
SLC39A14 Is a Potential Therapy Target and Prognostic Biomarker for Acute Myeloid Leukemia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
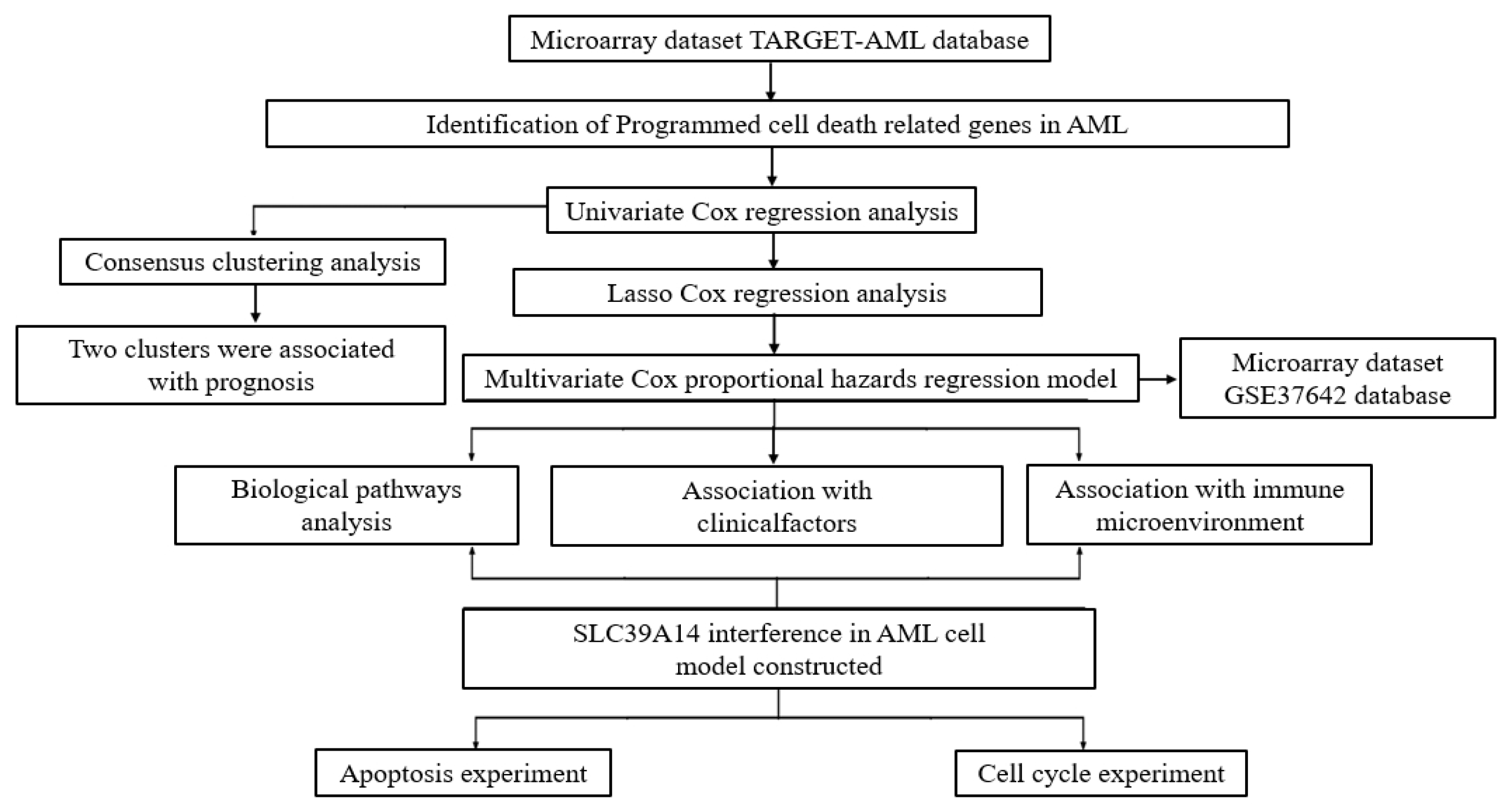
2. Materials and Methods
2.1. AML Dataset Preprocessing
2.2. Identification of PCDRGs with Prognostic Relevance in AML Patients
2.3. Consensus Clustering Analysis of Survival-Related PCDRGs
2.4. Construction and Validation of AML Prognostic Risk Score Model
2.5. Biological Pathway Analysis
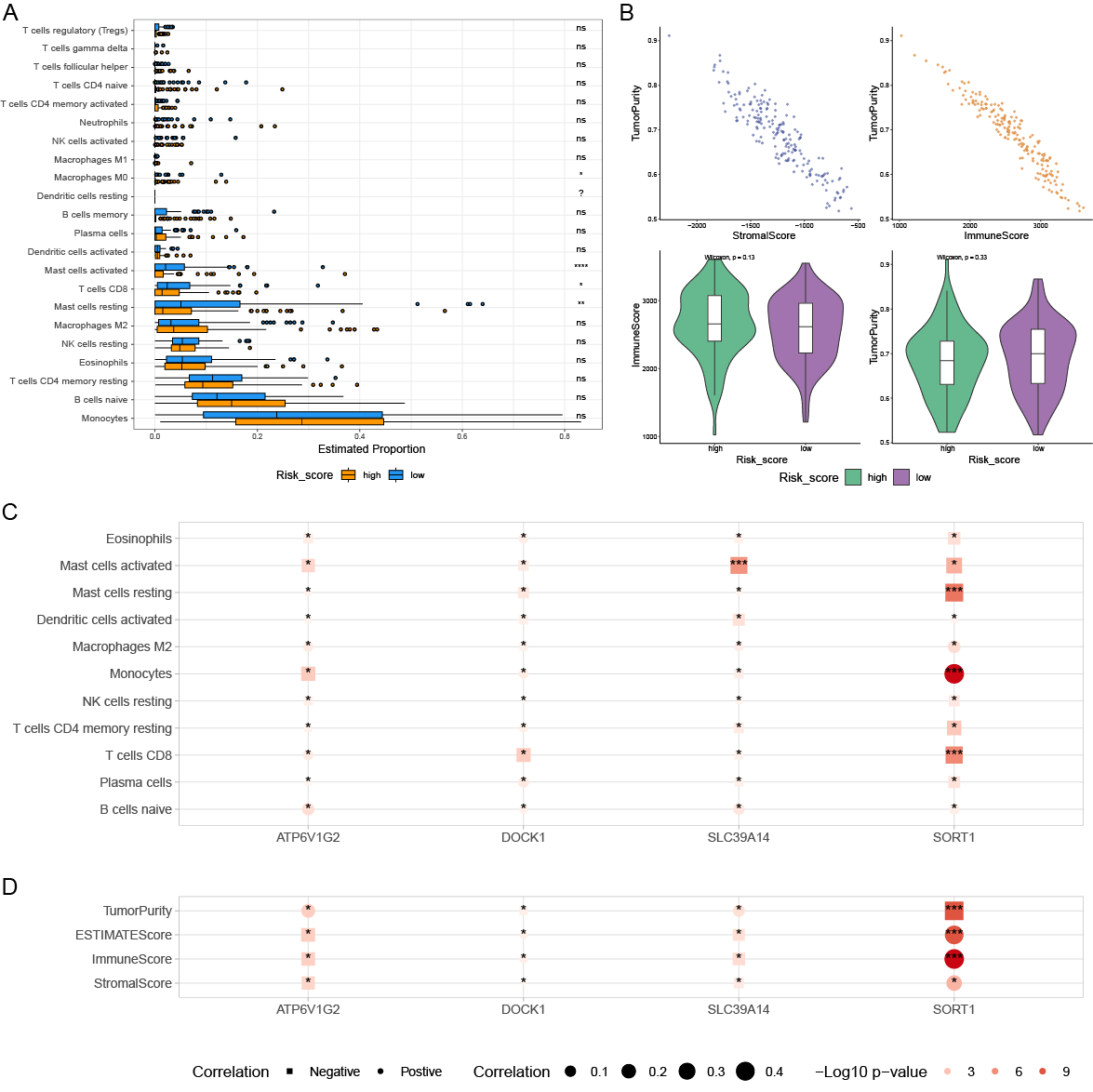
2.6. Analysis of Variations in Tumor Immune Cell Infiltration Across Distinct Risk Groups
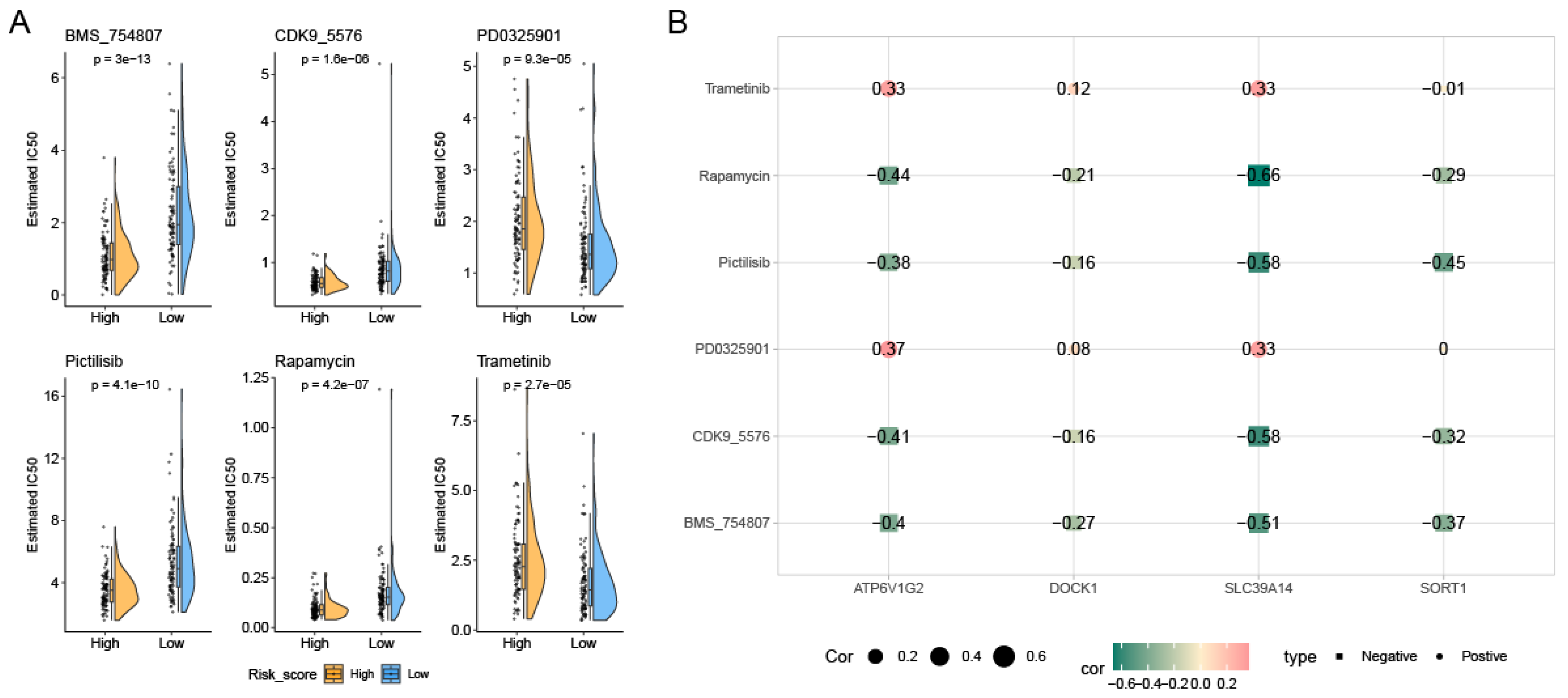
2.7. Estimation of Therapeutic Drugs
2.8. SLC39A14 Interference in Constructed AML Cell Model
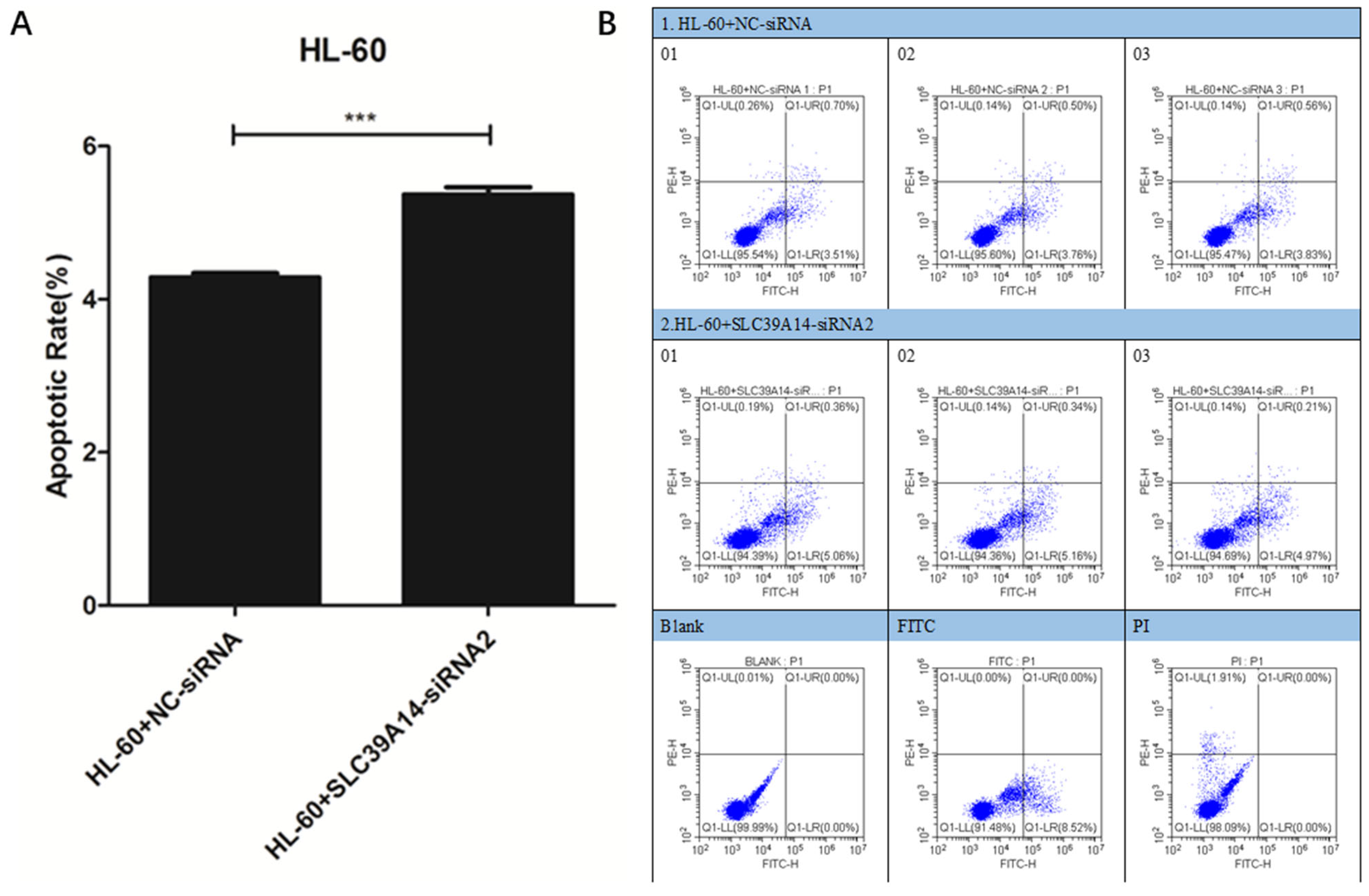
2.9. Apoptosis Experiment
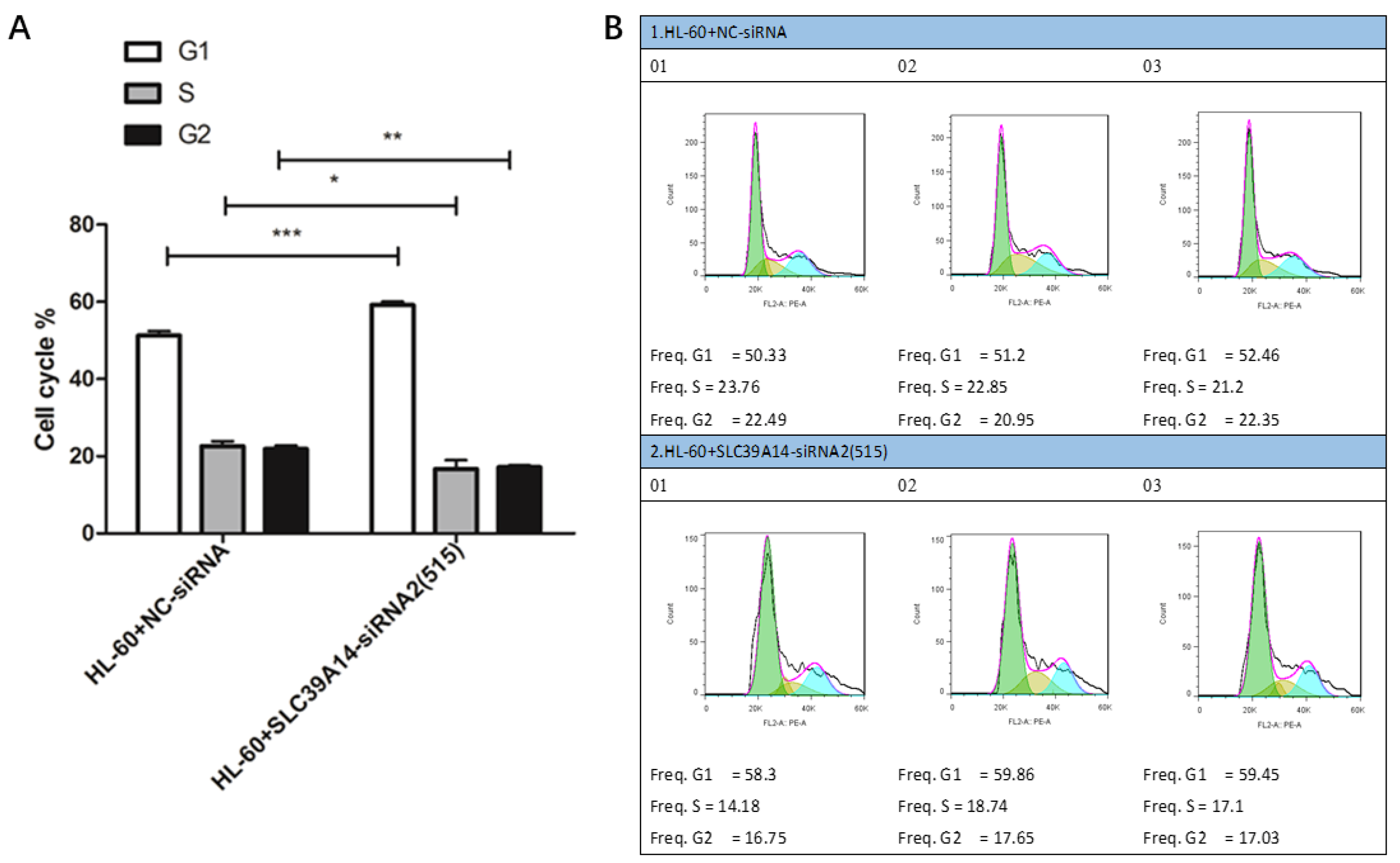
2.10. AML Cell Cycle Experiment
2.11. Statistical Analysis
3. Results
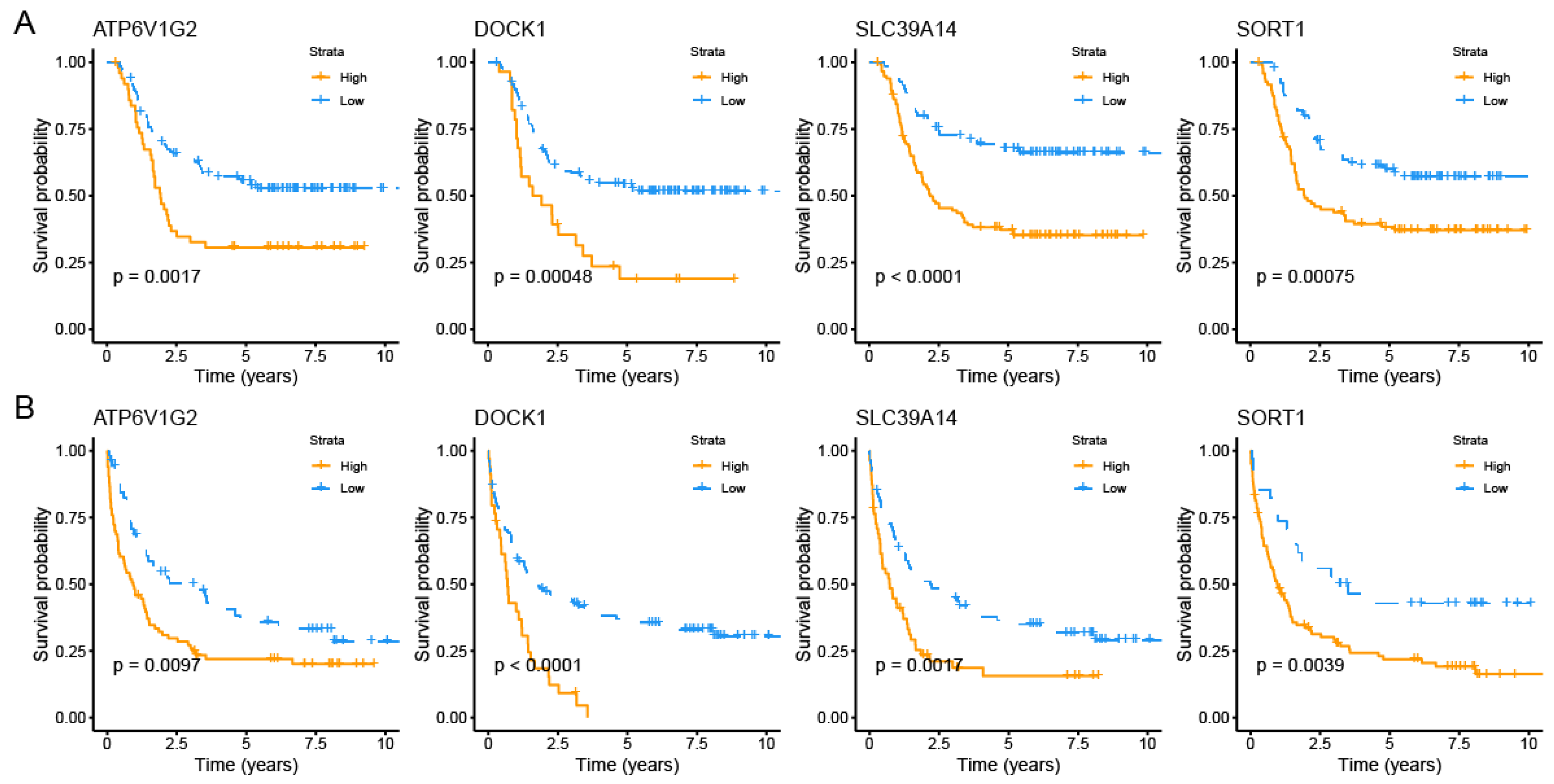
3.1. Identification of PCDRGs with Prognostic Relevance in AML Patients
3.2. Consensus Clustering Analysis Based on Prognostic Significance of PCDRGs
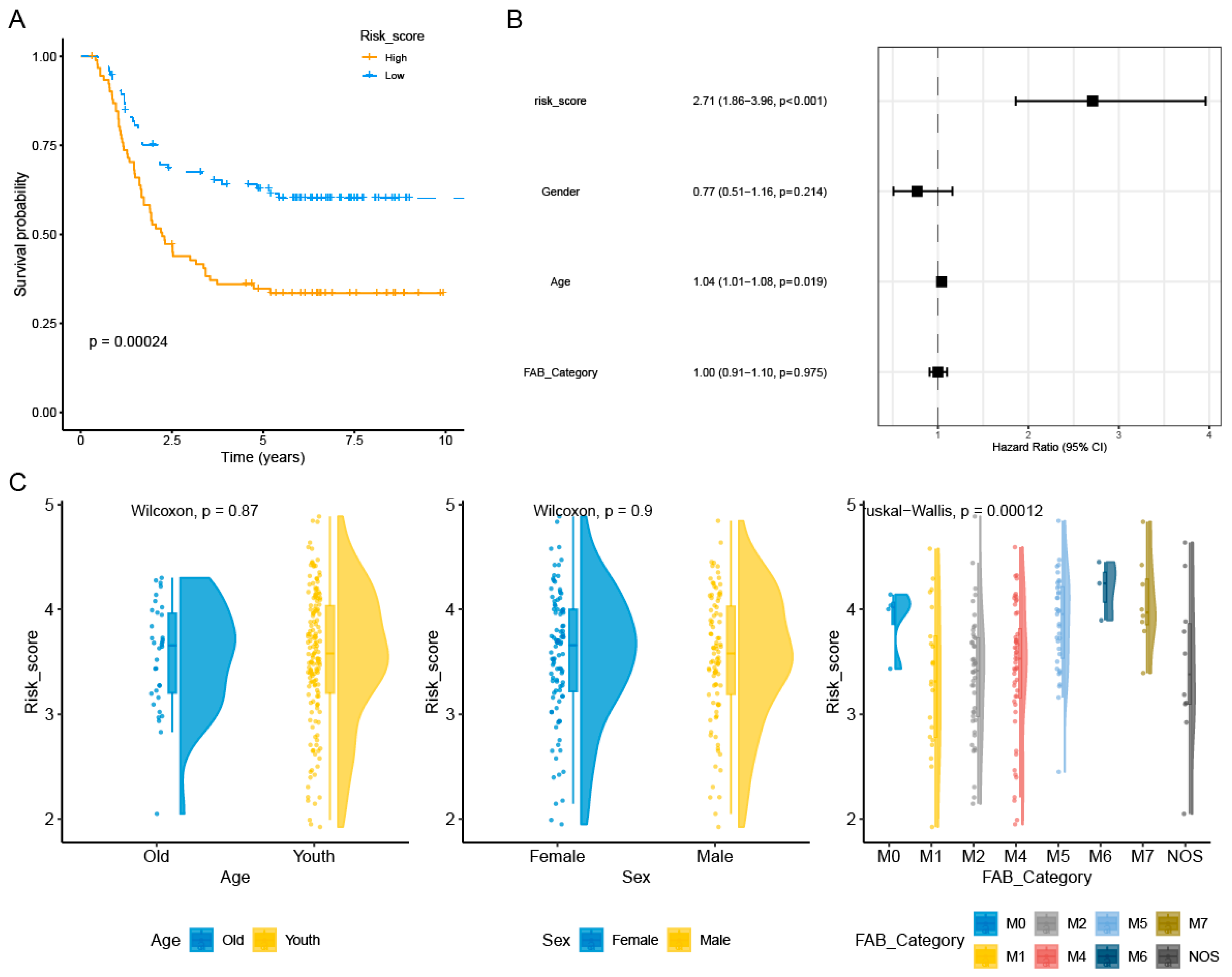
3.3. Construction and Validation of AML Prognostic Risk Score Model
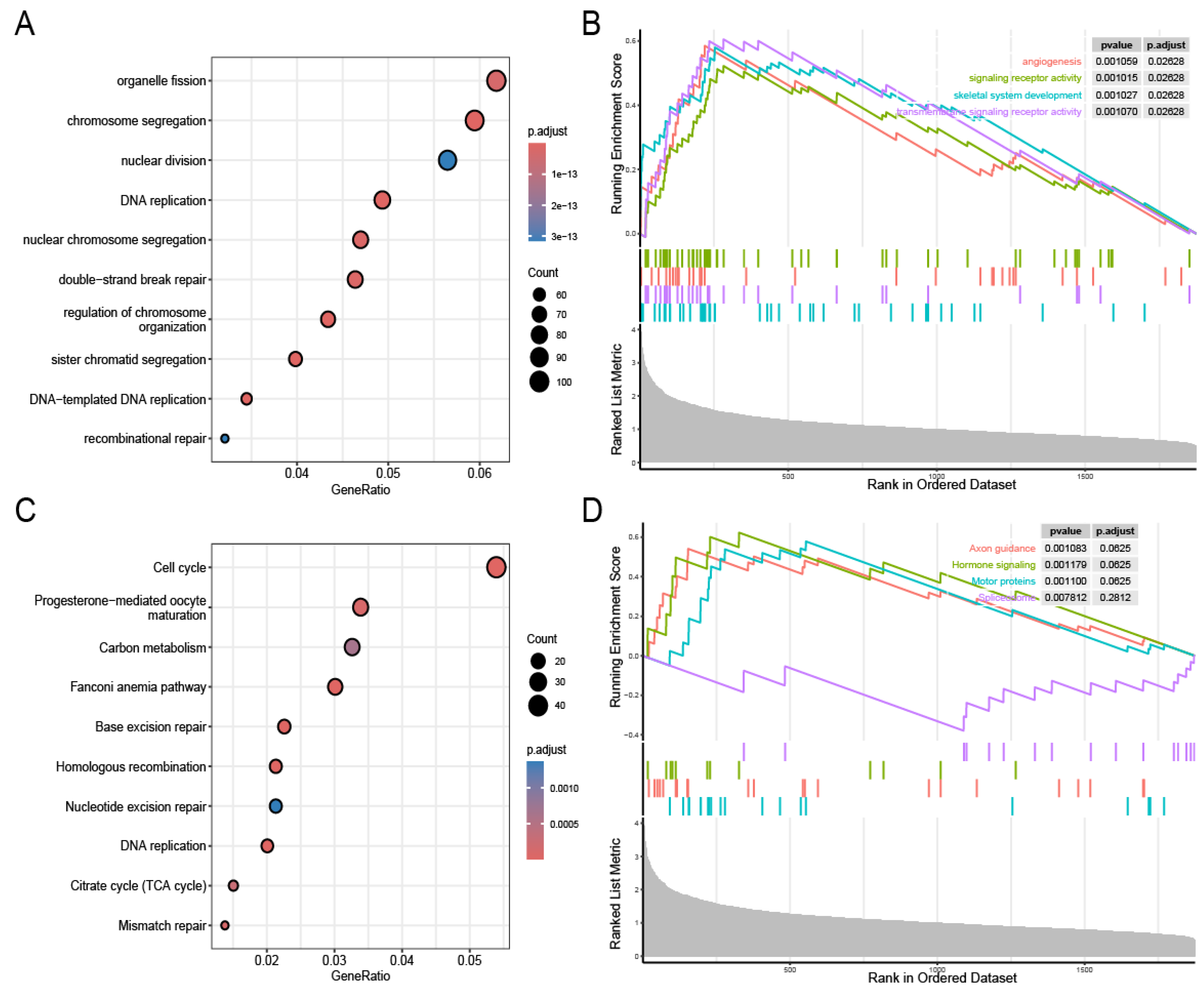
3.4. Biological Pathways
3.5. Variations in Tumor Immune Cell Infiltration Across Distinct Risk Groups and Their Association with Drug Efficacy
3.6. SLC39A14 Interference in Constructed AML Cell Model
3.7. Apoptosis Experiment
3.8. Cell Cycle Experiment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Park, M.; Ghoda, L.Y.; Zhao, D.; Valerio, M.; Nafie, E.; Gonzalez, A.; Ly, K.; Parcutela, B.; Choi, H.; et al. IL1RAP-specific T cell engager depletes acute myeloid leukemia stem cells. J. Hematol. Oncol. 2024, 17, 67. [Google Scholar] [CrossRef]
- Wachter, F.; Pikman, Y. Pathophysiology of Acute Myeloid Leukemia. Acta Haematol. 2024, 147, 229–246. [Google Scholar] [CrossRef]
- Chaudhary, S.; Chaudhary, P.; Ahmad, F.; Arora, N. Acute Myeloid Leukemia and Next-Generation Sequencing Panels for Diagnosis: A Comprehensive Review. J. Pediatr. Hematol. Oncol. 2024, 46, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Zha, C.; Yang, X.; Yang, J.; Zhang, Y.; Huang, R. Immunosuppressive microenvironment in acute myeloid leukemia: Overview, therapeutic targets and corresponding strategies. Ann. Hematol. 2024, 103, 4883–4899. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Zhang, B.; Wu, S.; Zhang, Y.; Xie, J.; Ning, W.; Jiang, H. Prognosis and Characterization of Immune Microenvironment in Acute Myeloid Leukemia Through Identification of an Autophagy-Related Signature. Front. Immunol. 2021, 12, 695865. [Google Scholar] [CrossRef]
- Cao, K.; Zhu, J.; Lu, M.; Zhang, J.; Yang, Y.; Ling, X.; Zhang, L.; Qi, C.; Wei, S.; Zhang, Y.; et al. Analysis of multiple programmed cell death-related prognostic genes and functional validations of necroptosis-associated genes in oesophageal squamous cell carcinoma. EBioMedicine 2024, 99, 104920. [Google Scholar] [CrossRef]
- Wu, X.; Cao, J.; Wan, X.; Du, S. Programmed cell death in hepatocellular carcinoma: Mechanisms and therapeutic prospects. Cell Death Discov. 2024, 10, 356. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, Y.; Zhou, G.; Zhang, Z.; Zhang, P. Leveraging mitochondrial-programmed cell death dynamics to enhance prognostic accuracy and immunotherapy efficacy in lung adenocarcinoma. J. Immunother. Cancer 2024, 12, e010008. [Google Scholar] [CrossRef]
- Qin, H.; Abulaiti, A.; Maimaiti, A.; Abulaiti, Z.; Fan, G.; Aili, Y.; Ji, W.; Wang, Z.; Wang, Y. Integrated machine learning survival framework develops a prognostic model based on inter-crosstalk definition of mitochondrial function and cell death patterns in a large multicenter cohort for lower-grade glioma. J. Transl. Med. 2023, 21, 588. [Google Scholar] [CrossRef]
- Bao, X.; Chen, Y.; Chang, J.; Du, J.; Yang, C.; Wu, Y.; Sha, Y.; Li, M.; Chen, S.; Yang, M.; et al. Machine learning-based bulk RNA analysis reveals a prognostic signature of 13 cell death patterns and potential therapeutic target of SMAD3 in acute myeloid leukemia. BMC Cancer 2025, 25, 273. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, H. A novel mitochondrial function-associated programmed cell death-related prognostic signature for predicting the prognosis of early breast cancer. Front. Genet. 2024, 15, 1406426. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.C.; Xu, Q.T.; Wang, H.B.; Ren, S.Y.; Zhang, Y. A novel prognostic signature related to programmed cell death in osteosarcoma. Front. Immunol. 2024, 15, 1427661. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, X.; Huang, Y.; Chen, C.; Wu, W.; Ma, X.; You, C.; Chen, L.; Huang, J. Prognostic assessment value of immune escape-related genes in patients with acute myeloid leukemia. Leuk. Lymphoma 2025, 66, 72–83. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Shen, X.; Li, L.; Zhang, N.; Wang, X.; Tang, B. A novel angiogenesis-related scoring model predicts prognosis risk and treatment responsiveness in diffuse large B-cell lymphoma. Clin. Exp. Med. 2023, 23, 3781–3797. [Google Scholar] [CrossRef]
- Sheng, L.; Li, T.; Li, Y.; Zhou, M.; Wang, J.; Lai, Y.; Zhang, Y.; Yi, P.; Mu, Q.; Ouyang, G. Prognostic and immunological characterization of diffuse large B-cell lymphoma evaluated by co-stimulatory molecular-related features. Heliyon 2023, 9, e19342. [Google Scholar] [CrossRef]
- Zhou, Z.; Liang, C. Construction of regulatory T cells specific genes predictive models of prostate cancer patients based on machine learning: A computational analysis and in vitro experiments. Discov. Oncol. 2025, 16, 178. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Wu, L.; Feng, Q.; Luo, Z.; Zeng, T.; Luo, J. A necroptosis gene signature predicts prostate cancer recurrence, and is linked to somatic mutation, therapeutic landscape, and immune infiltration. Am. J. Transl. Res. 2023, 15, 2460–2480. [Google Scholar]
- Zhang, R.; Liu, F. Cancer-associated fibroblast-derived gene signatures predict radiotherapeutic survival in prostate cancer patients. J. Transl. Med. 2022, 20, 453. [Google Scholar] [CrossRef]
- Liu, F.; Wang, Y.; Xia, L.; Sun, C.; Li, Y.; Xia, Y. Immunological characterization and prognostic of colon cancer evaluated by angiogenesis-related features: A computational analysis and in vitro experiments. Discov. Oncol. 2025, 16, 101. [Google Scholar] [CrossRef]
- Chen, C.; Li, T.; Li, Y.; Chen, Z.; Shi, P.; Li, Y.; Qian, S. GPX4 is a potential diagnostic and therapeutic biomarker associated with diffuse large B lymphoma cell proliferation and B cell immune infiltration. Heliyon 2024, 10, e24857. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Li, Y.; Zhang, Z.; Xia, L.; Jiang, J.; Chai, Y.; Wang, Z.; Wan, Y.; Li, T.; et al. SLC27A2 is a potential immune biomarker for hematological tumors and significantly regulates the cell cycle progression of diffuse large B-cell lymphoma. BMC Med. Genom. 2024, 17, 105. [Google Scholar] [CrossRef]
- Chen, C.; Li, Y.; Li, Y.; Chen, Z.; Shi, P.; Xie, Y.; Qian, S. SNCA is a potential therapeutic target for COVID-19 infection in diffuse large B-cell lymphoma patients. Apoptosis 2024, 29, 1454–1465. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Fu, J.; Chen, X. A combined immune and exosome-related risk signature as prognostic biomakers in acute myeloid leukemia. Hematology 2024, 29, 2300855. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Chen, X.; Shen, H.J.; Wang, Z.; Cai, X.; Jiang, N.; Hua, H. Comprehensive Mutation Profile in Acute Myeloid Leukemia Patients with RUNX1-RUNX1T1 or CBFB-MYH11 Fusions. Turk. J. Hematol. 2022, 39, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Korsos, V.; Miller, W.H., Jr. How retinoic acid and arsenic transformed acute promyelocytic leukemia therapy. J. Mol. Endocrinol. 2022, 69, T69–T83. [Google Scholar] [CrossRef]
- Dillon, L.W.; Gui, G.; Ravindra, N.; Andrew, G.; Mukherjee, D.; Wong, Z.C.; Huang, Y.; Gerhold, J.; Holman, M.; D’Angelo, J.; et al. Measurable Residual FLT3 Internal Tandem Duplication Before Allogeneic Transplant for Acute Myeloid Leukemia. JAMA Oncol. 2024, 10, 1104–1110. [Google Scholar] [CrossRef]
- Shahzad, M.; Amin, M.K.; Daver, N.G.; Shah, M.V.; Hiwase, D.; Arber, D.A.; Kharfan-Dabaja, M.A.; Badar, T. What have we learned about TP53-mutated acute myeloid leukemia ? Blood Cancer J. 2024, 14, 202. [Google Scholar] [CrossRef]
- Gao, Q.; Shen, K.; Xiao, M. TET2 mutation in acute myeloid leukemia: Biology, clinical significance, and therapeutic insights. Clin. Epigenetics 2024, 16, 155. [Google Scholar] [CrossRef]
- Makkar, H.; Majhi, R.K.; Goel, H.; Gupta, A.K.; Chopra, A.; Tanwar, P.; Seth, R. Acute myeloid leukemia: Novel mutations and their clinical implications. Am. J. Blood Res. 2023, 13, 12–27. [Google Scholar]
- Huang, Z.W.; Zhang, X.N.; Zhang, L.; Liu, L.L.; Zhang, J.W.; Sun, Y.X.; Xu, J.Q.; Liu, Q.; Long, Z.J. STAT5 promotes PD-L1 expression by facilitating histone lactylation to drive immunosuppression in acute myeloid leukemia. Signal Transduct. Target. Ther. 2023, 8, 391. [Google Scholar] [CrossRef]
- Mishra, S.K.; Millman, S.E.; Zhang, L. Metabolism in acute myeloid leukemia: Mechanistic insights and therapeutic targets. Blood 2023, 141, 1119–1135. [Google Scholar] [CrossRef]
- Bolkun, L.; Pienkowski, T.; Sieminska, J.; Godzien, J.; Pietrowska, K.; Kłoczko, J.; Wierzbowska, A.; Moniuszko, M.; Ratajczak, M.; Kretowski, A.; et al. Metabolomic profile of acute myeloid leukaemia parallels of prognosis and response to therapy. Sci. Rep. 2023, 13, 21809. [Google Scholar] [CrossRef]
- Wei, Q.; Jiang, X.; Miao, X.; Zhang, Y.; Chen, F.; Zhang, P. Molecular subtypes of lung adenocarcinoma patients for prognosis and therapeutic response prediction with machine learning on 13 programmed cell death patterns. J. Cancer Res. Clin. Oncol. 2023, 149, 11351–11368. [Google Scholar] [CrossRef] [PubMed]
- Morana, O.; Wood, W.; Gregory, C.D. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 1328. [Google Scholar] [CrossRef] [PubMed]
- Diepstraten, S.T.; Young, S.; La Marca, J.E.; Wang, Z.; Kluck, R.M.; Strasser, A.; Kelly, G.L. Lymphoma cells lacking pro-apoptotic BAX are highly resistant to BH3-mimetics targeting pro-survival MCL-1 but retain sensitivity to conventional DNA-damaging drugs. Cell Death Differ. 2023, 30, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Cindi, Z.; Kawuma, A.N.; Maartens, G.; Bradford, Y.; Venter, F.; Sokhela, S.; Chandiwana, N.; Wasmann, R.E.; Denti, P.; Wiesner, L.; et al. Pharmacogenetics of Dolutegravir Plasma Exposure Among Southern Africans With Human Immunodeficiency Virus. J. Infect. Dis. 2022, 226, 1616–1625. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, X.; Zhu, J.; Lu, R.; Ouyang, Y. Knockdown of SLC39A14 inhibits glioma progression by promoting erastin-induced ferroptosis SLC39A14 knockdown inhibits glioma progression. BMC Cancer 2023, 23, 1120. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Shan, L. SLC39A14 Is a Potential Therapy Target and Prognostic Biomarker for Acute Myeloid Leukemia. Genes 2025, 16, 887. https://doi.org/10.3390/genes16080887
Li Y, Shan L. SLC39A14 Is a Potential Therapy Target and Prognostic Biomarker for Acute Myeloid Leukemia. Genes. 2025; 16(8):887. https://doi.org/10.3390/genes16080887
Chicago/Turabian StyleLi, Yun, and Liming Shan. 2025. "SLC39A14 Is a Potential Therapy Target and Prognostic Biomarker for Acute Myeloid Leukemia" Genes 16, no. 8: 887. https://doi.org/10.3390/genes16080887
APA StyleLi, Y., & Shan, L. (2025). SLC39A14 Is a Potential Therapy Target and Prognostic Biomarker for Acute Myeloid Leukemia. Genes, 16(8), 887. https://doi.org/10.3390/genes16080887