Genome Size Diversity and Its Impact on the Evolution of Land Plants



Abstract
1. Introduction
2. Genome Size Diversity across Land Plants: What Do We Know?
3. Mechanisms Contributing to Genome Size Changes and its Impact on the Structure of the Genome
4. The Impact of Genome Size in Plant Diversity: From Species to Ecology
5. Concluding Remarks and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Greilhuber, J.; Doležel, J.; Lysak, M.A.; Bennett, M.D. The origin, evolution and proposed stabilization of the terms ‘Genome size’ and ‘C-value’ to describe nuclear DNA contents. Ann. Bot. 2005, 95, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Swift, H. The constancy of desoxyribose nucleic acid in plant nuclei. Proc. Natl. Acad. Sci. USA 1950, 36, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Leitch, I.J.; Johnston, E.; Pellicer, J.; Hidalgo, O.; Bennett, M.D. Angiosperm DNA C-values database (release 7.0). 2017. Available online: http://data.kew.org/cvalues/ (accessed on 29 November 2017).
- Gregory, T.R. Animal Genome Size Database. 2018. Available online: http://www.genomesize.com (accessed on 3 January 2018).
- Kullman, B.; Tamm, H.; Kullman, K. Fungal Genome Size Database. 2005. Available online: http://www.zbi.ee/fungal-genomesize (accessed on 14 November 2017).
- Hidalgo, O.; Pellicer, J.; Christenhusz, M.J.M.; Schneider, H.; Leitch, A.R.; Leitch, I.J. Is there an upper limit to genome size? Trends Plant Sci. 2017, 22, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.J.; Leitch, A.R.; Fay, M.F.; Renny-Byfield, S.; Pellicer, J.; Macas, J.; Leitch, I.J. Why size really matters when sequencing plant genomes. Plant Ecol. 2012, 5, 415–425. [Google Scholar] [CrossRef]
- Kenrick, P.; Crane, P.R. The origin and early evolution of plants on land. Nature 1997, 389, 33–39. [Google Scholar] [CrossRef]
- Wellman, C.H.; Osterloff, P.L.; Mohiuddin, U. Fragments of the earliest land plants. Nature 2003, 425, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Leitch, I.J.; Anadon-Rosell, A.; Canela, M.Á.; Gálvez, F.; Garnatje, T.; Gras, A.; Hidalgo, O.; Johnston, E.; Mas de Xaxars, G.; et al. Recent updates and developments to plant genome size databases. Nucl. Acids Res. 2014, 42, D1159–D1166. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.J.; Brummitt, N.; Govaerts, R.; Harman, K.; Hinchcliffe, S.; Allkin, B.; Lughadha, E.N. Towards Target 1 of the Global Strategy for Plant Conservation: A working list of all known plant species-progress and prospects. Taxon 2008, 57, 602–611. [Google Scholar]
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Leitch, I.J.; Leitch, A.R. Genome size diversity and evolution in land plants. In Plant Genome Diversity, Volume 2, Physical Structure, Behaviour and Evolution of Plant Genomes; Leitch, I.J., Greilhuber, J., Doležel, J., Wendel, J.F., Eds.; Springer-Verlag: Wien, Austria, 2013; pp. 307–322. [Google Scholar]
- Zanne, A.E.; Tank, D.C.; Cornwell, W.K.; Eastman, J.M.; Smith, S.A.; FitzJohn, R.G.; McGlinn, D.J.; O’Meara, B.C.; Moles, A.T.; Reich, P.B.; et al. Three keys to the radiation of angiosperms into freezing environments. Nature 2014, 506, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R. Genome size evolution in animals. In The Evolution of the Genome; Gregory, T.R., Ed.; Elsevier: San Diego, CA, USA, 2005; pp. 3–87. [Google Scholar]
- Fleischmann, A.; Michael, T.P.; Rivadavia, F.; Sousa, A.; Wang, W.; Temsch, E.M.; Greilhuber, J.; Müller, K.F.; Heubl, G. Evolution of genome size and chromosome number in the carnivorous plant genus Genlisea (Lentibulariaceae), with a new estimate of the minimum genome size in angiosperms. Ann. Bot. 2014, 114, 1651–1663. [Google Scholar] [CrossRef] [PubMed]
- Baniaga, A.E.; Arrigo, N.; Barker, M.S. The small nuclear genomes of Selaginella are associated with a low rate of genome size evolution. Genome Biol. Evol. 2016, 8, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Fay, M.F.; Leitch, I.J. The largest eukaryotic genome of them all? Bot. J. Linn. Soc. 2010, 164, 10–15. [Google Scholar] [CrossRef]
- Hidalgo, O.; Pellicer, J.; Christenhusz, M.J.M.; Schneider, H.; Leitch, I.J. Genomic gigantism in the whisk-fern family (Psilotaceae): Tmesipteris obliqua challenges record holder Paris japonica. Bot. J. Linn. Soc. 2017, 183, 509–514. [Google Scholar] [CrossRef]
- Kelly, L.J.; Renny-Byfield, S.; Pellicer, J.; Macas, J.; Novák, P.; Neumann, P.; Lysak, M.A.; Day, P.D.; Berger, M.; Fay, M.F.; et al. Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. New Phytol. 2015, 208, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Kovach, A.; Wegrzyn, J.L.; Parra, G.; Holt, C.; Bruening, G.E.; Loopstra, C.A.; Hartigan, J.; Yandell, M.; Langley, C.H.; Korf, I.; et al. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genomics 2010, 11, 420. [Google Scholar] [CrossRef] [PubMed]
- Zonneveld, B.J.M. New record holders for maximum genome size in eudicots and monocots. J. Bot. 2010, 2010, 527357. [Google Scholar] [CrossRef]
- Clark, J.; Hidalgo, O.; Pellicer, J.; Liu, H.; Marquardt, J.; Robert, Y.; Christenhusz, M.; Zhang, S.; Gibby, M.; Leitch, I.J.; et al. Genome evolution of ferns: Evidence for relative stasis of genome size across the fern phylogeny. New Phytol. 2016, 210, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Dodsworth, S.; Chase, M.W.; Leitch, A.R. Is post-polyploidization diploidization the key to the evolutionary success of angiosperms? Bot. J. Linn. Soc. 2016, 180, 1–5. [Google Scholar] [CrossRef]
- Leitch, I.J.; Bennett, M.D. Genome downsizing in polyploid plants. Biol. J. Linn. Soc. 2004, 82, 651–663. [Google Scholar] [CrossRef]
- Murray, B. Karyotype variation and evolution in gymnosperms. In Plant Genome Diversity, Volume 2, Physical Structure, Behaviour and Evolution of Plant Genomes; Leitch, I.J., Greilhuber, J., Doležel, J., Wendel, J.F., Eds.; Springer-Verlag: Wien, Austria, 2013; pp. 231–243. [Google Scholar]
- Wu, H.; Ma, Z.; Wang, M.-M.; Qin, A.-L.; Ran, J.-H.; Wang, X.-Q. A high frequency of allopolyploid speciation in the gymnospermous genus Ephedra and its possible association with some biological and ecological features. Mol. Evol. 2016, 25, 1192–1210. [Google Scholar]
- Ickert-Bond, S.M. Systematics of New World Ephedra L. (Ephedraceae): Integrating Morphological and Molecular Data; Arizona State University: Tempe, AZ, USA, 2003; p. 363. [Google Scholar]
- Romo, A.; Hidalgo, O.; Boratyński, A.; Sobierajska, K.; Jasińska, A.K.; Vallès, J.; Garnatje, T. Genome size and ploidy levels in highly fragmented habitats: the case of western Mediterranean Juniperus (Cupressaceae) with special emphasis on J. thurifera L. Tree Genet. 2013, 9, 587–599. [Google Scholar] [CrossRef]
- Šmarda, P.; Veselý, P.; Šmerda, J.; Bureš, P.; Knápek, O.; Chytrá, M. Polyploidy in a ‘living fossil’ Ginkgo biloba. New Phytol. 2016, 212, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Fozuar, B.S.; Libby, W.J. Chromosomes of Sequoia sempervirens; 8–Hydroxy-Quinoline-Castor Oil Pretreatment for Improving Preparation. Stain Tech. 1968, 43, 97–100. [Google Scholar] [CrossRef]
- Jaramillo-Correa, J.P.; Verdú, M.; González-Martínez, S.C. The contribution of recombination to heterozygosity differs among plant evolutionary lineages and life-forms. BMC Evol. Biol. 2010, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Recombination: The good, the bad and the variable. Philos. Trans. R. Soc. B. Biol. Sci. 2017, 372, 20170279. [Google Scholar] [CrossRef] [PubMed]
- Morse, A.M.; Peterson, D.G.; Islam-Faridi, M.N.; Smith, K.E.; Magbanua, Z.; Garcia, S.A.; Kubisiak, T.L.; Amerson, H.V.; Carlson, J.E.; Nelson, C.D.; et al. Evolution of genome size and complexity in Pinus. PLoS ONE 2009, 4, e4332. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Lin, B.; Zieve, J.; Dougherty, W.; Martinez-Garcia, P.; Koriabine, M.; Holtz-Morris, A.; de Jong, P.; Crepeau, M.; Langley, C.; et al. Insights into the loblolly pine genome: Characterization of BAC and fosmid sequences. PLoS ONE 2013, 8, e72439. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Zhao, Y.; Zhang, H.; Fan, G.; Liu, X.; Zhou, W.; Shi, C.; Wang, J.; Liu, W.; Liang, X.; et al. Draft genome of the living fossil Ginkgo biloba. GigaScience 2016, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Cossu, R.M.; Casola, C.; Giacomello, S.; Vidalis, A.; Scofield, D.G.; Zuccolo, A. LTR retrotransposons show row levels of unequal recombination and high rates of intraelement gene conversion in large plant genomes. Genome Biol. Evol. 2017, 9, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Liu, Z.-M.; Li, L.-F.; Leitch, A.R.; Leitch, I.J.; Lohaus, R.; Liu, Z.-J.; Xin, H.-P.; Gong, Y.-B.; Liu, Y.; et al. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 2018, 4, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Schubert, I.; Vu, G.T.H. Genome stability and evolution: Attempting a holistic view. Trends Plant Sci. 2016, 21, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Devos, K.M.; Brown, J.K.M.; Bennetzen, J.L. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002, 12, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Vu, G.T.H.; Schmutzer, T.; Bull, F.; Cao, H.X.; Fuchs, J.; Tran, T.D.; Jovtchev, G.; Pistrick, K.; Stein, N.; Pecinka, A.; et al. Comparative genome analysis reveals divergent genome size evolution in a carnivorous plant genus. The Plant Genome 2015, 8, 1–14. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Visger, C.J.; Soltis, P.S. The polyploidy revolution then…and now: Stebbins revisited. Amer. J. Bot. 2014, 101, 1057–1078. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.A.; Van de Peer, Y.; Maere, S. Significance and biological consequences of polyploidization in land plants. In Plant Genome Diversity, Volume 2, Physical Structure, Behaviour and Evolution of Plant Genomes; Leitch, I.J., Greilhuber, J., Doležel, J., Wendel, J.F., Eds.; Springer-Verlag: Wien, Austria, 2013; pp. 277–293. [Google Scholar]
- Macas, J.; Novák, P.; Pellicer, J.; Čížková, J.; Koblížková, A.; Neumann, P.; Fuková, I.; Doležel, J.; Kelly, L.J.; Leitch, I.J. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE 2015, 10, e0143424. [Google Scholar] [CrossRef] [PubMed]
- Renny-Byfield, S.; Kovarik, A.; Kelly, L.J.; Macas, J.; Novak, P.; Chase, M.W.; Nichols, R.A.; Pancholi, M.R.; Grandbastien, M.A.; Leitch, A.R. Diploidization and genome size change in allopolyploids is associated with differential dynamics of low- and high-copy sequences. Plant J. 2013, 74, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Dodsworth, S.; Jang, T.-S.; Struebig, M.; Chase, M.W.; Weiss-Schneeweiss, H.; Leitch, A.R. Genome-wide repeat dynamics reflect phylogenetic distance in closely related allotetraploid Nicotiana (Solanaceae). Plant. Syst. Evol. 2017, 303, 1013–1020. [Google Scholar] [CrossRef]
- Dodsworth, S.; Chase, M.W.; Särkinen, T.; Knapp, S.; Leitch, A.R. Using genomic repeats for phylogenomics: A case study in wild tomatoes (Solanum section Lycopersicon: Solanaceae). Biol. J. Linn. Soc. 2016, 117, 96–105. [Google Scholar] [CrossRef]
- Piednoël, M.; Aberer, A.J.; Schneeweiss, G.M.; Macas, J.; Novak, P.; Gundlach, H.; Temsch, E.M.; Renner, S.S. Next-generation sequencing reveals the impact of repetitive DNA across phylogenetically closely related genomes of Orobanchaceae. Mol. Biol. Evol. 2012, 29, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Dodsworth, S.; Chase, M.W.; Kelly, L.J.; Leitch, I.J.; Macas, J.; Novák, P.; Piednoël, M.; Weiss-Schneeweiss, H.; Leitch, A.R. Genomic repeat abundances contain phylogenetic signal. Syst. Biol. 2015, 64, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Hřibová, E.; Neumann, P.; Koblížková, A.; Doležel, J.; Macas, J. Genome-wide Analysis of repeat diversity across the family Musaceae. PLoS ONE 2014, 9, e98918. [Google Scholar] [CrossRef] [PubMed]
- Dodsworth, S.; Leitch, A.R.; Leitch, I.J. Genome size diversity in angiosperms and its influence on gene space. Curr. Opin. Genet. Dev. 2015, 35, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hatlen, A.; Kelly, L.J.; Becher, H.; Wang, W.; Kovarik, A.; Leitch, I.J.; Leitch, A.R. Angiosperms are unique among land plant lineages in the occurrence of key genes in the RNA-Directed DNA Methylation (RdDM) pathway. Genome Biol. Evol. 2015, 7, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Ausin, I.; Feng, S.; Yu, C.; Liu, W.; Kuo, H.Y.; Jacobsen, E.L.; Zhai, J.; Gallego-Bartolome, J.; Wang, L.; Egertsdotter, U.; et al. DNA methylome of the 20-gigabase Norway spruce genome. Proc. Natl. Acad. Sci. USA 2016, 113, E8106–E8113. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, N.V. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Becher, H.; Ma, L.; Kelly, L.J.; Kovarik, A.; Leitch, I.J.; Leitch, A.R. Endogenous pararetrovirus sequences associated with 24 nt small RNAs at the centromeres of Fritillaria imperialis L. (Liliaceae), a species with a giant genome. The Plant J. 2014, 80, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Jackson, S.A.; Meyers, B.C.; Wing, R.A. Evolution of plant genome architecture. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Greilhuber, J.; Leitch, I.J. Genome size and the phenotype. In Plant Genome Diversity, Volume 2, Physical Structure, Behaviour and Evolution of Plant Genomes; Leitch, I.J., Greilhuber, J., Doležel, J., Wendel, J.F., Eds.; Springer-Verlag: Wien, Austria, 2013; pp. 323–344. [Google Scholar]
- Suda, J.; Meyerson, L.A.; Leitch, I.J.; Pyšek, P. The hidden side of plant invasions: The role of genome size. New Phytol. 2015, 205, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Herben, T.; Suda, J.; Klimešová, J.; Mihulka, S.; Říha, P.; Šímová, I. Ecological effects of cell-level processes: Genome size, functional traits and regional abundance of herbaceous plant species. Ann. Bot. 2012, 110, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Guignard, M.S.; Nichols, R.A.; Knell, R.J.; Macdonald, A.; Romila, C.-A.; Trimmer, M.; Leitch, I.J.; Leitch, A.R. Genome size and ploidy influence angiosperm species’ biomass under nitrogen and phosphorus limitation. New Phytol. 2016, 210, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Bilinski, P.; Albert, P.S.; Berg, J.J.; Birchler, J.; Grote, M.; Lorant, A.; Quezada, J.; Swarts, K.; Yang, J.; Ross-Ibarra, J. Parallel altitudinal clines reveal adaptive evolution of genome size in Zea mays. 2017. Available online: https://www.biorxiv.org/content/early/2017/07/13/134528 (accessed on 21 November 2017).
- Lyu, H.; He, Z.; Wu, C.-I.; Shi, S. Convergent adaptive evolution in marginal environments: Unloading transposable elements as a common strategy among mangrove genomes. New Phytol. 2018, 217, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Bromham, L.; Hua, X.; Lanfear, R.; Cowman, P.F. Exploring the relationships between mutation rates, life history, genome size, environment, and species richness in flowering plants. Am. Nat. 2015, 185, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Puttick, M.N.; Clark, J.; Donoghue, P.C.J. Size is not everything: Rates of genome size evolution, not C-value, correlate with speciation in angiosperms. Proc. Natl. Acad. Sci. USA 2015, 282, 2015–2289. [Google Scholar] [CrossRef] [PubMed]
- Leitch, A.R.; Leitch, I.J. Ecological and genetic factors linked to contrasting genome dynamics in seed plants. New Phytol. 2012, 194, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.A. Mutational equilibrium model of genome size evolution. Theor. Popul. Biol. 2002, 61, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The origins of genome complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E. Compactness of human housekeeping genes: Selection for economy or genomic design? Trends Genet. 2004, 20, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Oliver, M.J.; Petrov, D.; Ackerly, D.; Falkowski, P.; Schofield, O.M. The mode and tempo of genome size evolution in eukaryotes. Genome Res. 2007, 17, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.A. Evolution of genome size: New approaches to an old problem. Trends Genet. 2001, 17, 23–28. [Google Scholar] [CrossRef]
- Simonin, K.A.; Roddy, A.B. Genome downsizing, physiological novelty, and the global dominance of flowering plants. PLoS Biol. 2018, 16, e2003706. [Google Scholar] [CrossRef] [PubMed]
- Veselý, P.; Bureš, P.; Šmarda, P.; Pavlicek, T. Genome size and DNA base composition of geophytes: The mirror of phenology and ecology? Ann. Bot. 2012, 109, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Veselý, P.; Bureš, P.; Šmarda, P. Nutrient reserves may allow for genome size increase: Evidence from comparison of geophytes and their sister non-geophytic relatives. Ann. Bot. 2013, 112, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Šmarda, P.; Hejcman, M.; Březinová, A.; Horová, L.; Steigerová, H.; Zedek, F.; Bureš, P.; Hejcmanová, P.; Schellberg, J. Effect of phosphorus availability on the selection of species with different ploidy levels and genome sizes in a long-term grassland fertilization experiment. New Phytol. 2013, 200, 911–921. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
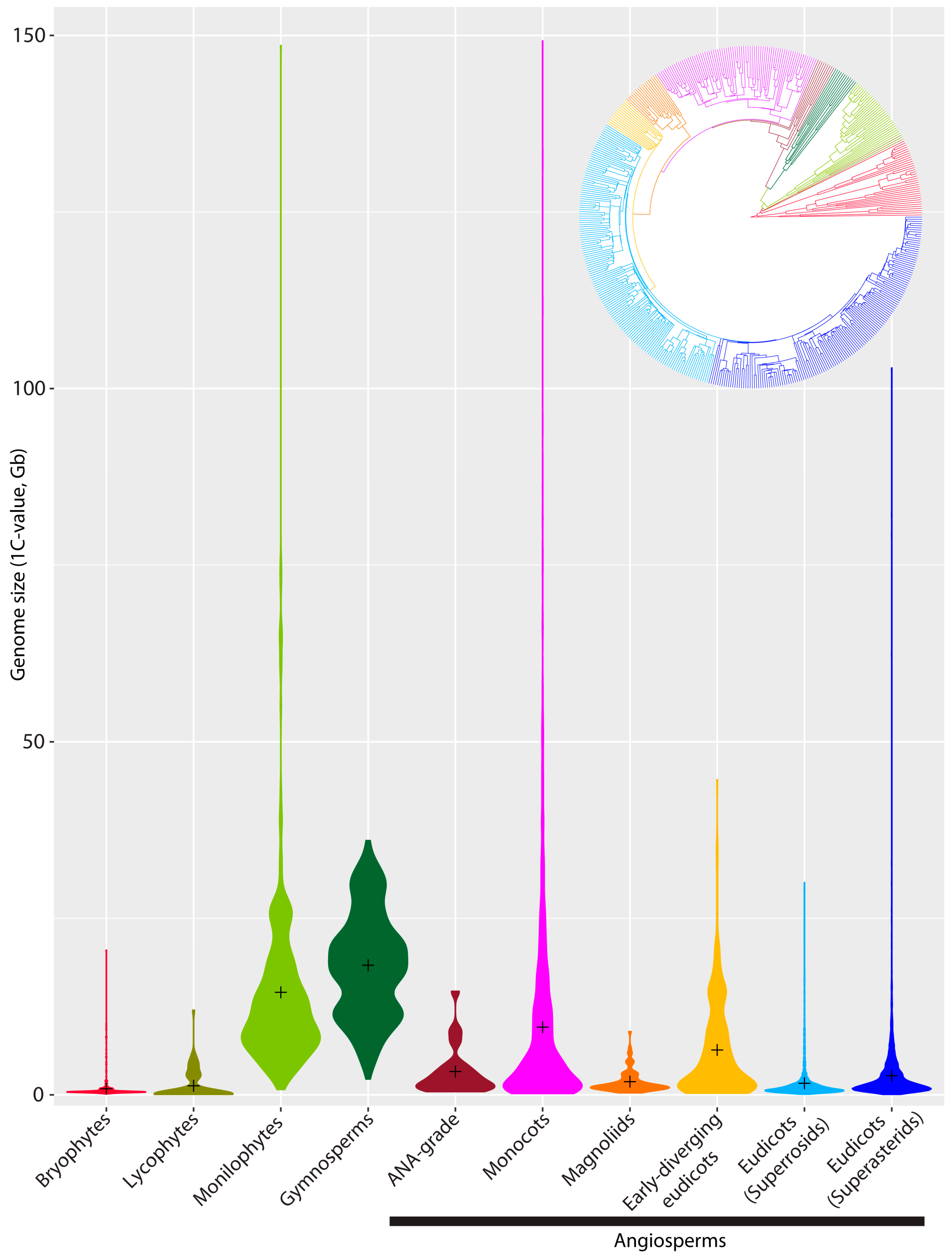{kind=link}
| Min. (Mb) | Max. (Mb) | Mean (Mb) | Mode (Mb) | Median (Mb) | Range in Absolute GS (Mb) | Range (Max./Min.) | Approx. no. of Species Recognised | No. of Species in the Plant DNA C-Values Database | Approx. % Species Representation in the Plant DNA C-Values Database | |
|---|---|---|---|---|---|---|---|---|---|---|
| Bryophytes | ||||||||||
| Hornworts | 156 | 714 | 244 | 176 | 205 | 558 | 4-fold | 250 | 23 | 9.2 |
| Liverworts | 206 | 20,010 | 1844 | 740 | 751 | 19,804 | 97-fold | 5,000 | 102 | 2.0 |
| Mosses | 170 | 2004 | 504 | 442 | 433 | 1834 | 12-fold | 12,000 | 184 | 1.7 |
| Pteridophytes | ||||||||||
| Lycophytes | 78 | 11,704 | 1165 | 117 | 127 | 11,626 | 150-fold | 900 | 57 | 6.3 |
| Monilophytes | 748 | 147,297 | 14,320 | 12,073 | 11,110 | 146,549 | 196-fold | 11,000 | 246 | 2.2 |
| Spermatophytes | ||||||||||
| Gymnosperms | 2201 | 35,208 | 17,947 | 21,614 | 21,614 | 33,007 | 16-fold | 1026 | 421 | 41.0 |
| Angiosperms | 61 | 148,852 | 5020 | 587 | 1663 | 148,791 | 2440-fold | 352,000 | 10,768 | 3.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes 2018, 9, 88. https://doi.org/10.3390/genes9020088
Pellicer J, Hidalgo O, Dodsworth S, Leitch IJ. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes. 2018; 9(2):88. https://doi.org/10.3390/genes9020088
Chicago/Turabian StylePellicer, Jaume, Oriane Hidalgo, Steven Dodsworth, and Ilia J. Leitch. 2018. "Genome Size Diversity and Its Impact on the Evolution of Land Plants" Genes 9, no. 2: 88. https://doi.org/10.3390/genes9020088
APA StylePellicer, J., Hidalgo, O., Dodsworth, S., & Leitch, I. J. (2018). Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes, 9(2), 88. https://doi.org/10.3390/genes9020088