
| Transcripts | 28,269 | 29,812 | 33,132 | 35,386 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kliver, S.; Rayko, M.; Komissarov, A.; Bakin, E.; Zhernakova, D.; Prasad, K.; Rushworth, C.; Baskar, R.; Smetanin, D.; Schmutz, J.; et al. Assembly of the Boechera retrofracta Genome and Evolutionary Analysis of Apomixis-Associated Genes. Genes 2018, 9, 185. https://doi.org/10.3390/genes9040185
Kliver S, Rayko M, Komissarov A, Bakin E, Zhernakova D, Prasad K, Rushworth C, Baskar R, Smetanin D, Schmutz J, et al. Assembly of the Boechera retrofracta Genome and Evolutionary Analysis of Apomixis-Associated Genes. Genes. 2018; 9(4):185. https://doi.org/10.3390/genes9040185
Chicago/Turabian StyleKliver, Sergei, Mike Rayko, Alexey Komissarov, Evgeny Bakin, Daria Zhernakova, Kasavajhala Prasad, Catherine Rushworth, R. Baskar, Dmitry Smetanin, Jeremy Schmutz, and et al. 2018. "Assembly of the Boechera retrofracta Genome and Evolutionary Analysis of Apomixis-Associated Genes" Genes 9, no. 4: 185. https://doi.org/10.3390/genes9040185
APA StyleKliver, S., Rayko, M., Komissarov, A., Bakin, E., Zhernakova, D., Prasad, K., Rushworth, C., Baskar, R., Smetanin, D., Schmutz, J., Rokhsar, D. S., Mitchell-Olds, T., Grossniklaus, U., & Brukhin, V. (2018). Assembly of the Boechera retrofracta Genome and Evolutionary Analysis of Apomixis-Associated Genes. Genes, 9(4), 185. https://doi.org/10.3390/genes9040185