Endogenous DNA Double-Strand Breaks during DNA Transactions: Emerging Insights and Methods for Genome-Wide Profiling


Abstract
1. Introduction
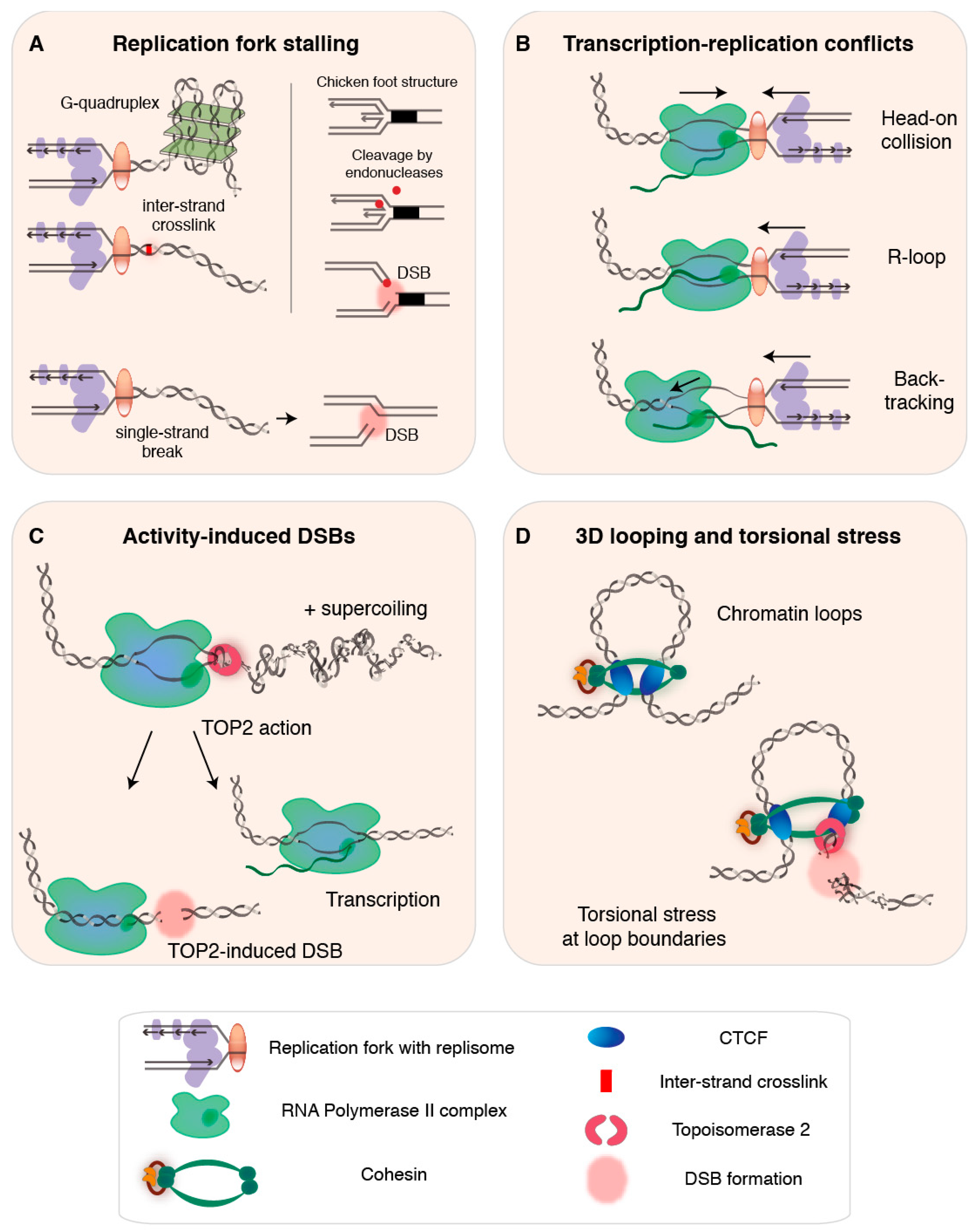
2. DNA Replication as a Source of Endogenous DSBs
2.1. Exhaustion of Replication Resources and Disturbed Replication Programs
2.2. Conflicts with Pre-Existing DNA Damage and Secondary Structures
2.3. Transcription-Replication Conflicts, R-Loops, and Backtracking
2.4. The Cellular Response to Replication Stress and the Impact of Replication Timing
2.4.1. Fork Stabilization and Restart
2.4.2. DSB Formation from Stalled Replication Forks
2.4.3. Common Fragile Sites
2.4.4. Replication Timing and Fragility
3. Transcription as a Source of Endogenous DSBs
3.1. DSBs Accumulate around Activated Genes
3.2. Transcription Activation through DNA Damage
3.3. Transcription Activation Assisted by TOP2-Induced DSBs
3.4. TOP2 Poisons Are Associated with Therapy-Related Acute Myeloid Leukemias
4. 3D Genome Architecture and DSBs
4.1. TOP2-Induced DSBs at Chromatin Loop Anchors
4.2. Intertwined Actions Predispose Regulatory Regions to Fragility
4.3. Special Cases of Genome Rewiring Require Programmed DSBs
5. Adverse Outcomes of DSB Repair
5.1. Mechanisms Underlying Structural Genomic Alterations
5.2. Repair Signatures in Cancer Genomes
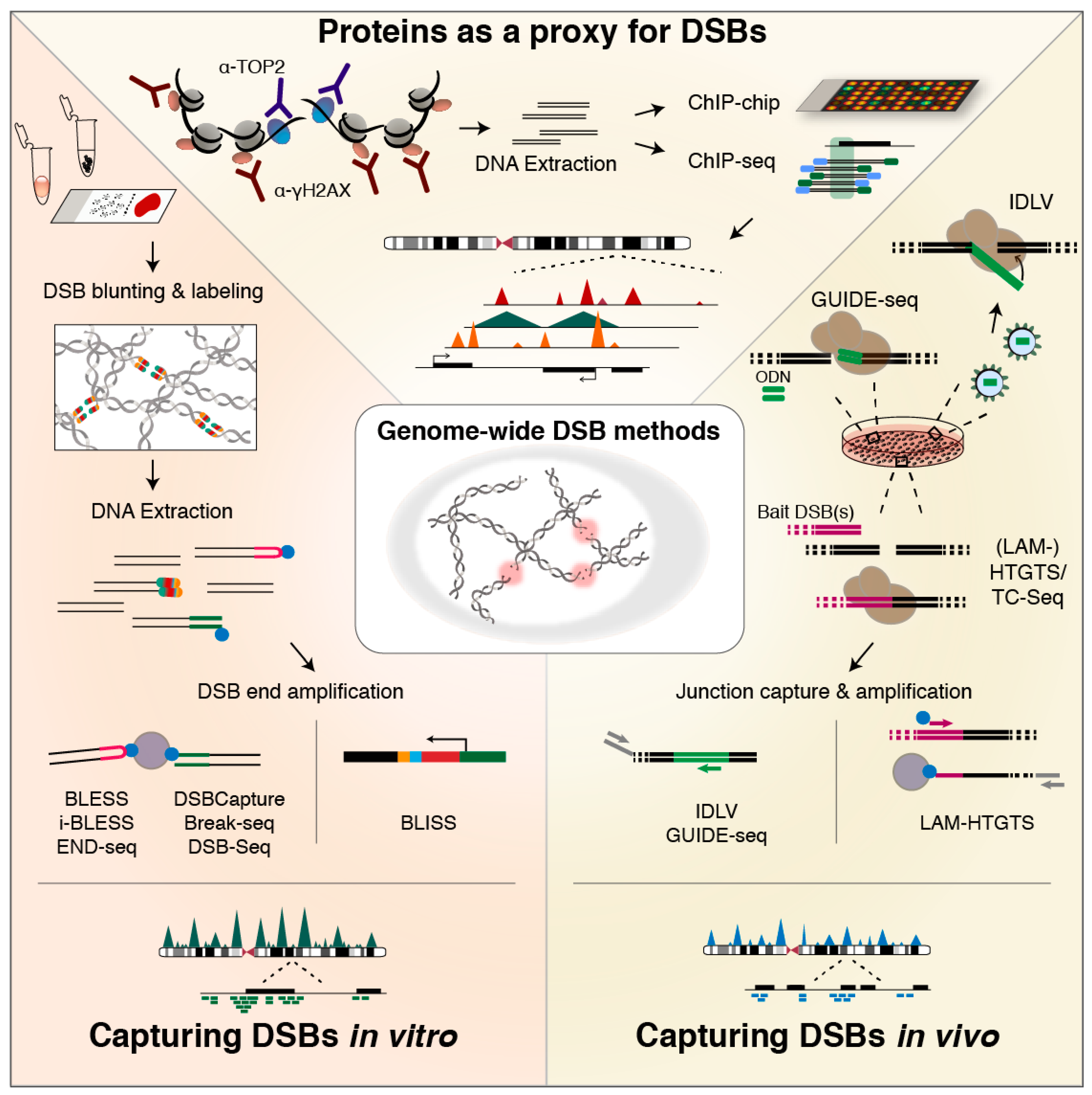
6. Methods for Genome-Wide DSB Profiling
6.1. Indirect Identification Based on Association of Recruited or Responsible Proteins
6.2. Methods for In Vivo DSB Capture
6.3. Methods for In Vitro Tagging of DSBs
6.4. On Assay Choice
7. Concluding Remarks and Outlook
7.1. Conclusions and Additional Remarks
7.1.1. Integrative Approaches and Confounding Factors
7.1.2. Compartmentalized DSB repair
7.1.3. Studying DSB Biology at Ectopically Induced Genome-Wide DSB Sites
7.2. Open Questions in the Field and Outlook
Funding
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Garinis, G.A.; van der Horst, G.T.J.; Vijg, J.; Hoeijmakers, J.H.J. DNA damage and ageing: new-age ideas for an age-old problem. Nat. Cell Biol. 2008, 10, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lindahl, T. Maintenance of Genome Stability. Genomics Proteomics Bioinform. 2016, 14, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [PubMed]
- van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J.; Caldecott, K.W. DNA strand break repair and human genetic disease. Annu. Rev. Genomics Hum. Genet. 2007, 8, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, G.; d’Adda di Fagagna, F. Transcription and DNA Damage: Holding Hands or Crossing Swords? J. Mol. Biol. 2017, 429, 3215–3229. [Google Scholar] [CrossRef] [PubMed]
- Borde, V.; de Massy, B. Programmed induction of DNA double strand breaks during meiosis: Setting up communication between DNA and the chromosome structure. Curr. Opin. Genet. Dev. 2013, 23, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed]
- Alt, F.W.; Schwer, B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 2018. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.C.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. TIG 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Frit, P.; Barboule, N.; Yuan, Y.; Gomez, D.; Calsou, P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair 2014, 17, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed]
- Little, R.D.; Platt, T.H.; Schildkraut, C.L. Initiation and termination of DNA replication in human rRNA genes. Mol. Cell. Biol. 1993, 13, 6600–6613. [Google Scholar] [CrossRef] [PubMed]
- Petryk, N.; Kahli, M.; d’Aubenton-Carafa, Y.; Jaszczyszyn, Y.; Shen, Y.; Silvain, M.; Thermes, C.; Chen, C.-L.; Hyrien, O. Replication landscape of the human genome. Nat. Commun. 2016, 7, 10208. [Google Scholar] [CrossRef] [PubMed]
- Fachinetti, D.; Bermejo, R.; Cocito, A.; Minardi, S.; Katou, Y.; Kanoh, Y.; Shirahige, K.; Azvolinsky, A.; Zakian, V.A.; Foiani, M. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol. Cell 2010, 39, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Dewar, J.M.; Walter, J.C. Mechanisms of DNA replication termination. Nat. Rev. Mol. Cell Biol. 2017, 18, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Gelot, C.; Magdalou, I.; Lopez, B.S. Replication stress in Mammalian cells and its consequences for mitosis. Genes 2015, 6, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, T.G.; Komseli, E.-S.; Papaioannou, M.; Vougas, K.; Polyzos, A.; Myrianthopoulos, V.; Mikros, E.; Trougakos, I.P.; Thanos, D.; Branzei, D.; et al. Exploring and exploiting the systemic effects of deregulated replication licensing. Semin. Cancer Biol. 2016, 37–38, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Pederson, K.D. Dormant origins as a built-in safeguard in eukaryotic DNA replication against genome instability and disease development. DNA Repair 2017, 56, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hills, S.A.; Diffley, J.F.X. DNA replication and oncogene-induced replicative stress. Curr. Biol. CB 2014, 24, R435–444. [Google Scholar] [CrossRef] [PubMed]
- Dellino, G.I.; Pelicci, P.G. Next-generation sequencing and DNA replication in human cells: The future has arrived. Future Oncol. Lond. Engl. 2014, 10, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Orr-Weaver, T.L. Replication fork instability and the consequences of fork collisions from rereplication. Genes Dev. 2016, 30, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability--an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Preventing replication fork collapse to maintain genome integrity. DNA Repair 2015, 32, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. MMBR 2007, 71, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Maizels, N.; Gray, L.T. The G4 genome. PLoS Genet. 2013, 9, e1003468. [Google Scholar] [CrossRef] [PubMed]
- Valton, A.-L.; Prioleau, M.-N. G-Quadruplexes in DNA Replication: A Problem or a Necessity? Trends Genet. TIG 2016, 32, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Westhorpe, F.G.; Straight, A.F. Functions of the centromere and kinetochore in chromosome segregation. Curr. Opin. Cell Biol. 2013, 25, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Langston, L.D.; O’Donnell, M. DNA replication: Keep moving and don’t mind the gap. Mol. Cell 2006, 23, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.G.; Yeeles, J.T.P. The Initial Response of a Eukaryotic Replisome to DNA Damage. Mol. Cell 2018, 70, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.M.; Newlon, C.S. DNA replication fork pause sites dependent on transcription. Science 1996, 272, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Azvolinsky, A.; Giresi, P.G.; Lieb, J.D.; Zakian, V.A. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 2009, 34, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; García-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gómez-González, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Samarabandu, J.; Devdhar, R.S.; Siegel, A.J.; Acharya, R.; Berezney, R. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Meryet-Figuiere, M.; Alaei-Mahabadi, B.; Ali, M.M.; Mitra, S.; Subhash, S.; Pandey, G.K.; Larsson, E.; Kanduri, C. Temporal separation of replication and transcription during S-phase progression. Cell Cycle Georget. Tex 2014, 13, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.M.; Ryan, M.; Kim, R.; Zakas, A.L.; Fu, H.; Lin, C.M.; Reinhold, W.C.; Davis, S.R.; Bilke, S.; Liu, H.; et al. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res. 2011, 21, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Huvet, M.; Nicolay, S.; Touchon, M.; Audit, B.; d’Aubenton-Carafa, Y.; Arneodo, A.; Thermes, C. Human gene organization driven by the coordination of replication and transcription. Genome Res. 2007, 17, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jinks-Robertson, S. Transcription as a source of genome instability. Nat. Rev. Genet. 2012, 13, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Berretta, J.; Morillon, A. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 2009, 10, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Nudler, E. RNA polymerase backtracking in gene regulation and genome instability. Cell 2012, 149, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Core, L.J.; Waterfall, J.J.; Lis, J.T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008, 322, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Westover, K.D.; Bushnell, D.A.; Kornberg, R.D. Structural basis of transcription: Separation of RNA from DNA by RNA polymerase II. Science 2004, 303, 1014–1016. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chédin, F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chédin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Chedin, F.; Hsieh, C.-L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Aguilera, A. Transcription-replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K. A critical role for topoisomerase IIb and DNA double strand breaks in transcription. Transcription 2016, 7, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.-Y.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Uusküla-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [PubMed]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.-R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome Organization Drives Chromosome Fragility. Cell 2017, 170, 507–521.e18. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.; Core, L.J.; Lis, J.T. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 2006, 7, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010, 11, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Ashley, A.K.; Hromas, R.; Nickoloff, J.A. More forks on the road to replication stress recovery. J. Mol. Cell Biol. 2011, 3, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Maehara, K.; Yoshida, K.; Ohkawa, Y.; Fujita, M. Genome-wide analysis of the spatiotemporal regulation of firing and dormant replication origins in human cells. Nucleic Acids Res. 2018, 46, 6683–6696. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. The DNA damage response during DNA replication. Curr. Opin. Cell Biol. 2005, 17, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Carr, A.M. Impediments to replication fork movement: Stabilisation, reactivation and genome instability. Chromosoma 2013, 122, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Lundin, C.; Erixon, K.; Arnaudeau, C.; Schultz, N.; Jenssen, D.; Meuth, M.; Helleday, T. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell. Biol. 2002, 22, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.-H.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Cohen, S.; Legube, G. Transcription-Coupled DNA Double-Strand Break Repair: Active Genes Need Special Care. J. Mol. Biol. 2017, 429, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Geijer, M.E.; Marteijn, J.A. What happens at the lesion does not stay at the lesion: Transcription-coupled nucleotide excision repair and the effects of DNA damage on transcription in cis and trans. DNA Repair 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, F.; Wu, X.; Rechkoblit, O.; Taylor, J.S.; Geacintov, N.E.; Wang, Z. Error-prone lesion bypass by human DNA polymerase eta. Nucleic Acids Res. 2000, 28, 4717–4724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, F.; Wu, X.; Wang, M.; Rechkoblit, O.; Taylor, J.S.; Geacintov, N.E.; Wang, Z. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 2000, 28, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. The checkpoint response to replication stress. DNA Repair 2009, 8, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Ledesma, F.; Aguilera, A. Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep. 2006, 7, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, A.; Ristic, D.; Dionne, I.; Liu, X.Z.; Wyman, C.; Wellinger, R.J.; Petrini, J.H.J. The Ku heterodimer and the metabolism of single-ended DNA double-strand breaks. Cell Rep. 2013, 3, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Tsantoulis, P.; Kotsinas, A.; Michalopoulos, I.; Townsend, P.; Gorgoulis, V.G. Are common fragile sites merely structural domains or highly organized “functional” units susceptible to oncogenic stress? Cell. Mol. Life Sci. CMLS 2014, 71, 4519–4544. [Google Scholar] [CrossRef] [PubMed]
- Franchitto, A. Genome instability at common fragile sites: Searching for the cause of their instability. BioMed Res. Int. 2013, 2013, 730714. [Google Scholar] [CrossRef] [PubMed]
- Fungtammasan, A.; Walsh, E.; Chiaromonte, F.; Eckert, K.A.; Makova, K.D. A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res. 2012, 22, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Michor, F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat. Biotechnol. 2011, 29, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Hupé, P.; Maciorowski, Z.; La Rosa, P.; Schleiermacher, G.; Pierron, G.; Liva, S.; Barillot, E.; Delattre, O. Preferential occurrence of chromosome breakpoints within early replicating regions in neuroblastoma. Cell Cycle Georget. Tex 2005, 4, 1842–1846. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Sima, J.; Gilbert, D.M. Complex correlations: Replication timing and mutational landscapes during cancer and genome evolution. Curr. Opin. Genet. Dev. 2014, 25, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect-part III. Curr. Opin. Cell Biol. 2016, 40, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Mesner, L.D.; Valsakumar, V.; Cieslik, M.; Pickin, R.; Hamlin, J.L.; Bekiranov, S. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res. 2013, 23, 1774–1788. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, G.; Rappailles, A.; Baker, A.; Chen, C.-L.; Arneodo, A.; Goldar, A.; d’Aubenton-Carafa, Y.; Thermes, C.; Audit, B.; Hyrien, O. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput. Biol. 2011, 7, e1002322. [Google Scholar] [CrossRef] [PubMed]
- Finn, K.J.; Li, J.J. Single-stranded annealing induced by re-initiation of replication origins provides a novel and efficient mechanism for generating copy number expansion via non-allelic homologous recombination. PLoS Genet. 2013, 9, e1003192. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Roukos, V.; Misteli, T. The biogenesis of chromosome translocations. Nat. Cell Biol. 2014, 16, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Wijchers, P.J.; de Laat, W. Genome organization influences partner selection for chromosomal rearrangements. Trends Genet. TIG 2011, 27, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Norton, H.K.; Phillips-Cremins, J.E. Crossed wires: 3D genome misfolding in human disease. J. Cell Biol. 2017, 216, 3441–3452. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; De Marzo, A.M.; Meeker, A.K.; Nelson, W.G.; Yegnasubramanian, S. Transcription-induced DNA double strand breaks: Both oncogenic force and potential therapeutic target? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 3858–3864. [Google Scholar] [CrossRef] [PubMed]
- Vitelli, V.; Galbiati, A.; Iannelli, F.; Pessina, F.; Sharma, S.; d’Adda di Fagagna, F. Recent Advancements in DNA Damage-Transcription Crosstalk and High-Resolution Mapping of DNA Breaks. Annu. Rev. Genomics Hum. Genet. 2017, 18, 87–113. [Google Scholar] [PubMed]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Mourad, R.; Ginalski, K.; Legube, G.; Cuvier, O. Predicting double-strand DNA breaks using epigenome marks or DNA at kilobase resolution. Genome Biol. 2018, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.-C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W.; Schwer, B. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016, 164, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Lensing, S.V.; Marsico, G.; Hänsel-Hertsch, R.; Lam, E.Y.; Tannahill, D.; Balasubramanian, S. DSBCapture: In situ capture and sequencing of DNA breaks. Nat. Methods 2016, 13, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Baranello, L.; Kouzine, F.; Wojtowicz, D.; Cui, K.; Przytycka, T.M.; Zhao, K.; Levens, D. DNA break mapping reveals topoisomerase II activity genome-wide. Int. J. Mol. Sci. 2014, 15, 13111–13122. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.-C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Wei, P.-C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W. Transcription-associated processes cause DNA double-strand breaks and translocations in neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2016, 113, 2258–2263. [Google Scholar] [CrossRef] [PubMed]
- Pankotai, T.; Bonhomme, C.; Chen, D.; Soutoglou, E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 2012, 19, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Capozzo, I.; Iannelli, F.; Francia, S.; d’Adda di Fagagna, F. Express or repress? The transcriptional dilemma of damaged chromatin. FEBS J. 2017, 284, 2133–2147. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Ali, S.O. Physiological Roles of DNA Double-Strand Breaks. J. Nucleic Acids 2017, 2017, 6439169. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Kozbial, P.; Li, W.; Tan, Y.; Liu, Z.; Suter, T.; Ohgi, K.A.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell 2015, 160, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Hartung, M.L.; Gruber, D.C.; Koch, K.N.; Grüter, L.; Rehrauer, H.; Tegtmeyer, N.; Backert, S.; Müller, A. H. pylori-Induced DNA Strand Breaks Are Introduced by Nucleotide Excision Repair Endonucleases and Promote NF-κB Target Gene Expression. Cell Rep. 2015, 13, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Toller, I.M.; Neelsen, K.J.; Steger, M.; Hartung, M.L.; Hottiger, M.O.; Stucki, M.; Kalali, B.; Gerhard, M.; Sartori, A.A.; Lopes, M.; et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14944–14949. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.-G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Panova, T.; Miller, G.; Volkov, A.; Porter, A.C.G.; Russell, J.; Panov, K.I.; Zomerdijk, J.C.B.M. Topoisomerase IIα promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 2013, 4, 1598. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R. The Roles of DNA Topoisomerase IIβ in Transcription. Int. J. Mol. Sci. 2018, 19, 1917. [Google Scholar] [CrossRef] [PubMed]
- Schaefer-Klein, J.L.; Murphy, S.J.; Johnson, S.H.; Vasmatzis, G.; Kovtun, I.V. Topoisomerase 2 Alpha Cooperates with Androgen Receptor to Contribute to Prostate Cancer Progression. PLoS ONE 2015, 10, e0142327. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ziv, R.; Voichek, Y.; Barkai, N. Chromatin dynamics during DNA replication. Genome Res. 2016, 26, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Gupta, A.; Baranello, L.; Wojtowicz, D.; Ben-Aissa, K.; Liu, J.; Przytycka, T.M.; Levens, D. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol. 2013, 20, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Naughton, C.; Avlonitis, N.; Corless, S.; Prendergast, J.G.; Mati, I.K.; Eijk, P.P.; Cockroft, S.L.; Bradley, M.; Ylstra, B.; Gilbert, N. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 2013, 20, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Corless, S.; Gilbert, N. Effects of DNA supercoiling on chromatin architecture. Biophys. Rev. 2016, 8, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Ashour, M.E.; Atteya, R.; El-Khamisy, S.F. Topoisomerase-mediated chromosomal break repair: An emerging player in many games. Nat. Rev. Cancer 2015, 15, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herreros, F.; Zagnoli-Vieira, G.; Ntai, I.; Martínez-Macías, M.I.; Anderson, R.M.; Herrero-Ruíz, A.; Caldecott, K.W. TDP2 suppresses chromosomal translocations induced by DNA topoisomerase II during gene transcription. Nat. Commun. 2017, 8, 233. [Google Scholar] [CrossRef] [PubMed]
- Sasanuma, H.; Tsuda, M.; Morimoto, S.; Saha, L.K.; Rahman, M.M.; Kiyooka, Y.; Fujiike, H.; Cherniack, A.D.; Itou, J.; Callen Moreu, E.; et al. BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II-DNA complexes. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.M.; Travis, L.B. Mechanisms of therapy-related carcinogenesis. Nat. Rev. Cancer 2005, 5, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.L.; Vaughan, A.T.M. A systematic description of MLL fusion gene formation. Crit. Rev. Oncol. Hematol. 2014, 91, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rowley, J.D. Chromatin structural elements and chromosomal translocations in leukemia. DNA Repair 2006, 5, 1282–1297. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.L.; Kircher, S.M.; Didwania, A.; Goel, M.S. Screening for Recurrence and Secondary Cancers. Med. Clin. North Am. 2017, 101, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Habash, M.; Bohorquez, L.C.; Kyriakou, E.; Kron, T.; Martin, O.A.; Blyth, B.J. Clinical and Functional Assays of Radiosensitivity and Radiation-Induced Second Cancer. Cancers 2017, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Austin, C.A. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public. Health 2012, 9, 2075–2091. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, E.; Tanay, A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 2011, 43, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Splinter, E.; Heath, H.; Kooren, J.; Palstra, R.-J.; Klous, P.; Grosveld, F.; Galjart, N.; de Laat, W. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2006, 20, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.S.; Cattoglio, C.; Darzacq, X.; Tjian, R. Recent evidence that TADs and chromatin loops are dynamic structures. Nucl. Austin Tex 2018, 9, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, V.B.; Semple, C.A. Chromatin loop anchors are associated with genome instability in cancer and recombination hotspots in the germline. Genome Biol. 2018, 19, 101. [Google Scholar] [CrossRef] [PubMed]
- Krefting, J.; Andrade-Navarro, M.A.; Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biol. 2018, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Kemp, C.J.; Henikoff, S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutat. Res. 2015, 773, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Grosveld, F.G.; Papantonis, A. Forces driving the three-dimensional folding of eukaryotic genomes. Mol. Syst. Biol. 2018, 14, e8214. [Google Scholar] [CrossRef] [PubMed]
- Gothe, H.J.; Bouwman, B.A.M.; Gusmao, E.G.; Piccinno, R.; Sayols, S.; Drechsel, O.; Petrosino, G.; Minneker, V.; Josipovic, N.; Mizi, A.; et al. Spatial chromosome folding and active transcription drive DNA fragility and formation of oncogenic MLL translocations. bioRxiv 2018, 485763. [Google Scholar]
- Roca, J. Transcriptional inhibition by DNA torsional stress. Transcription 2011, 2, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Johnson, J.; Bentivoglio, A.; Corless, S.; Gilbert, N.; Gonnella, G.; Marenduzzo, D. Stochastic Model of Supercoiling-Dependent Transcription. Phys. Rev. Lett. 2016, 117, 018101. [Google Scholar] [CrossRef] [PubMed]
- Tock, A.J.; Henderson, I.R. Hotspots for Initiation of Meiotic Recombination. Front. Genet. 2018, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Alt, F.W.; Zhang, Y.; Meng, F.-L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Paigen, K.; Petkov, P. Mammalian recombination hot spots: Properties, control and evolution. Nat. Rev. Genet. 2010, 11, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gostissa, M.; Hildebrand, D.G.; Becker, M.S.; Boboila, C.; Chiarle, R.; Lewis, S.; Alt, F.W. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv. Immunol. 2010, 106, 93–133. [Google Scholar] [PubMed]
- Ghosh, R.; Das, D.; Franco, S. The Role for the DSB Response Pathway in Regulating Chromosome Translocations. Adv. Exp. Med. Biol. 2018, 1044, 65–87. [Google Scholar] [PubMed]
- Willis, N.A.; Rass, E.; Scully, R. Deciphering the Code of the Cancer Genome: Mechanisms of Chromosome Rearrangement. Trends Cancer 2015, 1, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Lupski, J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Imielinski, M. Modeling cancer rearrangement landscapes. Curr. Opin. Syst. Biol. 2017, 1, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.R.; Grewal, D.S.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.M.; et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 2017, 49, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.-L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.W.; Chan, F.C.; Shah, S.P. Observing Clonal Dynamics across Spatiotemporal Axes: A Prelude to Quantitative Fitness Models for Cancer. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Szilard, R.K.; Jacques, P.-E.; Laramée, L.; Cheng, B.; Galicia, S.; Bataille, A.R.; Yeung, M.; Mendez, M.; Bergeron, M.; Robert, F.; et al. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat. Struct. Mol. Biol. 2010, 17, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Allis, C.D.; Nussenzweig, A. Phosphorylation of histone H2B at DNA double-strand breaks. J. Exp. Med. 2004, 199, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Massip, L.; Caron, P.; Iacovoni, J.S.; Trouche, D.; Legube, G. Deciphering the chromatin landscape induced around DNA double strand breaks. Cell Cycle Georget. Tex 2010, 9, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.S.; Pugh, B.F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 2011, 147, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Vrouwe, M.G.; Pines, A.; Overmeer, R.M.; Hanada, K.; Mullenders, L.H.F. UV-induced photolesions elicit ATR-kinase-dependent signaling in non-cycling cells through nucleotide excision repair-dependent and -independent pathways. J. Cell Sci. 2011, 124, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.-Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.-D.; Guan, H.; Zhang, S.-M.; Tang, Y.; Rang, W.-Q.; Zhou, P.-K. γH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef] [PubMed]
- Marti, T.M.; Hefner, E.; Feeney, L.; Natale, V.; Cleaver, J.E. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2006, 103, 9891–9896. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Giachino, C. Multiple facets of histone variant H2AX: A DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 2015, 43, 2489–2498. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Lombardo, A.; Arens, A.; Miller, J.C.; Genovese, P.; Kaeppel, C.; Nowrouzi, A.; Bartholomae, C.C.; Wang, J.; Friedman, G.; et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011, 29, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wu, X.; Wang, J.; Wang, Y.; Qiu, Z.; Chang, T.; Huang, H.; Lin, R.-J.; Yee, J.-K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015, 33, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Zhang, Y.; Frock, R.L.; Lewis, S.M.; Molinie, B.; Ho, Y.-J.; Myers, D.R.; Choi, V.W.; Compagno, M.; Malkin, D.J.; et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 2011, 147, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.-J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Meyers, R.M.; Dong, J.; Panchakshari, R.A.; Alt, F.W.; Frock, R.L. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 2016, 11, 853–871. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.A.; Resch, W.; Jankovic, M.; Oliveira, T.; Yamane, A.; Nakahashi, H.; Di Virgilio, M.; Bothmer, A.; Nussenzweig, A.; Robbiani, D.F.; et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 2011, 147, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef] [PubMed]
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol. Cell 2016, 63, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Zhu, Y.; Skrzypczak, M.; Forey, R.; Pardo, B.; Grzelak, M.; Nde, J.; Mitra, A.; Kudlicki, A.; Crosetto, N.; et al. i-BLESS is an ultra-sensitive method for detection of DNA double-strand breaks. Commun. Biol. 2018, 1, 181. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.A.; McCulley, A.; Haarer, B.; Arnak, R.; Feng, W. Break-seq reveals hydroxyurea-induced chromosome fragility as a result of unscheduled conflict between DNA replication and transcription. Genome Res. 2015, 25, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.J.; Webber, B.R.; Knipping, F.; Lonetree, C.; Tennis, N.; DeFeo, A.P.; McElroy, A.N.; Starker, C.G.; Lee, C.; Merkel, S.; et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Sánchez-Hernández, S.; Gutiérrez-Guerrero, A.; Pinedo-Gomez, J.; Benabdellah, K. Biased and Unbiased Methods for the Detection of Off-Target Cleavage by CRISPR/Cas9: An Overview. Int. J. Mol. Sci. 2016, 17, 1507. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.Y.; Resch, W.; Jankovic, M.; Casellas, R.; Nussenzweig, M.C.; Klein, I.A. Translocation capture sequencing: A method for high throughput mapping of chromosomal rearrangements. J. Immunol. Methods 2012, 375, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.-C.; Lee, C.-S.; Du, Z.; Schwer, B.; Zhang, Y.; Kao, J.; Zurita, J.; Alt, F.W. Three classes of recurrent DNA break clusters in brain progenitors identified by 3D proximity-based break joining assay. Proc. Natl. Acad. Sci. USA 2018, 115, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Galbiati, A.; Capozzo, I.; Nguyen, Q.; Magnuson, B.; Michelini, F.; D’Alessandro, G.; Cabrini, M.; Roncador, M.; Francia, S.; et al. A damaged genome’s transcriptional landscape through multilayered expression profiling around in situ-mapped DNA double-strand breaks. Nat. Commun. 2017, 8, 15656. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Baranello, L.; Kouzine, F.; Wojtowicz, D.; Cui, K.; Zhao, K.; Przytycka, T.M.; Capranico, G.; Levens, D. Mapping DNA Breaks by Next-Generation Sequencing. Methods Mol. Biol. Clifton NJ 2018, 1672, 155–166. [Google Scholar]
- Leduc, F.; Faucher, D.; Bikond Nkoma, G.; Grégoire, M.-C.; Arguin, M.; Wellinger, R.J.; Boissonneault, G. Genome-wide mapping of DNA strand breaks. PLoS ONE 2011, 6, e17353. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, M.-C.; Massonneau, J.; Leduc, F.; Arguin, M.; Brazeau, M.-A.; Boissonneault, G. Quantification and genome-wide mapping of DNA double-strand breaks. DNA Repair 2016, 48, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, M.-C.; Leduc, F.; Morin, M.H.; Cavé, T.; Arguin, M.; Richter, M.; Jacques, P.-É.; Boissonneault, G. The DNA double-strand “breakome” of mouse spermatids. Cell. Mol. Life Sci. CMLS 2018, 75, 2859–2872. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, Y.; Zhou, Y.; Tubbs, A.T.; Chen, B.-R.; Purman, C.; Lee, B.-S.; George, R.; Bredemeyer, A.L.; Zhao, J.-Y.; Sodergen, E.; et al. HCoDES reveals chromosomal DNA end structures with single-nucleotide resolution. Mol. Cell 2014, 56, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.G.; Ba, Z.; Du, Z.; Zhang, Y.; Hu, J.; Alt, F.W. Highly sensitive and unbiased approach for elucidating antibody repertoires. Proc. Natl. Acad. Sci. USA 2016, 113, 7846–7851. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dojer, N.; Biernacka, A.; Pardo, B.; Forey, R.; Skrzypczak, M.; Fongang, B.; Kengne, J.B.N.; Yousefi, R.; Pasero, P.; et al. Quantitative DSB sequencing (qDSB-Seq): A method for genome-wide accurate estimation of absolute DNA double-strand break frequencies per cell. bioRxiv 2017, 171405. [Google Scholar]
- Ross, W.E.; Shipley, N. Relationship between DNA damage and survival in formaldehyde-treated mouse cells. Mutat. Res. 1980, 79, 277–283. [Google Scholar] [CrossRef]
- Mirzazadeh, R.; Kallas, T.; Bienko, M.; Crosetto, N. Genome-Wide Profiling of DNA Double-Strand Breaks by the BLESS and BLISS Methods. Methods Mol. Biol. Clifton NJ 2018, 1672, 167–194. [Google Scholar]
- Gao, L.; Cox, D.B.T.; Yan, W.X.; Manteiga, J.C.; Schneider, M.W.; Yamano, T.; Nishimasu, H.; Nureki, O.; Crosetto, N.; Zhang, F. Engineered Cpf1 variants with altered PAM specificities. Nat. Biotechnol. 2017, 35, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Halász, L.; Karányi, Z.; Boros-Oláh, B.; Kuik-Rózsa, T.; Sipos, É.; Nagy, É.; Mosolygó-L, Á.; Mázló, A.; Rajnavölgyi, É.; Halmos, G.; et al. RNA-DNA hybrid (R-loop) immunoprecipitation mapping: An analytical workflow to evaluate inherent biases. Genome Res. 2017, 27, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Schoenfelder, S.; Furlan-Magaril, M.; Mifsud, B.; Tavares-Cadete, F.; Sugar, R.; Javierre, B.-M.; Nagano, T.; Katsman, Y.; Sakthidevi, M.; Wingett, S.W.; et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015, 25, 582–597. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813.e6. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046.
- de Wit, E.; de Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, C.; Soutoglou, E. DSB (Im)mobility and DNA repair compartmentalization in mammalian cells. J. Mol. Biol. 2015, 427, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Kalousi, A.; Soutoglou, E. Nuclear compartmentalization of DNA repair. Curr. Opin. Genet. Dev. 2016, 37, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Legube, G. Organizing DNA repair in the nucleus: DSBs hit the road. Curr. Opin. Cell Biol. 2017, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dryden, N.H.; Broome, L.R.; Dudbridge, F.; Johnson, N.; Orr, N.; Schoenfelder, S.; Nagano, T.; Andrews, S.; Wingett, S.; Kozarewa, I.; et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014, 24, 1854–1868. [Google Scholar] [CrossRef] [PubMed]
- Guénolé, A.; Legube, G. A meeting at risk: Unrepaired DSBs go for broke. Nucl. Austin Tex 2017, 8, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Caridi, C.P.; Delabaere, L.; Tjong, H.; Hopp, H.; Das, D.; Alber, F.; Chiolo, I. Quantitative Methods to Investigate the 4D Dynamics of Heterochromatic Repair Sites in Drosophila Cells. Methods Enzymol. 2018, 601, 359–389. [Google Scholar] [PubMed]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, T.J.; Bouwman, B.; Mirzazadeh, R.; Garnerone, S.; Crosetto, N.; Semple, C. Modelling double strand break susceptibility to interrogate structural variation in cancer. bioRxiv 2018, 441832. [Google Scholar]



{kind=link}
{kind=link}
| Method | Detection | Main Features | Sample (Input) | Reported Applications |
|---|---|---|---|---|
| GUIDE-seq [194] | Indirect | In vivo incorporation of dsODN through NHEJ. | Transfected live cells | |
| IDLV capture [195] | Indirect | In vivo random incorporation of integration defective lentiviral vectors, through NHEJ. | Transduced live cells | |
| TC-Seq [200,207], (LAM-) HTGTS [197,199], and3D-proximity based break joining assay [208] | Indirect | Sequencing of translocation products between DSBs ends and a bait DSB, produced via NHEJ. | Live cells treated to induce translocations | |
| ChIP-chip and ChIP-seq [183,187,188] | Indirect | Capture of chromatin marked by DSB markers or associated with DSB-inducing enzymes. | Fixed cells (at least 107) | |
| BLISS [201] | Direct | In situ blunting and ligation of an adapter containing a T7 promoter, UMI and sample barcode. IVT to selectively, linearly amplify DSB ends. | Fixed cells or tissue sections (at least 103 cells) | |
| BLESS [122] and i-BLESS [203] | Direct | In situ or in agarose blunting and ligation of biotinylated adapters. DSB capture on streptavidin, then PCR amplification. | Fixed cells (at least 106) for BLESS, i-BLESS can use non-fixed cells | |
| DSBCapture [125] | Direct | In situ blunting and A-tailing, ligation of adapters with Illumina sequences. | Fixed cells (at least 106) |
|
| End-Seq [202] | Direct | In vivo blunting and A-tailing in agarose plugs. Labeling with adapters that contain Illumina sequences. | Live cells (at least 107) | |
| Break-Seq [204] | Direct | Biotin labeling of DSB ends in HMW gDNA in agarose, then capture and sequencing. | Live cells embedded in agarose (106) |
|
| DSB-Seq [126] | Direct | Biotin labeling of DSB (and SSB) ends in HMW gDNA, then capture and sequencing. | 500 μg HMW gDNA (extracted from 108 cells) |
|
| dDIP [214] and DBrIC [215] | Direct | Biotin labeling of DNA ends in gDNA, then IP and qPCR. | 0.5–1 μg extracted DNA | |
| HCoDES [217] | Direct | Hairpin capture of ssDNA-ligated DSB ends, then PCR and sequencing. | 10 μg gDNA for ssDNA ligation |
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouwman, B.A.M.; Crosetto, N. Endogenous DNA Double-Strand Breaks during DNA Transactions: Emerging Insights and Methods for Genome-Wide Profiling. Genes 2018, 9, 632. https://doi.org/10.3390/genes9120632
Bouwman BAM, Crosetto N. Endogenous DNA Double-Strand Breaks during DNA Transactions: Emerging Insights and Methods for Genome-Wide Profiling. Genes. 2018; 9(12):632. https://doi.org/10.3390/genes9120632
Chicago/Turabian StyleBouwman, Britta A. M., and Nicola Crosetto. 2018. "Endogenous DNA Double-Strand Breaks during DNA Transactions: Emerging Insights and Methods for Genome-Wide Profiling" Genes 9, no. 12: 632. https://doi.org/10.3390/genes9120632
APA StyleBouwman, B. A. M., & Crosetto, N. (2018). Endogenous DNA Double-Strand Breaks during DNA Transactions: Emerging Insights and Methods for Genome-Wide Profiling. Genes, 9(12), 632. https://doi.org/10.3390/genes9120632