Chromatin as a Platform for Modulating the Replication Stress Response


{kind=link}
{kind=link}
Abstract
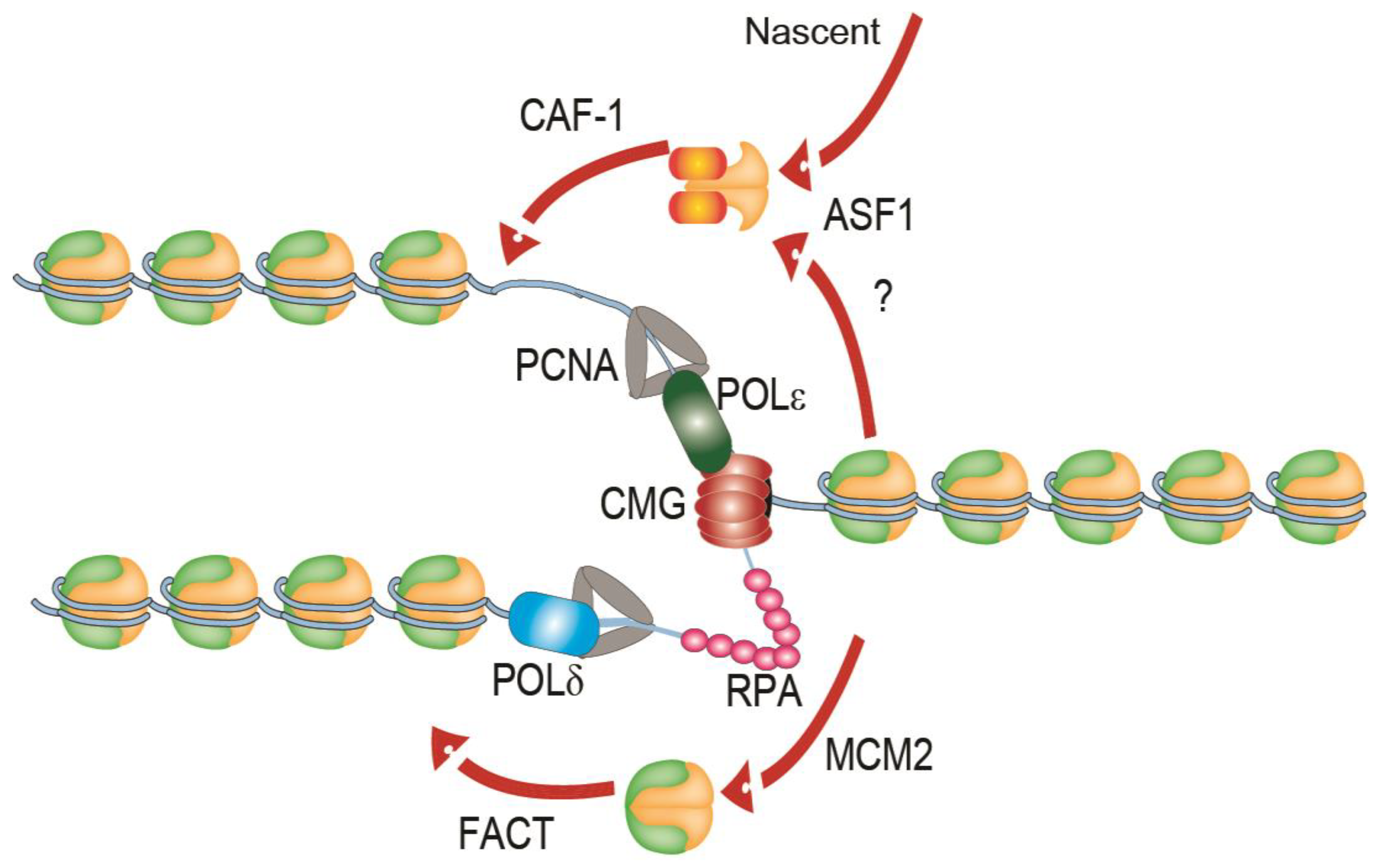
1. Replication of Chromatin
2. Replication Stress and Fork Stalling
3. Histone Modifications in Replication Stress Responses
3.1. H2A Ubiquitination
3.2. H2B Ubiquitination
3.3. EZH2 and H3K27me3
3.4. SETD1A, MLL3/4 and H3K4 Methylation
4. Histone Variants in Replication Stress Responses
4.1. H2A.X
4.2. MACRO-H2A
4.3. H2A.Z and Its Regulators
5. Readers, Remodellers and Chaperones in Replication Stress
5.1. INO80
5.2. TONSL-MMS22L
5.3. SUMO2-Modified PCNA Recruitment of Histone Chaperones
5.4. ASF1-CAF1
5.5. SWI/SNF and Related Chromatin Remodellers
5.6. ALC1/CHD1L
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Horard, B.; Sapey-Triomphe, L.; Bonnefoy, E.; Loppin, B. ASF1 is required to load histones on the HIRA complex in preparation of paternal chromatin assembly at fertilization. Epigenet. Chromatin 2018, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Serra-Cardona, A.; Zhang, Z. Replication-Coupled Nucleosome Assembly in the Passage of Epigenetic Information and Cell Identity. Trends Biochem. Sci. 2018, 43, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Maya, D. Regulation of Replication Fork Advance and Stability by Nucleosome Assembly. Genes 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.M.; Stromme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Boil. 2017, 18, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef]
- Clement, C.; Almouzni, G. MCM2 binding to histones H3-H4 and ASF1 supports a tetramer-to-dimer model for histone inheritance at the replication fork. Nat. Struct. Mol. Biol. 2015, 22, 587–589. [Google Scholar] [CrossRef]
- Das, C.; Lucia, M.S.; Hansen, K.C.; Tyler, J.K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009, 459, 113–117. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Feng, J.; Leng, H.; Li, S.; Xiao, J.; Liu, S.; Xu, Z.; Xu, J.; Li, D.; et al. The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Rep. 2016, 14, 1128–1141. [Google Scholar] [CrossRef]
- Khurana, S.; Oberdoerffer, P. Replication Stress: A Lifetime of Epigenetic Change. Genes 2015, 6, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, R.; Belan, O.; Pye, V.E.; Clement, C.; Maslen, S.L.; Skehel, J.M.; Cherepanov, P.; Almouzni, G.; Boulton, S.J. POLE3-POLE4 Is a Histone H3–H4 Chaperone that Maintains Chromatin Integrity during DNA Replication. Mol. Cell 2018, 72, 112–126.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xu, Z.; Leng, H.; Zheng, P.; Yang, J.; Chen, K.; Feng, J.; Li, Q. RPA binds histone H3–H4 and functions in DNA replication-coupled nucleosome assembly. Science 2017, 355, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Foltman, M.; Evrin, C.; De Piccoli, G.; Jones, R.C.; Edmondson, R.D.; Katou, Y.; Nakato, R.; Shirahige, K.; Labib, K. Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep. 2013, 3, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Petryk, N.; Dalby, M.; Wenger, A.; Stromme, C.B.; Strandsby, A.; Andersson, R.; Groth, A. MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 2018, 361, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.F.; Peterson, C.L. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res. 1999, 27, 2022–2028. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.A.; Kwong, T.J.; Tsukiyama, T. ATP-dependent chromatin remodeling shapes the DNA replication landscape. Nat. Struct. Mol. Boil. 2008, 15, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; West, S.C. MUS81-EME2 promotes replication fork restart. Cell Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Georgoulis, A.; Vorgias, C.E.; Chrousos, G.P.; Rogakou, E.P. Genome Instability and γH2AX. Int. J. Mol. Sci. 2017, 18, 1979. [Google Scholar] [CrossRef] [PubMed]
- Al-Hakim, A.; Escribano-Diaz, C.; Landry, M.C.; O’Donnell, L.; Panier, S.; Szilard, R.K.; Durocher, D. The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair (Amst) 2010, 9, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Endo, T.A.; Endoh, T.; Isono, K.; Sharif, J.; Ohara, O.; Toyoda, T.; Ito, T.; Eskeland, R.; Bickmore, W.A.; et al. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet. 2012, 8, e1002774. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kajitani, T.; Togo, S.; Masuko, N.; Ohdan, H.; Hishikawa, Y.; Koji, T.; Matsuyama, T.; Ikura, T.; Muramatsu, M.; et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008, 22, 37–49. [Google Scholar] [CrossRef]
- Bravo, M.; Nicolini, F.; Starowicz, K.; Barroso, S.; Cales, C.; Aguilera, A.; Vidal, M. Polycomb RING1A- and RING1B-dependent histone H2A monoubiquitylation at pericentromeric regions promotes S-phase progression. J. Cell Sci. 2015, 128, 3660–3671. [Google Scholar] [CrossRef]
- Schmid, J.A.; Berti, M.; Walser, F.; Raso, M.C.; Schmid, F.; Krietsch, J.; Stoy, H.; Zwicky, K.; Ursich, S.; Freire, R.; et al. Histone Ubiquitination by the DNA Damage Response Is Required for Efficient DNA Replication in Unperturbed S Phase. Mol. Cell 2018, 71, 897–910.e8. [Google Scholar] [CrossRef]
- Vitaliano-Prunier, A.; Babour, A.; Herissant, L.; Apponi, L.; Margaritis, T.; Holstege, F.C.; Corbett, A.H.; Gwizdek, C.; Dargemont, C. H2B ubiquitylation controls the formation of export-competent mRNP. Mol. Cell 2012, 45, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Pirngruber, J.; Shchebet, A.; Schreiber, L.; Shema, E.; Minsky, N.; Chapman, R.D.; Eick, D.; Aylon, Y.; Oren, M.; Johnsen, S.A. CDK9 directs H2B monoubiquitination and controls replication-dependent histone mRNA 3’-end processing. EMBO Rep. 2009, 10, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Herissant, L.; Moehle, E.A.; Bertaccini, D.; Van Dorsselaer, A.; Schaeffer-Reiss, C.; Guthrie, C.; Dargemont, C. H2B ubiquitylation modulates spliceosome assembly and function in budding yeast. Boil. Cell 2014, 106, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Kao, C.F.; Krogan, N.J.; Sun, Z.W.; Greenblatt, J.F.; Osley, M.A.; Strahl, B.D. Histone H2B ubiquitylation is associated with elongating RNA polymerase II. Mol. Cell. Biol. 2005, 25, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.B.; Kao, C.F.; Hillyer, C.; Pikaart, M.; Osley, M.A. H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol. Cell 2008, 31, 57–66. [Google Scholar] [CrossRef]
- Weake, V.M.; Workman, J.L. Histone ubiquitination: Triggering gene activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef]
- Chernikova, S.B.; Razorenova, O.V.; Higgins, J.P.; Sishc, B.J.; Nicolau, M.; Dorth, J.A.; Chernikova, D.A.; Kwok, S.; Brooks, J.D.; Bailey, S.M.; et al. Deficiency in mammalian histone H2B ubiquitin ligase Bre1 (Rnf20/Rnf40) leads to replication stress and chromosomal instability. Cancer Res. 2012, 72, 2111–2119. [Google Scholar] [CrossRef]
- Trujillo, K.M.; Osley, M.A. A role for H2B ubiquitylation in DNA replication. Mol. Cell 2012, 48, 734–746. [Google Scholar] [CrossRef]
- Lin, C.Y.; Wu, M.Y.; Gay, S.; Marjavaara, L.; Lai, M.S.; Hsiao, W.C.; Hung, S.H.; Tseng, H.Y.; Wright, D.E.; Wang, C.Y.; et al. H2B mono-ubiquitylation facilitates fork stalling and recovery during replication stress by coordinating Rad53 activation and chromatin assembly. PLoS Genet. 2014, 10, e1004667. [Google Scholar] [CrossRef]
- Hung, S.H.; Wong, R.P.; Ulrich, H.D.; Kao, C.F. Monoubiquitylation of histone H2B contributes to the bypass of DNA damage during and after DNA replication. Proc. Natl. Acad. Sci. USA 2017, 114, E2205–E2214. [Google Scholar] [CrossRef]
- Nicassio, F.; Corrado, N.; Vissers, J.H.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.B.; Vermeulen, W.; Di Fiore, P.P.; et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr. Biol. 2007, 17, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Pradhan, S. Mammalian epigenetic mechanisms. IUBMB Life 2014, 66, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Muller, C.W.; Vermeulen, M.; Muller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Boil. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yucel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Boil. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Sato, K.; Reynolds, J.J.; Begum, S.; Bayley, R.; Goula, A.; Vernet, A.; Paquin, K.L.; Skalnik, D.G.; Kobayashi, W.; et al. Histone Methylation by SETD1A Protects Nascent DNA through the Nucleosome Chaperone Activity of FANCD2. Mol. Cell 2018, 71, 25–41.e6. [Google Scholar] [CrossRef] [PubMed]
- Faucher, D.; Wellinger, R.J. Methylated H3K4, a transcription-associated histone modification, is involved in the DNA damage response pathway. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Munoz, I.M.; Rouse, J. Control of histone methylation and genome stability by PTIP. EMBO Rep. 2009, 10, 239–245. [Google Scholar] [CrossRef]
- Wang, X.; Takenaka, K.; Takeda, S. PTIP promotes DNA double-strand break repair through homologous recombination. Genes Cells 2010, 15, 243–254. [Google Scholar] [CrossRef]
- Cho, Y.W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Tomaszowski, K.H.; Luzwick, J.W.; Park, S.; Li, J.; Murphy, M.; Schlacher, K. p53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLtheta pathways. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pichardo, D.; Canas, J.C.; Garcia-Rubio, M.L.; Gomez-Gonzalez, B.; Rondon, A.G.; Aguilera, A. Histone Mutants Separate R Loop Formation from Genome Instability Induction. Mol. Cell 2017, 66, 597–609.e5. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Pozo, M.; Santos-Pereira, J.M.; Rondon, A.G.; Barroso, S.; Andujar, E.; Perez-Alegre, M.; Garcia-Muse, T.; Aguilera, A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 2013, 52, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef]
- Ward, I.M.; Minn, K.; Chen, J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J. Biol. Chem. 2004, 279, 9677–9680. [Google Scholar] [CrossRef]
- Chanoux, R.A.; Yin, B.; Urtishak, K.A.; Asare, A.; Bassing, C.H.; Brown, E.J. ATR and H2AX cooperate in maintaining genome stability under replication stress. J. Biol. Chem. 2009, 284, 5994–6003. [Google Scholar] [CrossRef]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008, 453, 682–686. [Google Scholar] [CrossRef]
- Kim, J.; Sturgill, D.; Sebastian, R.; Khurana, S.; Tran, A.D.; Edwards, G.B.; Kruswick, A.; Burkett, S.; Hosogane, E.K.; Hannon, W.W.; et al. Replication Stress Shapes a Protective Chromatin Environment across Fragile Genomic Regions. Mol. Cell 2018, 69, 36–47.e7. [Google Scholar] [CrossRef]
- Herrera-Moyano, E.; Mergui, X.; Garcia-Rubio, M.L.; Barroso, S.; Aguilera, A. The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014, 28, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Kurat, C.F.; Yeeles, J.T.P.; Patel, H.; Early, A.; Diffley, J.F.X. Chromatin Controls DNA Replication Origin Selection, Lagging-Strand Synthesis, and Replication Fork Rates. Mol. Cell 2017, 65, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, C.B.; Hauer, M.H.; Gasser, S.M. INO80-C and SWR-C: Guardians of the genome. J. Mol. Boil. 2015, 427, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Brahma, S.; Udugama, M.I.; Kim, J.; Hada, A.; Bhardwaj, S.K.; Hailu, S.G.; Lee, T.H.; Bartholomew, B. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nat. Commun. 2017, 8, 15616. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, G.; Shen, X.; Landry, J.; Wu, W.H.; Sen, S.; Wu, C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 2004, 303, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, A.; Li, B.Z.; Szakal, B.; Branzei, D.; Putnam, C.D.; Kolodner, R.D. The Swr1 chromatin-remodeling complex prevents genome instability induced by replication fork progression defects. Nat. Commun. 2018, 9, 3680. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Watanabe, S.; Rando, O.J.; Peterson, C.L. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 2011, 144, 200–213. [Google Scholar] [CrossRef]
- Huh, M.S.; Ivanochko, D.; Hashem, L.E.; Curtin, M.; Delorme, M.; Goodall, E.; Yan, K.; Picketts, D.J. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 2016, 7, e2220. [Google Scholar] [CrossRef]
- Simoneau, A.; Ricard, E.; Wurtele, H. An interplay between multiple sirtuins promotes completion of DNA replication in cells with short telomeres. PLoS Genet. 2018, 14, e1007356. [Google Scholar] [CrossRef]
- Frey, A.; Listovsky, T.; Guilbaud, G.; Sarkies, P.; Sale, J.E. Histone H3.3 is required to maintain replication fork progression after UV damage. Curr. Biol. 2014, 24, 2195–2201. [Google Scholar] [CrossRef] [PubMed]
- Mathew, V.; Pauleau, A.L.; Steffen, N.; Bergner, A.; Becker, P.B.; Erhardt, S. The histone-fold protein CHRAC14 influences chromatin composition in response to DNA damage. Cell Rep. 2014, 7, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Oma, Y.; Schleker, T.; Kugou, K.; Ohta, K.; Harata, M.; Gasser, S.M. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr. Biol. 2008, 18, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Peterson, C.L. The Ino80 chromatin-remodeling enzyme regulates replisome function and stability. Nat. Struct. Mol. Boil. 2008, 15, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Falbo, K.B.; Alabert, C.; Katou, Y.; Wu, S.; Han, J.; Wehr, T.; Xiao, J.; He, X.; Zhang, Z.; Shi, Y.; et al. Involvement of a chromatin remodeling complex in damage tolerance during DNA replication. Nat. Struct. Mol. Boil. 2009, 16, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Lee, S.A.; Hur, S.K.; Seo, J.W.; Kwon, J. Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat. Commun. 2014, 5, 5128. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, K.; Chang, D.Y.; Kang, H.B.; Shin, E.C.; Kwon, J.; Choi, J.K. Genome-wide reorganization of histone H2AX toward particular fragile sites on cell activation. Nucleic Acids Res. 2014, 42, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Poli, J.; Gerhold, C.B.; Tosi, A.; Hustedt, N.; Seeber, A.; Sack, R.; Herzog, F.; Pasero, P.; Shimada, K.; Hopfner, K.P.; et al. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev 2016, 30, 337–354. [Google Scholar] [CrossRef]
- Saredi, G.; Huang, H.; Hammond, C.M.; Alabert, C.; Bekker-Jensen, S.; Forne, I.; Reveron-Gomez, N.; Foster, B.M.; Mlejnkova, L.; Bartke, T.; et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 2016, 534, 714–718. [Google Scholar] [CrossRef]
- Burgess, R.J.; Zhang, Z. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 2013, 20, 14–22. [Google Scholar] [CrossRef]
- Duro, E.; Lundin, C.; Ask, K.; Sanchez-Pulido, L.; MacArtney, T.J.; Toth, R.; Ponting, C.P.; Groth, A.; Helleday, T.; Rouse, J. Identification of the MMS22L–TONSL complex that promotes homologous recombination. Mol. Cell 2010, 40, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Piwko, W.; Mlejnkova, L.J.; Mutreja, K.; Ranjha, L.; Stafa, D.; Smirnov, A.; Brodersen, M.M.; Zellweger, R.; Sturzenegger, A.; Janscak, P.; et al. The MMS22L–TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J. 2016, 35, 2584–2601. [Google Scholar] [CrossRef] [PubMed]
- Campos, E.I.; Smits, A.H.; Kang, Y.H.; Landry, S.; Escobar, T.M.; Nayak, S.; Ueberheide, B.M.; Durocher, D.; Vermeulen, M.; Hurwitz, J.; et al. Analysis of the Histone H3.1 Interactome: A Suitable Chaperone for the Right Event. Mol. Cell 2015, 60, 697–709. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Adamson, B.; Lydeard, J.R.; Sowa, M.E.; Ciccia, A.; Bredemeyer, A.L.; Schlabach, M.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L–NFKBIL2 complex required for genomic stability. Mol. Cell 2010, 40, 645–657. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.; Panier, S.; Wildenhain, J.; Tkach, J.M.; Al-Hakim, A.; Landry, M.C.; Escribano-Diaz, C.; Szilard, R.K.; Young, J.T.; Munro, M.; et al. The MMS22L–TONSL complex mediates recovery from replication stress and homologous recombination. Mol. Cell 2010, 40, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xu, X.; Chang, C.W.; Zheng, L.; Shen, B.; Liu, Y. SUMO2 conjugation of PCNA facilitates chromatin remodeling to resolve transcription-replication conflicts. Nat. Commun. 2018, 9, 2706. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, Y.; Inoue, K.; Oikawa, M.; Kamimura, S.; Ogonuki, N.; Kodama, E.N.; Ohkawa, Y.; Tsukada, Y.; Ogura, A. Histone chaperone CAF-1 mediates repressive histone modifications to protect preimplantation mouse embryos from endogenous retrotransposons. Proc. Natl. Acad. Sci. USA 2015, 112, 14641–14646. [Google Scholar] [CrossRef]
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res. 2012, 40, 6049–6059. [Google Scholar] [CrossRef]
- Jasencakova, Z.; Scharf, A.N.; Ask, K.; Corpet, A.; Imhof, A.; Almouzni, G.; Groth, A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol. Cell 2010, 37, 736–743. [Google Scholar] [CrossRef]
- Loyola, A.; Tagami, H.; Bonaldi, T.; Roche, D.; Quivy, J.P.; Imhof, A.; Nakatani, Y.; Dent, S.Y.; Almouzni, G. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 2009, 10, 769–775. [Google Scholar] [CrossRef]
- Rivera, C.; Saavedra, F.; Alvarez, F.; Diaz-Celis, C.; Ugalde, V.; Li, J.; Forne, I.; Gurard-Levin, Z.A.; Almouzni, G.; Imhof, A.; et al. Methylation of histone H3 lysine 9 occurs during translation. Nucleic Acids Res. 2015, 43, 9097–9106. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.A.; Lam, W.M.; Burgers, P.M.; Kaufman, P.D. Histone deposition protein ASF1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 2005, 19, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.; Orsi, G.A.; Gatto, A.; Boyarchuk, E.; Forest, A.; Hajj, B.; Mine-Hattab, J.; Garnier, M.; Gurard-Levin, Z.A.; Quivy, J.P.; et al. High-resolution visualization of H3 variants during replication reveals their controlled recycling. Nat. Commun. 2018, 9, 3181. [Google Scholar] [CrossRef] [PubMed]
- Hoek, M.; Stillman, B. Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 12183–12188. [Google Scholar] [CrossRef] [PubMed]
- Minard, L.V.; Lin, L.J.; Schultz, M.C. SWI/SNF and ASF1 independently promote derepression of the DNA damage response genes under conditions of replication stress. PLoS ONE 2011, 6, e21633. [Google Scholar] [CrossRef] [PubMed]
- Niimi, A.; Chambers, A.L.; Downs, J.A.; Lehmann, A.R. A role for chromatin remodellers in replication of damaged DNA. Nucleic Acids Res. 2012, 40, 7393–7403. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Chastain, P.D., 2nd; Rosson, G.B.; Groh, B.S.; Weissman, B.E.; Kaufman, D.G.; Bultman, S.J. BRG1 co-localizes with DNA replication factors and is required for efficient replication fork progression. Nucleic Acids Res. 2010, 38, 6906–6919. [Google Scholar] [CrossRef]
- Espana-Agusti, J.; Warren, A.; Chew, S.K.; Adams, D.J.; Matakidou, A. Loss of PBRM1 rescues VHL dependent replication stress to promote renal carcinogenesis. Nat. Commun. 2017, 8, 2026. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Ooka, M.; Abe, T.; Cho, K.; Koike, K.; Takeda, S.; Hirota, K. Chromatin remodeler ALC1 prevents replication-fork collapse by slowing fork progression. PLoS ONE 2018, 13, e0192421. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.F.; Hu, L.; Fung, J.M.; Xie, D.; Zheng, B.J.; Chen, L.; Tang, D.J.; Fu, L.; Wu, Z.; Chen, M.; et al. Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 2008, 47, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.J.; Li, H.L.; Yao, Y.; Liu, S.C.; Yin, C.G.; Ma, X.Z. Chromodomain Helicase/ATPase DNA-Binding Protein 1-Like Gene (CHD1L) Expression and Implications for Invasion and Metastasis of Breast Cancer. PLoS ONE 2015, 10, e0143030. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fournier, L.-A.; Kumar, A.; Stirling, P.C. Chromatin as a Platform for Modulating the Replication Stress Response. Genes 2018, 9, 622. https://doi.org/10.3390/genes9120622
Fournier L-A, Kumar A, Stirling PC. Chromatin as a Platform for Modulating the Replication Stress Response. Genes. 2018; 9(12):622. https://doi.org/10.3390/genes9120622
Chicago/Turabian StyleFournier, Louis-Alexandre, Arun Kumar, and Peter C. Stirling. 2018. "Chromatin as a Platform for Modulating the Replication Stress Response" Genes 9, no. 12: 622. https://doi.org/10.3390/genes9120622
APA StyleFournier, L.-A., Kumar, A., & Stirling, P. C. (2018). Chromatin as a Platform for Modulating the Replication Stress Response. Genes, 9(12), 622. https://doi.org/10.3390/genes9120622