
Panel-Based Genetic Testing in a Consecutive Series of Individuals with Inherited Retinal Diseases in Australia: Identifying Predictors of a Diagnosis

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics and Participants
2.2. Genetic Testing
2.3. Clinical and Phenotype Characteristics

{kind=link}
{kind=link}
{kind=link}
| Human Phenotype Ontology (HPO) Term Identifier | Human Phenotype Ontology (HPO) Term Name | Additional Descriptive Features Used in the Classification of Retinal Images in This Study |
|---|---|---|
| Widespread Retinal Dystrophy a | ||
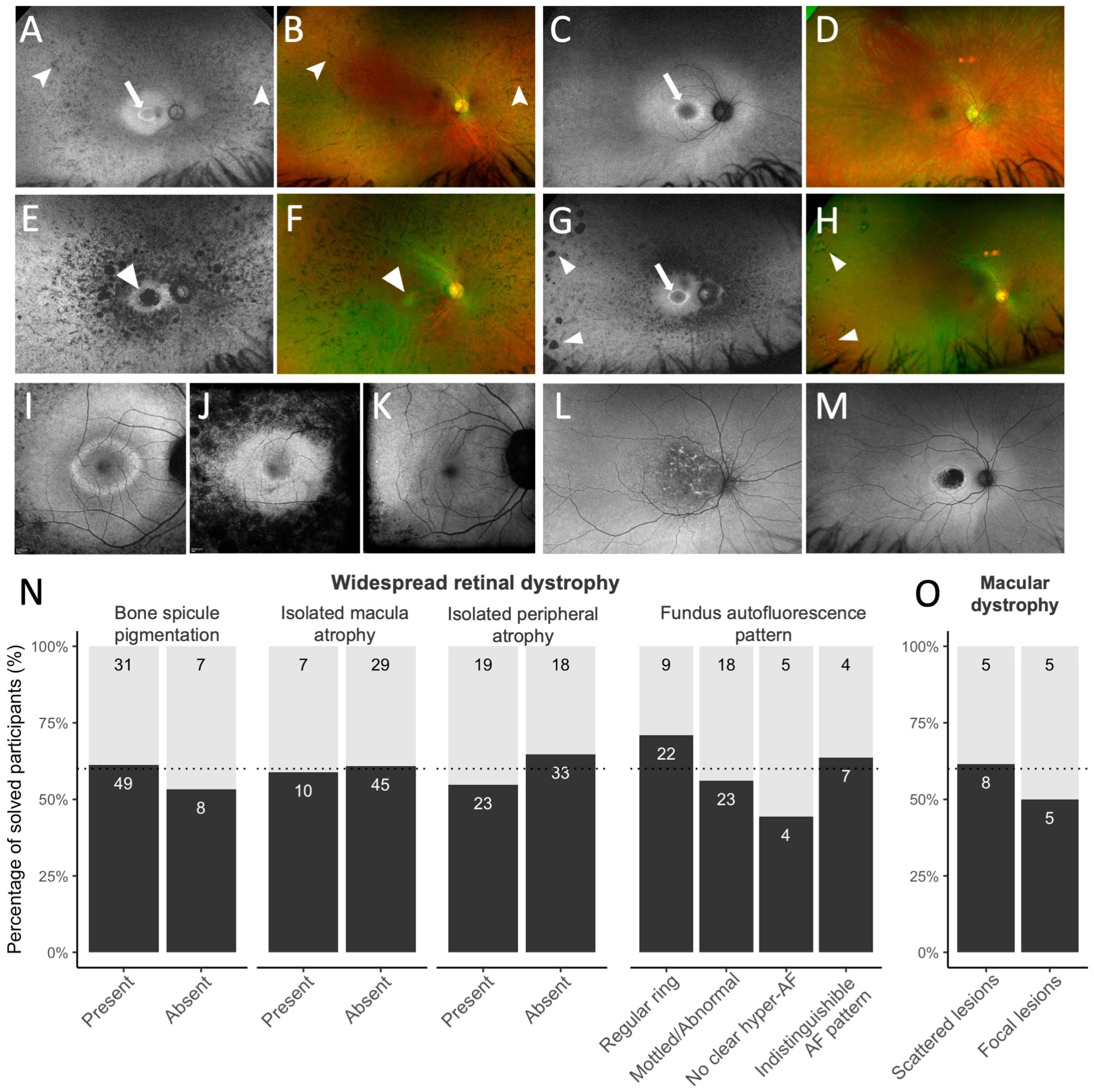
| HP:0007737 | Bone spicule pigmentation of the retina | Presence of distinct bone spicule pigmentation in any degrees of confluence (shown in Figure 1A,B) |
| HP:0007401 | Macular atrophy | Presence of macular atrophy, reflecting loss of the retinal pigment epithelium with associated retinal photoreceptor loss, in the central macular isolated from any mid-peripheral atrophy (shown in Figure 1E,F) |
| HP:0200070 | Peripheral retinal atrophy | Presence of discrete islands of sharply demarcated peripheral chorioretinal atrophy, interspersed by adjacent normal retina (shown in Figure 1G,H) |
| HP:0030602 | Abnormal fundus autofluorescence imaging | Hyperfluorescent ring at the posterior pole, graded as: (i) regular perifoveal ring of hyperautofluorescence, seen as closed rings with an ellipsoid/round shape and regular borders (Figure 1I); (ii) mottled, irregular, or abnormal central macular autofluorescence without a distinct, regular ring (Figure 1J); (iii) no visible hyperautofluorescence in the macular or peri-macular region; (iv) Indistinguishable hyperautofluorescence pattern due to extensive atrophy (Figure 1K) |
| Macular Dystrophy a | ||
| HP:0030500 | Yellow/white lesions of the macula | (i) Scattered macular lesions, including flecks, fundus flavimaculatus dystrophy, and speckled, reticular, and patterned hyperautofluorescence lesions, extending past the retinal arcades (shown in Figure 1L); (ii) Focal/isolated macular atrophy or lesions limited to the macula (shown in Figure 1M) |
2.4. Statistical Analysis
3. Results
3.1. Study Population
3.2. Diagnostic Outcomes
3.3. Diagnostic Utility
3.4. Diagnostic Yield Based on Clinical Diagnosis and Imaging Features
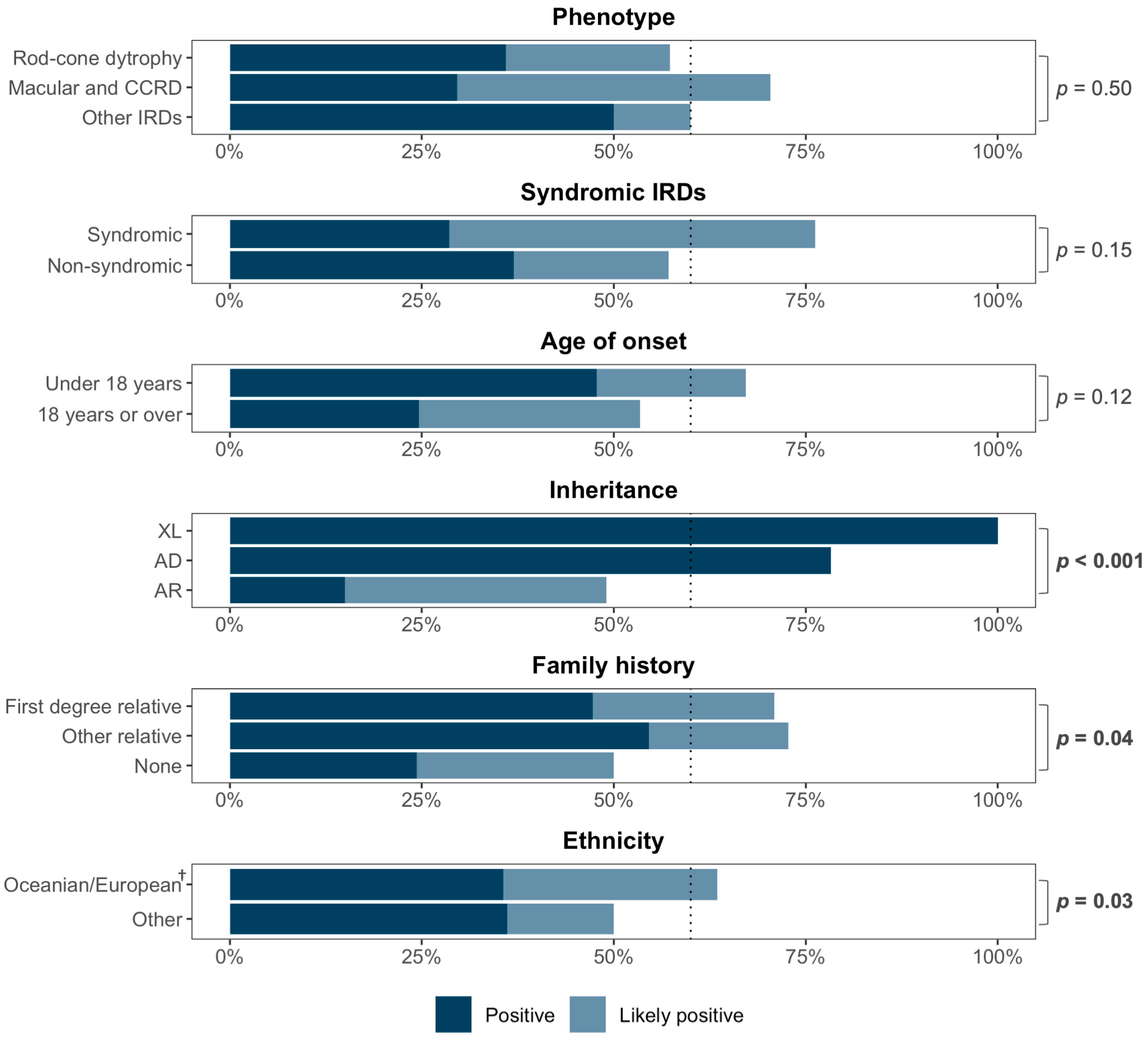
3.5. Predictors of Diagnosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FAF | Fundus autofluorescence |
| HPO | Human Phenotype Ontology |
| IRD | Inherited retinal disease |
| OCT | Optical coherence tomography |
| RCD | Rod–cone dystrophy |
| VENTURE | Victorian Evolution of inherited retinal diseases NaTUral history REgistry |
| VUS | Variant of Uncertain Significance |
Appendix A
| Genes on Both Blueprint and Invitae Targeted Panels | |||||
| ACO2 | ADAM9 | ADAMTS18 | ADGRV1 | ADIPOR1 | AGBL5 |
| AHI1 | AIPL1 | ALMS1 | ARHGEF18 | ARL13B | ARL2BP |
| ARL3 | ARL6 | ARMC9 | ARSG | ATF6 | ATOH7 |
| B9D1 | BBIP1 | BBS1 † | BBS10 | BBS12 | BBS2 |
| BBS4 † | BBS5 † | BBS7 | BBS9 | BEST1 † | C1QTNF5 |
| C2ORF71/PCARE | C5ORF42/CPLANE1 | C8ORF37/CFAP418 | CA4 | CABP4 | CACNA1F |
| CACNA2D4 | CAPN5 | CC2D2A | CDH23 | CDH3 | CDHR1 |
| CEP164 | CEP19 | CEP250 | CEP290 †,§ | CEP41 | CEP78 |
| CEP83 * | CERKL | C21ORF2 †/CFAP410 | CHM † | CIB2 | CISD2 |
| CLN3 † | CLN5 *,‡ | CLN6 *,†,‡ | CLN8 *,‡ | CLRN1 † | CNGA1 |
| CNGA3 † | CNGB1 | CNGB3 | CNNM4 | COL11A1 † | COL11A2 |
| COL18A1 | COL2A1 † | COL9A1 | COL9A2 | COL9A3 | CRB1 |
| CRX | CSPP1 | CTNNA1 | CTSD *,‡ | CWC27 | CYP4V2 |
| DHDDS † | DHX38 | DRAM2 | DTHD1 | EFEMP1 | ELOVL4 |
| EMC1 | EXOSC2 * | EYS † | FAM161A | FLVCR1 | FRMD7 † |
| FZD4 | GNAT1 | GNAT2 † | GNB3 | GNPTG † | GPR143 *,† |
| GPR179 | GRM6 | GUCA1A | GUCY2D † | HARS | HGSNAT † |
| HK1 † | HMX1 | IDH3A | IDH3B | IFT140 † | IFT172 |
| IFT27 | IFT81 | IMPDH1 | IMPG1 | IMPG2 | INPP5E |
| INVS | IQCB1 | JAG1 † | KCNJ13 | KCNV2 | KIAA0586 |
| KIAA1549 | KIF11 | KIF7 | KIZ | KLHL7 | LCA5 |
| LRAT † | LRIT3 | LRP2 | LRP5 | LZTFL1 | MAK |
| MERTK | MFN2 | MFRP | MFSD8 ‡ | MKKS | MKS1 |
| MTTP † | MYO7A † | NAGLU *,‡ | NDP † | NEK2 | NMNAT1 † |
| NPHP1 | NPHP3 | NPHP4 | NR2E3 | NR2F1 | NRL |
| NYX | OAT | OCA2 *,† | OFD1 † | OPA1 † | OPA3 |
| OPN1SW * | OTX2 | P3H2 | PAX2 | PCDH15 † | PCYT1A |
| PDE6A | PDE6B | PDE6C † | PDE6D | PDE6G | PDE6H |
| PDZD7 | PEX1 | PEX10 | PEX11B | PEX12 | PEX13 |
| PEX14 | PEX16 | PEX19 | PEX2 | PEX26 | PEX3 |
| PEX5 | PEX6 † | PEX7 † | PHYH | PITPNM3 | PLA2G5 |
| PLK4 * | PNPLA6 | POC1B | POMGNT1 | PPT1 *,†,‡,§ | PRCD |
| PRDM13 † | PROM1 † | PRPF3 | PRPF31 † | PRPF4 | PRPF6 |
| PRPF8 | PRPH2 | PRPS1 | RAB28 | RAX2 | RBP3 |
| RBP4 | RCBTB1 | RD3 | RDH11 | RDH12 | RDH5 † |
| REEP6 | RGR | RGS9 | RGS9BP | RHO | RIMS1 |
| RLBP1 | ROM1 | RP1 | RP1L1 | RP2 | RPE65 † |
| RPGR (including ORF15) †,‡ | RPGRIP1 † | RPGRIP1L | RS1 | RTN4IP1 | SAG |
| SAMD11 | SCLT1 | SDCCAG8 | SEMA4A | SGSH *,†,‡ | SLC24A1 |
| SLC45A2 * | SLC7A14 | SNRNP200 | SPATA7 | SPP2 | TCTN1 |
| TCTN2 | TCTN3 | TEAD1 | TIMM8A † | TIMP3 | TMEM107 |
| TMEM126A | TMEM138 | TMEM216 | TMEM231 † | TMEM237 | TMEM67 |
| TOPORS | TPP1 *,† | TRAF3IP1 | TREX1 | TRIM32 | TRPM1 |
| TSPAN12 | TTC21B | TTC8 | TTLL5 | TTPA | TUB |
| TUBGCP4 * | TUBGCP6 * | TULP1 | TYR *,† | TYRP1 * | USH1C |
| USH1G | USH2A † | VCAN | VPS13B | WDPCP | WDR19 |
| WFS1 † | ZNF408 | ZNF423 | ZNF513 | ||
| Genes Only on Blueprint Retinal Dystrophy Panel | |||||
| B9D2 | CEP104 | CEP120 | CPE | CTC1 | CTNNB1 |
| DFNB31 | ESPN | FDXR | GRK1 | KIAA0556 | KIAA0753 |
| MMACHC | MT-ATP6 | MT-ATP8 | MT-CO1 | MT-CO2 | MT-CO3 |
| MT-CYB | MT-ND1 | MT-ND2 | MT-ND3 | MT-ND4 | MT-ND4L |
| MT-ND5 | MT-ND6 | MT-RNR1 | MT-RNR2 | MT-TA | MT-TC |
| MT-TD | MT-TE | MT-TF | MT-TG | MT-TH | MT-TI |
| MT-TK | MT-TL1 | MT-TL2 | MT-TM | MT-TN | MT-TP |
| MT-TQ | MT-TR | MT-TS1 | MT-TS2 | MT-TT | MT-TV |
| MT-TW | MT-TY | MVK † | PANK2 † | PISD | SCAPER |
| SLC25A46 | SRD5A3 | TUBB4B | YME1L1 | ABCD1 * | AMACR * |
| ARR3 * | COQ2 * | DNAJC5 * | DYNC2H1 *,† | ISPD * | LAMA1 * |
| PDSS1 * | PDSS2 * | ||||
| Genes Only on Invitae Inherited Retinal Disorders Panel | |||||
| ACBD5 | ADGRA3 | AHR | ASRGL1 | CCT2 | CLCC1 |
| CLUAP1 | FSCN2 | GDF6 | GUCA1B | IFT43 | IFT80 |
| MAPKAPK3 | MPDZ | NEUROD1 | PAX6 | TRNT1 | UNC119 |
| WHRN | ADAMTSL4 ‡ | C10orf11 ‡/LRMDA | C12orf65 ‡/MTRFR | DHX32 ‡ | DNAJC17 ‡ |
| DSCAML1 ‡ | ERCC6 ‡ | FBLN5 ‡ | GNS ‡ | GPR45 ‡ | GRN ‡ |
| HMCN1 ‡ | IFT74 ‡ | IFT88 ‡ | ITM2B ‡ | LYST ‡ | MIR204 ‡ |
| MTPAP ‡ | NBAS ‡ | OR2W3 ‡ | POC5 ‡ | RBP1 ‡ | RP9 ‡ |
| SIX6 ‡ | SLC24A5 ‡ | TMED7 ‡ | WDR34 ‡ | ||
References
- Heath Jeffery, R.C.; Mukhtar, S.A.; McAllister, I.L.; Morgan, W.H.; Mackey, D.A.; Chen, F.K. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. 2021, 42, 431–439. [Google Scholar] [CrossRef]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, 004015. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Blanco-Kelly, F.; Sanchez-Navarro, I.; Lorda-Sanchez, I.; Tahsin-Swafiri, S.; Avila-Fernandez, A.; Martin-Merida, I.; Trujillo-Tiebas, M.J.; Lopez-Rodriguez, R.; Rodriguez de Alba, M.; et al. NGS and phenotypic ontology-based approaches increase the diagnostic yield in syndromic retinal diseases. Hum. Genet. 2021, 140, 1665–1678. [Google Scholar] [CrossRef]
- Daiger, S.P.; Rossiter, B.J.; Greenberg, J.; Christoffels, A.; Hide, W. RetNet-Retinal Information Network. Data Services and Software for Identifying Genes and Mutations Causing Retinal Degeneration. Available online: https://sph.uth.edu/RetNet/ (accessed on 28 June 2021).
- Britten-Jones, A.C.; Jin, R.; Gocuk, S.A.; Cichello, E.; O’Hare, F.; Hickey, D.G.; Edwards, T.L.; Ayton, L.N. The safety and efficacy of gene therapy treatment for monogenic retinal and optic nerve diseases: A systematic review. Genet. Med. 2021, 24, 521–534. [Google Scholar] [CrossRef]
- Britten-Jones, A.C.; Schultz, J.; Mack, H.G.; Kearns, L.S.; Huq, A.J.; Ruddle, J.B.; Mackey, D.A.; Hewitt, A.W.; Edwards, T.L.; Ayton, L.N. Patient experiences and perceived value of genetic testing in inherited retinal diseases: A cross-sectional survey. Sci. Rep. 2024, 14, 5403. [Google Scholar] [CrossRef]
- Black, G.C.; Sergouniotis, P.; Sodi, A.; Leroy, B.P.; Van Cauwenbergh, C.; Liskova, P.; Grønskov, K.; Klett, A.; Kohl, S.; Taurina, G.; et al. The need for widely available genomic testing in rare eye diseases: An ERN-EYE position statement. Orphanet J. Rare Dis. 2021, 16, 142. [Google Scholar] [CrossRef]
- Britten-Jones, A.C.; Gocuk, S.A.; Goh, K.L.; Huq, A.; Edwards, T.L.; Ayton, L.N. The diagnostic yield of next generation sequencing in inherited retinal diseases: A systematic review and meta-analysis. Am. J. Ophthalmol. 2022, 249, 57–73. [Google Scholar] [CrossRef]
- RANZCO. The Royal Australian and New Zealand College of Ophthalmologists (RANZCO): Guidelines for the Assessment and Management of Patients with Inherited Retinal Diseases (IRD). Published 6 May 2020. Available online: https://ranzco.edu/home/policies-and-guidelines/ (accessed on 1 August 2024).
- Stone, E.M.; Aldave, A.J.; Drack, A.V.; Maccumber, M.W.; Sheffield, V.C.; Traboulsi, E.; Weleber, R.G. Recommendations for genetic testing of inherited eye diseases: Report of the American Academy of Ophthalmology task force on genetic testing. Ophthalmology 2012, 119, 2408–2410. [Google Scholar] [CrossRef]
- Zhao, P.Y.; Branham, K.; Schlegel, D.; Fahim, A.T.; Jayasundera, K.T. Association of No-Cost Genetic Testing Program Implementation and Patient Characteristics With Access to Genetic Testing for Inherited Retinal Degenerations. JAMA Ophthalmol. 2021, 139, 449–455. [Google Scholar] [CrossRef]
- McClard, C.K.; Pollalis, D.; Jamshidi, F.; Kingsley, R.; Lee, S.Y. Utility of No-Charge Panel Genetic Testing for Inherited Retinal Diseases in a Real-World Clinical Setting. J. Vitreoretin. Dis. 2022, 6, 351–357. [Google Scholar] [CrossRef]
- Bao, Y.K.; Situ, B.A.; Runner, M.; Moshfeghi, A.; Ameri, H. Comparison of The Results of Sponsored Genetic Testing Panels for Inherited Retinal Diseases. J. Clin. Med. 2024, 13, 3118. [Google Scholar] [CrossRef]
- Mustafi, D.; Hisama, F.M.; Huey, J.; Chao, J.R. The Current State of Genetic Testing Platforms for Inherited Retinal Diseases. Ophthalmol. Retin. 2022, 6, 702–710. [Google Scholar] [CrossRef]
- Lidder, A.; Modi, Y.; Dedania, V.S.; Brodie, S.E. DNA testing for inherited retinal disease (IRD): Initial experience with the SPARK/Invitae ‘ID your IRD’ genetic testing panel. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1539. [Google Scholar]
- Mansfield, B.C.; Yerxa, B.R.; Branham, K.H. Implementation of a registry and open access genetic testing program for inherited retinal diseases within a non-profit foundation. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 838–845. [Google Scholar] [CrossRef]
- Sheck, L.H.N.; Esposti, S.D.; Mahroo, O.A.; Arno, G.; Pontikos, N.; Wright, G.; Webster, A.R.; Khan, K.N.; Michaelides, M. Panel-based genetic testing for inherited retinal disease screening 176 genes. Mol. Genet. Genom. Med. 2021, 9, 1663. [Google Scholar] [CrossRef]
- Taylor, R.L.; Parry, N.R.A.; Barton, S.J.; Campbell, C.; Delaney, C.M.; Ellingford, J.M.; Hall, G.; Hardcastle, C.; Morarji, J.; Nichol, E.J.; et al. Panel-Based Clinical Genetic Testing in 85 Children with Inherited Retinal Disease. Ophthalmology 2017, 124, 985–991. [Google Scholar] [CrossRef]
- Carrigan, M.; Duignan, E.; Malone, C.P.G.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-Based Population Next-Generation Sequencing for Inherited Retinal Degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef]
- Shah, M.; Shanks, M.; Packham, E.; Williams, J.; Haysmoore, J.; MacLaren, R.E.; Németh, A.H.; Clouston, P.; Downes, S.M. Next generation sequencing using phenotype-based panels for genetic testing in inherited retinal diseases. Ophthalmic Genet. 2020, 41, 331–337. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Oishi, A.; Müller, P.L.; Herrmann, P.; Holz, F.G.; Mangold, E.; Knapp, M.; Bolz, H.J.; Charbel Issa, P. Genetic testing in patients with retinitis pigmentosa: Features of unsolved cases. Clin. Exp. Ophthalmol. 2019, 47, 779–786. [Google Scholar] [CrossRef]
- Marques, J.P.; Marta, A.; Geada, S.; Carvalho, A.L.; Menéres, P.; Murta, J.; Saraiva, J.; Silva, R. Clinical/Demographic Functional Testing and Multimodal Imaging Differences between Genetically Solved and Unsolved Retinitis Pigmentosa. Ophthalmologica 2022, 245, 134–143. [Google Scholar] [CrossRef]
- Gocuk, S.A.; Jiao, Y.; Britten-Jones, A.C.; Kerr, N.M.; Lim, L.; Skalicky, S.; Stawell, R.; Ayton, L.N.; Mark, H.G. Genetic Testing of Inherited Retinal Disease in Australian Private Tertiary Ophthalmology Practice. Clin. Ophthalmol. 2022, 16, 1127–1138. [Google Scholar] [CrossRef]
- Britten-Jones, A.C.; O’Hare, F.; Edwards, T.L.; Ayton, L.N.; Consortium, T.V.S. Victorian Evolution of Inherited Retinal Diseases Natural History Registry (VENTURE study): Rationale, methodology, and initial participant characteristics. Clin. Exp. Ophthalmol. 2022, 50, 768–780. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef]
- Blueprint Genetics. A Guide to Understanding Variant Classification [White Paper]. Available online: https://blueprintgenetics.com/resources/classify-genetic-variants-interpreting-patients-results/ (accessed on 1 October 2023).
- Sergouniotis, P.I.; Maxime, E.; Leroux, D.; Olry, A.; Thompson, R.; Rath, A.; Robinson, P.N.; Dollfus, H. An ontological foundation for ocular phenotypes and rare eye diseases. Orphanet J. Rare Dis. 2019, 14, 8. [Google Scholar] [CrossRef]
- Kalaw, F.G.P.; Wagner, N.E.; de Oliveira, T.B.; Everett, L.A.; Yang, P.; Pennesi, M.E.; Borooah, S. Using Multimodal Imaging to Refine the Phenotype of PRPH2-associated Retinal Degeneration. Ophthalmol. Retin. 2024, 9, 69–77. [Google Scholar] [CrossRef]
- Jauregui, R.; Chan, L.; Oh, J.K.; Cho, A.; Sparrow, J.R.; Tsang, S.H. Disease asymmetry and hyperautofluorescent ring shape in retinitis pigmentosa patients. Sci. Rep. 2020, 10, 3364. [Google Scholar] [CrossRef]
- Charng, J.; Xiao, D.; Mehdizadeh, M.; Attia, M.S.; Arunachalam, S.; Lamey, T.M.; Thompson, J.A.; McLaren, T.L.; De Roach, J.N.; Mackey, D.A.; et al. Deep learning segmentation of hyperautofluorescent fleck lesions in Stargardt disease. Sci. Rep. 2020, 10, 16491. [Google Scholar] [CrossRef]
- Georgiou, M.; Robson, A.G.; Fujinami, K.; de Guimarães, T.A.C.; Fujinami-Yokokawa, Y.; Daich Varela, M.; Pontikos, N.; Kalitzeos, A.; Mahroo, O.A.; Webster, A.R.; et al. Phenotyping and genotyping inherited retinal diseases: Molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, leber congenital amaurosis, and cone dysfunction syndromes. Prog. Retin. Eye Res. 2024, 100, 101244. [Google Scholar] [CrossRef]
- Hariri, A.H.; Gui, W.; Datoo O’Keefe, G.A.; Ip, M.S.; Sadda, S.R.; Gorin, M.B. Ultra-Widefield Fundus Autofluorescence Imaging of Patients with Retinitis Pigmentosa: A Standardized Grading System in Different Genotypes. Ophthalmol. Retin. 2018, 2, 735–745. [Google Scholar] [CrossRef]
- Australian Bureau of Statistics. Australian Standard Classification of Cultural and Ethnic Groups (ASCCEG). Available online: https://www.abs.gov.au/statistics/classifications/australian-standard-classification-cultural-and-ethnic-groups-ascceg/latest-release (accessed on 1 September 2023).
- Truong, P.; Mack, H.G.; Metha, A.B.; Deen, N.; Hickey, D.G.; Huq, A.; Britten-Jones, A.C.; Ayton, L.N. Forty-year odyssey to Refsum disease diagnosis: Impact of diagnostic delay on effective treatment. Clin. Exp. Optom. 2024, 1–4. [Google Scholar] [CrossRef]
- Goetz, K.E.; Reeves, M.J.; Gagadam, S.; Blain, D.; Bender, C.; Lwin, C.; Naik, A.; Tumminia, S.J.; Hufnagel, R.B. Genetic testing for inherited eye conditions in over 6,000 individuals through the eyeGENE network. Am. J. Med. Genet. Part C: Semin. Med. Genet. 2020, 184, 828–837. [Google Scholar] [CrossRef]
- Ramkumar, H.L.; Gudiseva, H.V.; Kishaba, K.T.; Suk, J.J.; Verma, R.; Tadimeti, K.; Thorson, J.A.; Ayyagari, R. A Report on Molecular Diagnostic Testing for Inherited Retinal Dystrophies by Targeted Genetic Analyses. Genet. Test. Mol. Biomark. 2017, 21, 66–73. [Google Scholar] [CrossRef]
- Clark, M.M.; Stark, Z.; Farnaes, L.; Tan, T.Y.; White, S.M.; Dimmock, D.; Kingsmore, S.F. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 2018, 3, 16. [Google Scholar] [CrossRef]
- Shanks, M.E.; Downes, S.M.; Copley, R.R.; Lise, S.; Broxholme, J.; Hudspith, K.A.; Kwasniewska, A.; Davies, W.I.; Hankins, M.W.; Packham, E.R.; et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur. J. Hum. Genet. 2013, 21, 274–280. [Google Scholar] [CrossRef]
- Pucel, J.; Briere, L.C.; Reuter, C.; Gochyyev, P.; LeBlanc, K. Exome and genome sequencing in a heterogeneous population of patients with rare disease: Identifying predictors of a diagnosis. Genet. Med. 2024, 26, 101115. [Google Scholar] [CrossRef]
- Stanwyck, L.K.; Place, E.M.; Comander, J.; Huckfeldt, R.M.; Sobrin, L. Predictive value of genetic testing for inherited retinal diseases in patients with suspected atypical autoimmune retinopathy. Am. J. Ophthalmol. Case Rep. 2019, 15, 100461. [Google Scholar] [CrossRef]
- Kersten, E.; Geerlings, M.J.; Pauper, M.; Corominas, J.; Bakker, B.; Altay, L.; Fauser, S.; de Jong, E.K.; Hoyng, C.B.; den Hollander, A.I. Genetic screening for macular dystrophies in patients clinically diagnosed with dry age-related macular degeneration. Clin. Genet. 2018, 94, 569–574. [Google Scholar] [CrossRef]
- Lin, S.; Vermeirsch, S.; Pontikos, N.; Martin-Gutierrez, M.P.; Daich Varela, M.; Malka, S.; Schiff, E.; Knight, H.; Wright, G.; Jurkute, N.; et al. Spectrum of Genetic Variants in the Most Common Genes Causing Inherited Retinal Disease in a Large Molecularly Characterized United Kingdom Cohort. Ophthalmol. Retin. 2024, 8, 699–709. [Google Scholar] [CrossRef]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef]
- Schlottmann, P.G.; Luna, J.D.; Labat, N.; Yadarola, M.B.; Bainttein, S.; Esposito, E.; Ibañez, A.; Barbaro, E.I.; Álvarez Mendiara, A.; Picotti, C.P.; et al. Nationwide genetic analysis of more than 600 families with inherited eye diseases in Argentina. NPJ Genom. Med. 2023, 8, 8. [Google Scholar] [CrossRef]
- Ma, D.J.; Lee, H.S.; Kim, K.; Choi, S.; Jang, I.; Cho, S.H.; Yoon, C.K.; Lee, E.K.; Yu, H.G. Whole-exome sequencing in 168 Korean patients with inherited retinal degeneration. BMC Med. Genom. 2021, 14, 74. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Chen, N.; Wang, L.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes 2018, 9, 360. [Google Scholar] [CrossRef]
- Liu, X.; Tao, T.; Zhao, L.; Li, G.; Yang, L. Molecular diagnosis based on comprehensive genetic testing in 800 Chinese families with non-syndromic inherited retinal dystrophies. Clin. Exp. Ophthalmol. 2021, 49, 46–59. [Google Scholar] [CrossRef]
- Suga, A.; Yoshitake, K.; Minematsu, N.; Tsunoda, K.; Fujinami, K.; Miyake, Y.; Kuniyoshi, K.; Hayashi, T.; Mizobuchi, K.; Ueno, S.; et al. Genetic characterization of 1210 Japanese pedigrees with inherited retinal diseases by whole-exome sequencing. Hum. Mutat. 2022, 43, 2251–2264. [Google Scholar] [CrossRef]
- Black, G.C.; MacEwen, C.; Lotery, A.J. The integration of genomics into clinical ophthalmic services in the UK. Eye 2020, 34, 993–996. [Google Scholar] [CrossRef]
- Paudel, N.; Daly, A.; Moran, E.M.; Stratieva, P. The Landscape of Genomic Services for Inherited Retinal Degenerations (IRDs) Across Europe. Clin. Ophthalmol. 2024, 18, 2217–2224. [Google Scholar] [CrossRef]
- Lorenz, B.; Tavares, J.; van den Born, L.I.; Marques, J.P.; Pilotto, E.; Stingl, K.; Charbel Issa, P.; Leroux, D.; Dollfus, H.; Scholl, H.P.N. Current management of Inherited Retinal Degenerations (IRD) patients in Europe. Results of a 2 years follow-up multinational survey by the European Vision Institute Clinical Research Network-EVICR.net. Ophthalmol. Vis. Sci. 2023, 61, 3040. [Google Scholar] [CrossRef]
- Stephenson, K.A.J.; Zhu, J.; Dockery, A.; Whelan, L.; Burke, T.; Turner, J.; O’Byrne, J.J.; Farrar, G.J.; Keegan, D.J. Clinical and Genetic Re-Evaluation of Inherited Retinal Degeneration Pedigrees following Initial Negative Findings on Panel-Based Next Generation Sequencing. Int. J. Mol. Sci. 2022, 23, 995. [Google Scholar] [CrossRef]

| Characteristic (n = 140) | Summary |
|---|---|
| Age at genetic testing, years | 49 ± 19 |
| IQR | 33–66 |
| Sex | |
| Male | 81 (58%) |
| Female | 59 (42%) |
| Age at symptom onset, years | 23 ± 19 |
| IQR | 8–36 |
| Mean disease duration, years | 27 ± 18 |
| IQR | 12–39 |
| Family history of IRD | |
| Parent and/or sibling | 53 (38%) |
| Other relative | 13 (9%) |
| No known family history | 74 (53%) |
| Ethnicity a | |
| North African and Middle Eastern | 10 (7%) |
| North-West European | 11 (8%) |
| Oceanian | 82 (59%) |
| Peoples of The Americas | 1 (1%) |
| South-East Asian | 7 (5%) |
| Southern and Central Asian | 10 (7%) |
| Southern and Eastern European | 11 (8%) |
| Sub-Saharan African | 2 (1%) |
| Mixed ethnicities | 6 (4%) |
| Inherited retinal disease diagnosis | |
| Rod–cone dystrophy/Retinitis pigmentosa | 84 (60%) |
| Macular dystrophy | 19 (14%) |
| Syndromic IRD | 21 (15%) |
| Cone/cone–rod dystrophy | 8 (6%) |
| Other IRD (achromatopsia, congenital stationary night blindness, Leber congenital amaurosis) | 8 (6%) |
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| OR (95% CI) | p-Value | OR (95% CI) | p-Value | |
| Age at symptom onset (<30 years) | 2.57 (1.24, 5.41) | 0.012 * | 3.06 (1.34, 7.18) | 0.009 ** |
| Laboratory (Invitae) | 0.62 (0.31, 1.22) | 0.17 | - | |
| Sex (male) | 1.19 (0.60, 2.35) | 0.62 | - | |
| Ethnicity (European) | 2.57 (1.16, 5.79) | 0.020 * | 2.16 (0.86, 5.53) | 0.10 |
| Positive family history | 2.47 (1.24, 5.06) | 0.011 * | 2.87 (1.27, 6.78) | 0.013 * |
| Time between symptom onset and clinical diagnosis (years) | 0.99 (0.94, 1.03) | 0.54 | - | |
| Time between clinical and genetic diagnosis (years) | 1.00 (0.98, 1.02) | 0.82 | - | |
| Hearing impairment | 0.89 (0.41, 1.95) | 0.76 | - | |
| Potential indicators for phenocopies a | 0.40 (0.15, 1.00) | 0.051 | - | |
| Atypical phenotype b | 0.28 (0.08, 0.85) | 0.03 * | 0.26 (0.07, 0.85) | 0.031 * |
| Disease stage by visual acuity c | ||||
| Early/mild | Ref | - | ||
| Moderate | 2.01 (0.82, 5.26) | 0.14 | - | |
| Advanced | 1.29 (0.56, 3.03) | 0.55 | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Britten-Jones, A.C.; Hickey, D.G.; Edwards, T.L.; Ayton, L.N. Panel-Based Genetic Testing in a Consecutive Series of Individuals with Inherited Retinal Diseases in Australia: Identifying Predictors of a Diagnosis. Genes 2025, 16, 888. https://doi.org/10.3390/genes16080888
Britten-Jones AC, Hickey DG, Edwards TL, Ayton LN. Panel-Based Genetic Testing in a Consecutive Series of Individuals with Inherited Retinal Diseases in Australia: Identifying Predictors of a Diagnosis. Genes. 2025; 16(8):888. https://doi.org/10.3390/genes16080888
Chicago/Turabian StyleBritten-Jones, Alexis Ceecee, Doron G. Hickey, Thomas L. Edwards, and Lauren N. Ayton. 2025. "Panel-Based Genetic Testing in a Consecutive Series of Individuals with Inherited Retinal Diseases in Australia: Identifying Predictors of a Diagnosis" Genes 16, no. 8: 888. https://doi.org/10.3390/genes16080888
APA StyleBritten-Jones, A. C., Hickey, D. G., Edwards, T. L., & Ayton, L. N. (2025). Panel-Based Genetic Testing in a Consecutive Series of Individuals with Inherited Retinal Diseases in Australia: Identifying Predictors of a Diagnosis. Genes, 16(8), 888. https://doi.org/10.3390/genes16080888